
Abstract
Understanding the mechanisms triggering hepatogenic differentiation of stem/progenitor cells would be useful for studying postnatal liver regeneration and development of liver cell therapies. Many evidences support the involvement of Sox9 transcription factor in liver development. Here, we investigate the possibility of liver mesenchymal stem/progenitor cells to constitutively express Sox9 by using reverse transcription–quantitative polymerase chain reaction, immunocytochemistry, and western blotting. The involvement of Sox9 in hepatogenic differentiation was assessed by following its expression at different steps of the process, evaluating the impact of its altered expression, and analyzing its expression in human liver disease specimen. Liver mesenchymal stem/progenitor cells constitutively express Sox9 at both the mRNA and protein levels. Upon hepatogenic differentiation, Sox9 expression is downregulated mainly in the maturation step after oncostatin M treatment. Induction of Sox9 expression using transforming growth factor beta is accompanied with a decrease of the quality of hepatogenic differentiation. Blunting Sox9 expression using specific ShRNA clearly alters the levels of several hepatic markers, an effect confirmed in HepG2 cells. In human liver disease specimen, Sox9 expression is enhanced at both the mRNA and protein levels compared with healthy donors. The current data demonstrate that Sox9 may play a pivotal role in hepatocyte lineage development, including adult liver mesenchymal stem/progenitor cells. Further studies on the identification of pathways regulated by or regulating Sox9 will certainly gain insight into the molecular networks controlling hepatogenic differentiation.
Introduction
T
TGFβ constitutes a family of secretory polypeptides—including bone morphogenic proteins, Activins and Nodals—that elicits different cellular effects depending on the type and state of the cell [8,9]. By activating several cellular signals and transcription factors, TGFβ regulates the development of many cell lineages and tissues. During liver development, a gradient of TGFβ/activin has been shown to direct hepatoblasts to differentiate toward hepatocytes or biliary cells [10]. In the adult regenerating liver, TGFβ remains inefficient in inducing apoptosis of adult regenerating hepatocytes [11] and is involved in the activation and expansion of progenitor cell compartments [12]. In addition, TGFβ has been shown to promote liver fibrogenesis via an activation of the epithelial–mesenchymal transition process [13,14].
The transcription factor, Sox9, a member of the SRY (sex-determining region on the Y chromosome) family, expresses an high mobility group DNA binding domain and is widely accepted as a master regulator of chondrogenesis [15,16]. Modulation of its expression is involved in the normal development of several other cells and/or organs among which pancreas and intestine [17,18]. Sox9 has also been documented as a proto-oncogene directly involved in tumorigenesis [19 –21]. Recently, Sox9 overexpression has been reported in hepatocellular carcinoma tissue and associated to clinical outcome [22].
Current studies demonstrated that the expression of Sox9 is under the direct control of TGFβ and its downstream factors. In the developing chick limb bud model system, TGFβ induces chondrogenesis after 12 h of exposure, with Sox9 expression being detected after the first 30 min [23]. As chondrogenesis proceeds from the condensation of mesenchymal cells toward commitment to the chondrogenic lineage and subsequent formation of chondrocytes [24], the question arose in the current work is whether Sox9 is involved in the regulation of mesenchymal-derived epithelial hepatic lineage. At the hepatic level, Sox9 expression is absent in both fetal and adult hepatocytes [25]. Nevertheless, it has been recently reported that mature hepatocytes are supplied from Sox9-expressing progenitor cells as demonstrated using Cre-based lineage tracing tools [26]. In the adult liver, Sox9-induced expression, like after TGFβ treatment, occurs when fibrogenic cells are ativated and is principally responsible for the induction of the major fibrotic extracellular matrix, type 1 collagen [25].
We and others demonstrated that mesenchymal stem cells (MSCs) of extra- and intrahepatic origins have the potential to differentiate into hepatocyte-like cells [27 –29], which may potentially increase the pool of hepatocytic cells to be used for liver cell therapy development. Because MSC transplantation has been proposed as an attractive alternative approach to restore liver mass and function, it is important to determine the molecular networks that govern an efficient hepatogenic differentiation both in vitro and in vivo. In the present work, we address the role of Sox9 transcription factor in hepatogenic differentiation in vitro. The recent data suggesting that Sox9 is a biliary marker and is involved in bile duct formation [30] prompt us to investigate its expression during in vitro hepatocytic differentiation of MSCs.
The data of the current article indicate that Sox9 constitutive expression in liver mesenchymal stem/progenitor cells is downregulated after in vitro hepatogenic differentiation. The decrease of Sox9 expression mainly occurs during the late maturation phase. A defective in vitro hepatogenic differentiation of liver mesenchymal stem/progenitor cells is noted when TGFβ was added during the whole differentiation protocol or only at the maturation step. Knock down of Sox9 expression using ShRNA seems to improve the mRNA expression levels of several hepatic markers as demonstrated in early differentiated liver mesenchymal stem/progenitor cells and confirmed in epithelial HepG2 cells. In human liver disease specimen, Sox9 expression is mostly upregulated in hepatocytes as revealed at both the protein and mRNA levels.
Materials and Methods
Chemicals
The following chemicals were used: Ethanol (Merck), Tris[hydroxymethyl]aminomethane (Tris-acetate; Sigma),
Cell isolation and culture
All experiments of the current study were approved by the ethical committees of St-Luc Hospital and the Faculty of Medicine of the Université Catholique de Louvain.
Primary human hepatocytes were obtained from the Hepatocytes and Hepatic Stem Cells Bank of Cliniques Saint-Luc. Hepatocytes were isolated from deceased donors' livers by two-step collagenase perfusion as previously described [31].
Bone marrow-mesenchymal stem cells
Briefly, bone marrow (BM) aspirates were collected into heparinized syringes containing 10% Hank's balanced salt solution (HBSS; Invitrogen). Mononuclear cells were thereafter separated by centrifugation at 400 g for 20 min at 20°C after careful loading of the BM aspirate on Ficoll-Paque™ PLUS (Amersham Biosciences). Cells were washed twice with Dulbecco's-phosphate-buffered saline (D-PBS; Lonza) and seeded at a density of 1.6×106 cells/cm2. Expansion medium consisted of minimum essential medium alpha (Invitrogen) supplemented with 10% fetal calf serum (FCS; PAA Laboratories) and 1% penicillin/streptomycin (PS; Invitrogen). First medium change was performed after 48–72 h and then twice weekly. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. When reaching 70% confluence, cells were detached using 0.05% trypsin-EDTA (Invitrogen) and replated at a density of 5,000 cells/cm2. Obtained bone marrow-mesenchymal stem cells (BM-MSCs) were characterized by their ability to express mesenchymal markers and to differentiate as previously well described [32].
Umbilical cord-mesenchymal stem cells
Umbilical cords (UCs) were obtained from consenting patients delivering full-term infants. Cords are collected in 0.9% NaCl sterile solution, cut into pieces of 3–4 cm, and sections were incised along its length to expose underlying Wharton's Jelly. The recovered scrapped mesenchymal tissue was centrifuged at 250 g for 5 min at room temperature. The pellet was suspended in HBSS with calcium and magnesium (Invitrogen) containing 1 mg/mL collagenase type I (Sigma), 1% PS, and transferred to a 75-cm2 flask with ventilated cap (Greiner Bio-One). After 18–24 h incubation at 37°C and 5% CO2 atmosphere, the homogenate was diluted in D-PBS (Lonza) and centrifuged at 600 g for 15 min. The cell pellet was resuspended in DMEM 1 g/L
Plated cells were cultured in the expansion medium at 37°C 5% CO2 in a fully humidified atmosphere. At 80–90% of confluence, cells were detached using 0.05% trypsin/EDTA solution (Invitrogen) and replated at 8,000 cells/cm2 in the expansion medium. As for BM-MSCs, medium change was performed twice a week. Obtained umbilical cord-mesenchymal stem cells (UCMSCs) were characterized by their ability to express mesenchymal markers and to differentiate as previously well described [27].
Adult-derived human liver stem/progenitor cells
After liver cell isolation and parenchymal fraction primary culture, emerging adult-derived human liver stem/progenitor cells (ADHLSCs) become predominant after the second passage according to our previous documented observations [28]. The purity of the ADHLSC population was confirmed at the morphology, hepato-mesenchymal phenotype (albumin+, alpha smooth muscle actin+), and absence of epithelial markers, such as cytokeratins 7, 8, 18, and 19 as well as CD133 using immunocytochemistry, reverse transcription–polymerase chain reaction (RT-PCR), and flow cytometry.
Hepatogenic differentiation
For differentiation studies, cells were seeded (104 cells/cm2) on six-well plates coated with rat tail collagen type I. The procedure for hepatogenic differentiation was performed in vitro after sequential incubation with growth factors and cytokines as previously described [28,33]. When TGFβ was used, half of the plates were exposed to 10 ng/mL TGFβ (R&D Systems) throughout the differentiation procedure. The cells were microscopically followed at a regular basis, and the medium was replaced every 3 days throughout the differentiation protocol. At the end of the differentiation process, cells were harvested for subsequent analyses.
CYP3A activity
Metabolism of compounds and chemicals is a major function of the liver. Undifferentiated and differentiated ADHLSCs were trypsinized and seeded at a density of 105 cells per well on collagen-coated 48-well plates. CYP3A activity was then analyzed using P450-Glo™ assays according to the manufacturer's instructions as previously described [33]. Briefly, an exogenous substrate luciferin-6′-pentafluoro-benzyl ether (PFBE) was added in the culture medium of undifferentiated and differentiated ADHLSCs. The metabolization of this substrate allows the measurement of luminescence intensity, which proportionally reflected the activity of this protein.
Glucose-6-phosphatase activity assay
The aim of this assay is to evaluate glucose 6-phosphatase activity, the final key enzymatic step for glucose production, a major function of liver cells. For this purpose, cells were washed two times with PBS and incubated for 4 h in 2 mL of Tris-acetate buffer 0.1 M, pH 6.5, containing 2.08 mM glucose-6 phosphate and 2.4 mM nitric lead at 37°C in a humidified atmosphere containing 5% CO2. Glucose-6-phosphatase transformed glucose-6 phosphate into glucose, and the reaction leads to the production of intracellular lead nitrate precipitates. The supernatant was eliminated and cells were rinsed for 10 s in 1 mL of ammonium sulfide 5% to convert lead nitrate into brown-colored lead sulfate. Cells were examined using an HP50 inverted microscope coupled to a DFC camera (Leica), and digital images were acquired using Leica IM50 Image Manager Software.
Quantitative real-time reverse transcription–polymerase chain reaction
First-strand cDNA was synthesized using a superscript II cDNA synthesis kit according to the manufacturer's instructions (Invitrogen) and subsequently diluted with nuclease-free water (Invitrogen) to 10 ng/μL cDNA. PCR amplification mixtures (25 μL) contained 25 ng template cDNA, Master Mix buffer (12.5 μL; Applied Biosystems), and 300 nM forward and reverse primer PCRs were run in duplicate and performed on a StepOnePlus Real-time PCR (Applied Biosystems). The cycling conditions comprised 10 min polymerase activation at 95°C and 40 cycles at 95°C for 15 s and 60°C for 20 s and 72°C for 10 min. Each PCR was followed by a melting curve analysis. Relative quantification was normalized against the house keeping gene GAPDH and/or PPIA. The AB primers and probes used for the current study are listed in Table 1.
Immunocytochemistry
Plated cells were fixed using paraformaldehyde 4%. After washing with PBS, cells were permeabilized for 15 min using 1% Triton X-100 (Sigma) in PBS buffer at room temperature. Endogenous peroxidase was eliminated using methanol containing hydrogen peroxide 0.5% for 1 min. Nonspecific immunostaining was prevented by 1 h incubation in PBS buffer containing 1% bovine serum albumin. The cells were incubated with primary antibodies (Table 2) for 1 h at room temperature. Detection was performed after incubating with peroxidase-labeled polymer and substrate chromogen (Envision-DAB System; Dako). The nuclei were revealed by hematoxylin staining.
Immunoblotting
Cells were lysed in RIPA buffer [20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 0.1% sodium dodeyl sulfate (SDS), 1% deoxycholate sodium, and 0.1% protease inhibitor cocktail; Sigma-Aldrich]. After extraction, protein concentration was estimated by the Bradford method (Biorad Laboratories) with bovine serum albumin as standard. Total protein extracts were subjected to SDS-polyacrylamide gel electrophoresis and electroblotted onto nitrocellulose membranes (Hybond™-ECL). The membranes were blocked with 5% bovine serum albumin and probed overnight at 4°C with primary antibodies (Table 2). Membranes were thereafter incubated with corresponding anti-rabbit (Chemicon; AP132P) and anti-mouse (Dako; P0260) peroxidase-coupled secondary antibodies for 2 h at room temperature. Immunoreactive bands were detected using enhanced chemiluminescence with protein A-horseradish peroxidase and the SuperSignal® chemiluminescent system (Pierce). Band intensities were quantified by scanning films and processing image intensities with the program Image J.
Growth factors/cytokines treatment
After detachment of confluent cultures using 0.05% Trypsin-EDTA, ADHLSCs were suspended in the expansion medium and seeded in six-well plates at a density of 10,000 cells/cm2. Twenty-four hours later, cells were incubated with EGF (20 ng/mL), FGF-2 (10 ng/mL), HGF (20 ng/mL), oncostatin M (OSM) (20 ng/mL), and TGFβ (10 ng/mL) in IMDM serum-free medium for 3 and 7 days. Thereafter, total RNA was recovered for Sox9 mRNA expression analysis.
Virus production
Lentiviral vectors were produced by transient transfection of 293T cells as previously described [34]. Forty-eight hours before transfection, 20×106 293T cells per T175-cm2 flask were seeded in a total of eight flasks in DMEM supplemented with 10% FBS. Calcium phosphate-mediated transfection in the presence of chloroquine was performed on cells that reached 50–70% confluency. Cells were transfected during 4–6 h with a mix of VSVG-expressing envelope plasmid pMD2.G, packaging plasmid pCMVΔR8.74, and pTRIP (containing the SOX9si sequence under control of the inverted EF1α promoter and the reporter Ds-Red2 under control of the CMV-promoter) at a ratio of 1:2:2 (15 μg pMD2.G+30 μg pCMVΔR8.9+30 μg pTRIP per flask). The conditioned media were collected after 48 and after 72 h, pooled, cleared by centrifugation, and filtered through a 0.22-μm pore-size filter. After ultracentrifugation, the pellet was dissolved in PBS and determination of the viral titer was done as previously described [35] by real-time PCR for the minus strong-stop cDNA (U5/R region) that is present within lentiviral capsids.
Transduction procedure
Cells were seeded at 104 cells/cm2 in collagen-coated 75-cm2 flasks using DMEM supplemented with 10% FCS and P/S (100 IU/mL and 100 μg/mL, respectively). Twenty-four hours later, cells were transduced at multiplicity of infection (MOI) 30 in a volume of 7 mL expansion medium per flask. The medium was changed the next day and cells were grown to confluency by changing the media every 2 days. The reporter expression (Ds-Red2) was closely assessed using fluorescence microscopy. The cells were then detached and plated for subsequent analyses.
Immunohistochemistry analyses
Five micrometer liver sections were deparaffinized and rehydrated in graded alcohol series. Endogenous peroxidase activity was blocked by incubation for 30 min in a 3% hydrogen peroxide methanol solution. Antigen retrieval was performed by incubating the sections in citric acid monohydrate solution (pH 6.0) at 97°C for 30 min. Nonspecific immunostaining was prevented by 1 h incubation in PBS buffer containing 5% bovine serum albumin at room temperature. Slices were incubated for 1 h with polyclonal anti-human Sox9 (Millipore; 1/1,000), monoclonal anti-human KRT-19 (Dako; 1/100) in 0.5% bovine serum albumin at 37°C. Staining detection was visualized by Envision Dako anti-rabbit and anti-mouse, respectively (Dako), using diaminobenzidine (Sigma) as chromogenic substrate. The nuclei were counterstained using Mayer's hematoxylin for 10 min and mounted using Neo-Entellan® (Merck) for microscopy analysis.
Statistical analysis
Results are expressed as mean±standard error of the mean. Statistical differences were determined by Student's t-test for two groups' comparison or by one-way analysis of variance followed by the Tukey post hoc test for multiple comparisons between more than two groups. Differences were considered significant when P-values were *P<0.05, **P<0.01, ***P<0.001.
Results
SOX9 is constitutively expressed in human liver mesenchymal stem/progenitor cells
Based on the recent data demonstrating that mature hepatocytes are generated from Sox9-expressing liver progenitor cells, we analyzed the constitutive expression pattern of Sox9 in ADHLSCs obtained from expansion culture conditions. We first evaluated the Sox9 mRNA expression using RT-quantitative polymerase chain reaction (qPCR) and compared its level with those of multipotent MSCs of extra-hepatic origin as well as of isolated mature human hepatocytes. We found that Sox9 mRNA is expressed in ADHLSCs at similar levels than mature human hepatocytes (Fig. 1A). We also observed that Sox9 mRNA levels in both liver cells remained 70% lower than those of extra-hepatic BM-MSCs and UCMSCs (Fig. 1A).
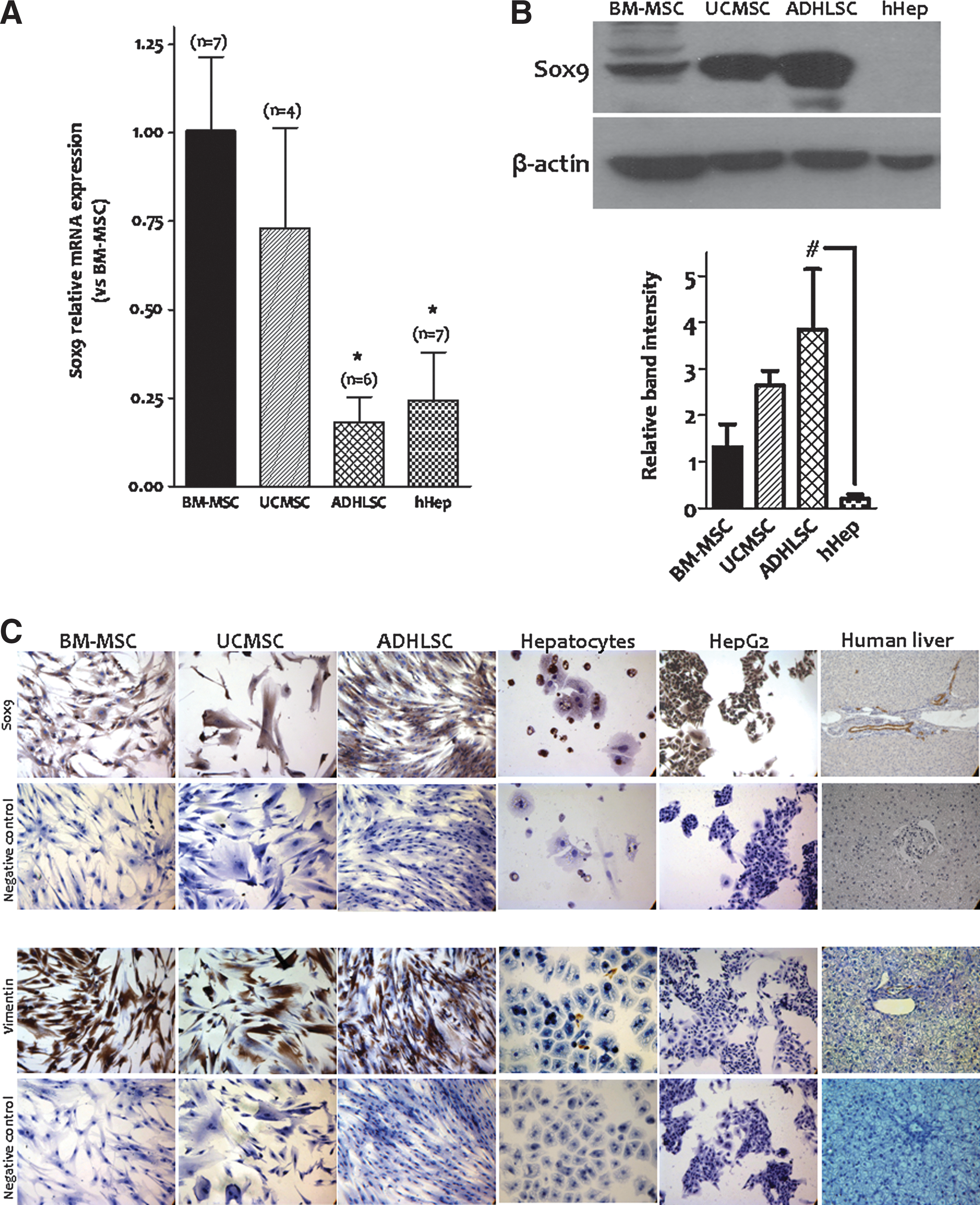
Basal expression of Sox9 in mesenchymal stem/progenitor cells. Cells from expansion cultures were analyzed for basal Sox9 expression.
Analysis of protein expression revealed that Sox9, at the optimized antibody concentration used, is positively expressed in all analyzed mesenchymal stem/progenitor cells (ADHLSCs, BM-MSCs, and UCMSCs). Conversely, no staining was observed in mature hepatocytes either freshly isolated, in primary cultures and in human liver slices (Fig. 1B, C). Furthermore, Sox9 protein level was highly present in ADHLSCs when compared with other MSCs (Fig. 1B). Using immunocytochemistry, we also demonstrated that all the studied MSCs expressed Sox9 at the cytoplasmic level (Fig. 1C). HepG2, a human hepatocellular liver carcinoma cell line, highly expressed this transcription factor at both the cytoplasmic and nuclear levels as for biliary cells (Fig. 1C). Mesenchymal and epithelial shape of the studied cells has been confirmed using vimentin-positive and -negative staining, respectively (Fig. 1C).
SOX9 expression is downregulated in differentiated human liver mesenchymal stem/progenitor cells
To investigate Sox9 expression after in vitro hepatogenic differentiation, ADHLSCs were submitted to four-step hepatogenic differentiation protocol. At the end of the differentiation process, total RNA was extracted from both undifferentiated and differentiated cells to analyze the expression of Sox9. In such samples, we first confirmed the quality of hepatogenic differentiation by demonstrating a significant upregulation of hepatocytic markers gene expression, like albumin, CYP3A4, TDO, TAT, PAH, and Onecut1, and a downregulation of the biliary marker KRT-19 (Fig. 2A). At the protein level, we also demonstrated that hepatogenic differentiation of ADHLSC is accompanied with an augmentation of CYP3A4, KRT-8, and connexin-32 expression concomitantly to a decrease of mesenchymal marker protein levels like for vimentin, alpha smooth muscle actin, and fibronectin (Fig. 2B, C). All these data confirmed the hepatogenic differentiation of ADHLSCs as demonstrated at both the morphological and functional levels (Supplementary Fig. S1; Supplementary Data are available online at
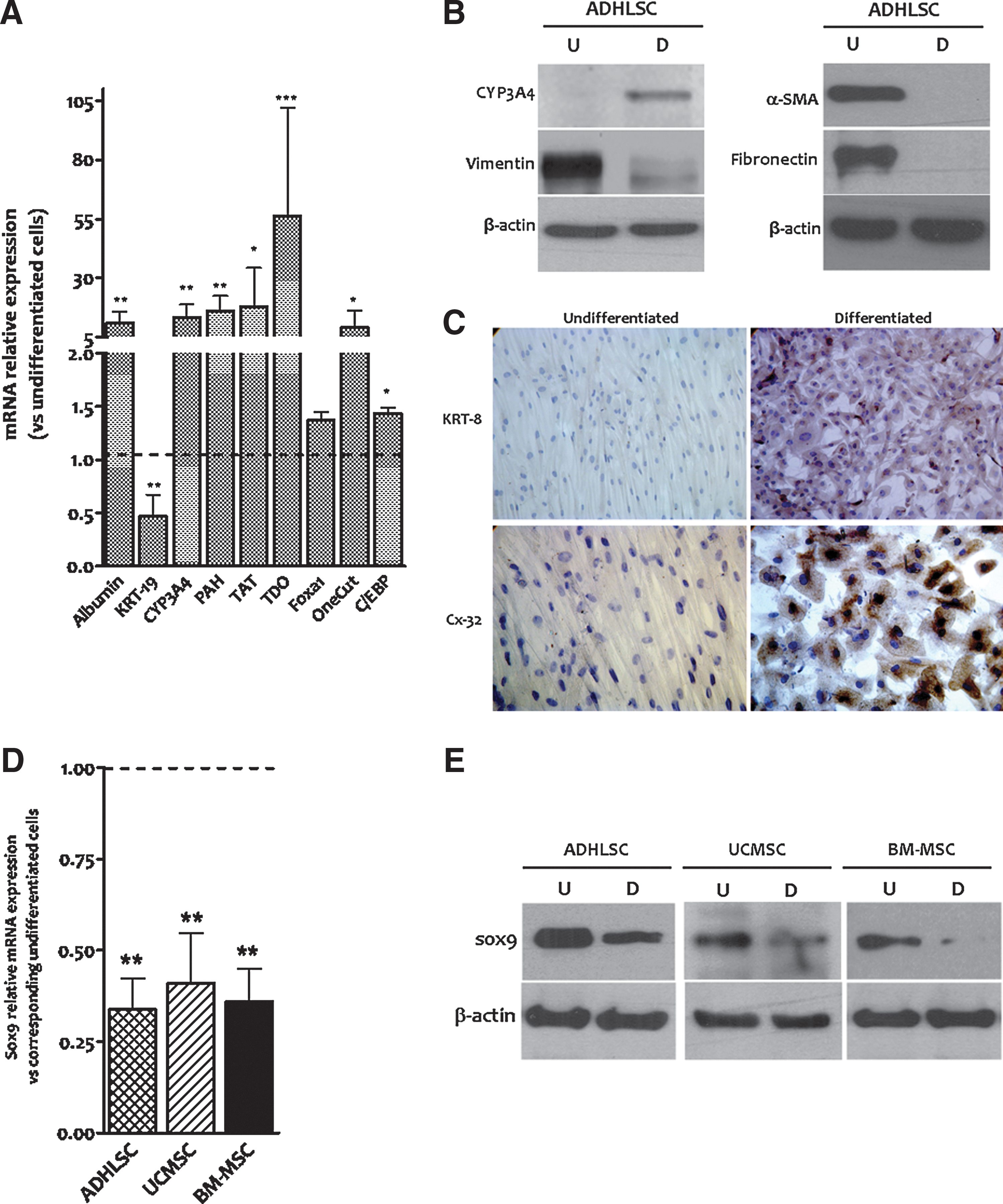
Downregulation of Sox9 mRNA expression after in vitro hepatogenic differentiation of mesenchymal stem/progenitor cells. Mesenchymal stem/progenitor cells were submitted to an in vitro hepatogenic differentiation process and processed for Sox9 expression analysis.
In vitro hepatogenic differentiation protocol we routinely use in our laboratory is based on specific growth factors and cytokines that are sequentially applied in four steps. To narrow down the step at which the decrease of Sox9 expression occurred, total RNA was extracted from undifferentiated and differentiated ADHLSCs recovered at different steps of the hepatogenic differentiation process. RT-qPCR analysis of the samples revealed that Sox9 mRNA expression was first upregulated by ∼4-fold after the first step (Fig. 3A). Sox9 mRNA expression was thereafter diminished by ∼45% after the second step of differentiation and remained as such up to the last step of the differentiation protocol (Fig. 3A). Compared with UCMSCs, the modulation of Sox9 mRNA expression profile in ADHLSCs seemed to be different in these extra-hepatic MSCs. Indeed, strong upregulation of Sox9 mRNA level was demonstrated after the first step of the differentiation protocol and maintained up to the second step (Fig. 2D). The decrease of Sox9 mRNA was only noticed after the last maturation step (Fig. 3B).
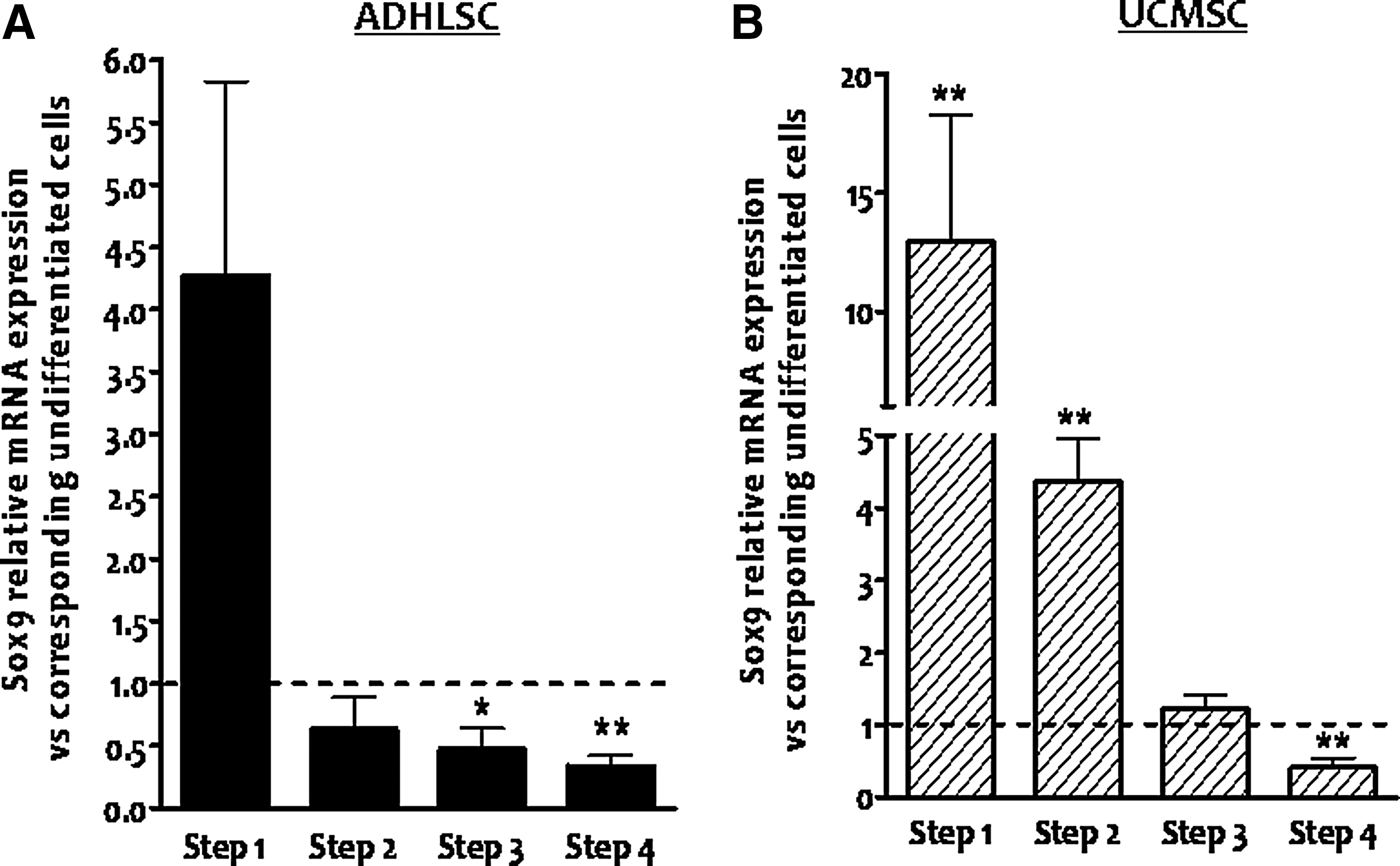
Downregulation of Sox9 mRNA expression occurs at the late maturation step of the in vitro hepatogenic differentiation protocol. The kinetic of Sox9 mRNA expression after in vitro hepatogenic differentiation process in
Therefore, we investigated the identity of the growth factor/cytokine that induces Sox9 mRNA downregulation. To do so, ADHLSCs were treated with each growth factor/cytokine for 3–7 days in serum-free conditions using the same basal medium as for differentiation studies. EGF and FGF-2 confirmed their inducing effect on Sox9 mRNA expression at both times of treatment, whereas HGF did not show any effect (Fig. 4). Only OSM was able to decrease Sox9 mRNA expression from the third day of treatment (Fig. 4). TGFβ was used as a positive control.
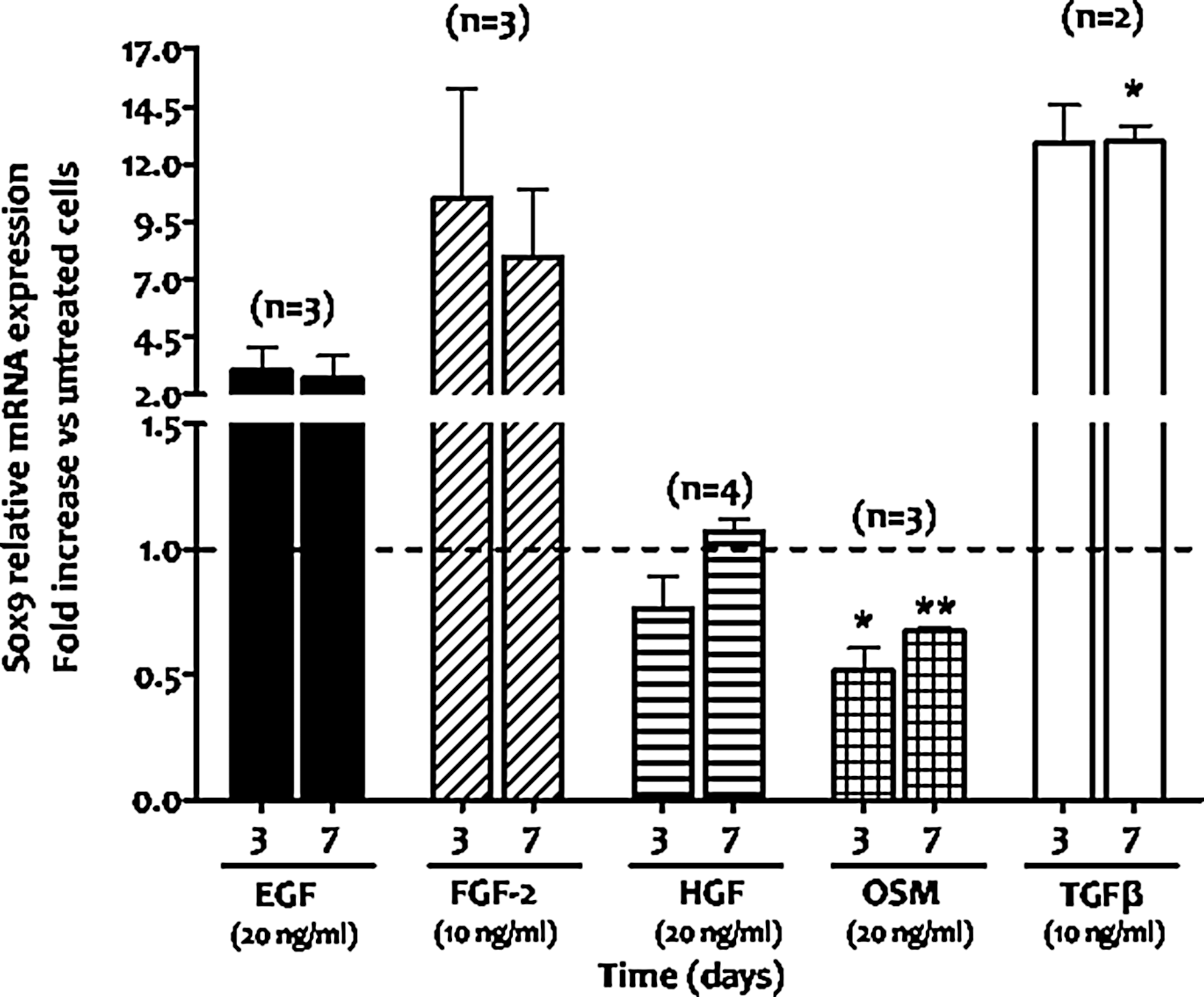
Downregulation of Sox9 expression in ADHLSCs mainly arises during the maturation step of the differentiation protocol under oncostatin M treatment. ADHLSCs were treated with one or the other growth factor/cytokine used in the hepatogenic differentiation protocol. Cells were treated for 3 and 7 days using the same basal medium as for differentiation experiments and under serum-free conditions. Total RNA for each group was extracted and retrotranscribed for Sox9 mRNA analysis using RT-qPCR. Data shown represent the mean±SEM of at least two different experiments as indicated in the graph (*P<0.05; **P<0.01 vs. corresponding untreated cells, unpaired t-test).
TGFβ-induced Sox9 expression is associated with an inhibition of hepatogenic differentiation potential of ADHLSCs
To investigate the involvement of Sox9 expression during hepatogenic differentiation of ADHLSC, we differentiated the cells in presence of TGFβ, a potent well-known inducer of this transcription factor (Fig. 4). We confirmed that this growth factor upregulates Sox9 expression in ADHLSCs when (1) incubated in the presence of 1% serum (undifferentiated cells group) (Supplementary Fig. S2) and (2) applied during the two first steps of the hepatogenic differentiation protocol (incubation with EGF and FGF-2 then with FGF2 and HGF). Indeed, TGFβ strongly maintained the Sox9-induced expression up to sixfold compared with differentiated untreated cells (Supplementary Fig. S2). Thereafter, we evaluated whether maintaining Sox9 expression induced by adding TGFβ is critical for the hepatogenic differentiation process. Hence, ADHLSCs were submitted to the four-step differentiation protocol with or without TGFβ and differentiated cells were evaluated at the morphology and functional levels. As shown in Fig. 5A, TGFβ treatment significantly altered the morphological differentiation of ADHLSCs, which maintained their fibroblastic morphology and failed to adopt polygonal shape compared to cells differentiated without TGFβ. To assess whether these morphological alterations were accompanied with functional hepatogenic differentiation failure, we investigated the ability of ADHLSCs differentiated with and without TGFβ to display active glucose 6-phosphatase (the final key enzyme of glucose synthesis) and CYP3A cytochrome (s).

Transforming growth factor beta (TGFβ) treatment inhibits the ADHLSCs acquired features after hepatogenic differentiation.
Using cytochemistry [33], we demonstrated that when incubated with glucose 6-phosphate substrate, differentiated ADHLSCs (in the absence of TGFβ) presented higher levels of specific positive staining (intracellular brownish precipitates of ammonium phosphate) compared with undifferentiated cells (Fig. 5A). When TGFβ was added to the hepatogenic differentiation cocktail, both undifferentiated and differentiated cells were faintly stained.
Exhibition of phase I drug metabolism activity is an indicator of ADHLSC advanced hepatogenic differentiation. We assessed the effect of TGFβ treatment on CYP3A activity. At the end of the hepatogenic differentiation process, luciferin-PFBE substrate was added in the culture medium to undifferentiated and differentiated ADHLSCs. Once the substrate is metabolized, luminescence proportionally related to the activity of CYP3A can be measured. ADHLSCs differentiated in the presence of TGFβ—same as for undifferentiated cells—were unable to significantly metabolize luciferin-PFBE substrate when compared with cells differentiated in the absence of TGFβ (Fig. 5B). Finally, we investigated at which step the effect of TGFβ in altering the quality of acquired hepatogenic differentiation is more potent. To do so, TGFβ was only added in each single step and then removed. At the end of differentiation, cells from each group were collected for CYP3A activity analysis. As shown in Fig. 5C, increased CYP3A activity acquired after in vitro hepatogenic differentiation was mostly altered when differentiated cells were incubated with TGFβ during the last maturation step.
Blunting of the Sox9 expression alters the expression of hepatocytic markers
To strive whether Sox9 plays critical role in hepatocytic differentiation, ADHLSCs and HepG2 cells were transduced with recombinant lentivirus vector expressing shRNA specifically designed to knock down Sox9. Using dsRed-related fluorescence, we demonstrated that MOI30 was efficient as more than 90% of transduced ADHLSCs were positive. Analysis of Sox9 mRNA expression confirmed that ∼69% decrease is noticed in transduced ADHLSCs compared with control ones (31±4%, **P<0.01 n=3). Thereafter, ADHLSCs were submitted to the first step of the hepatogenic differentiation process and cells were recovered for RNA analysis. As shown in Fig. 6A, stable silencing of Sox9 inhibited its own upregulation by EGF and FGF2 growth factors compared with cells transduced with control construct. We also noticed that upregulated TDO and downregulated KRT-19 expression was more pronounced in ShSox9 transduced ADHLSCs as compared with control cells (Fig. 6A). For the other investigated markers, no modification was remarked after hepatogenic differentiation between both groups of cells.
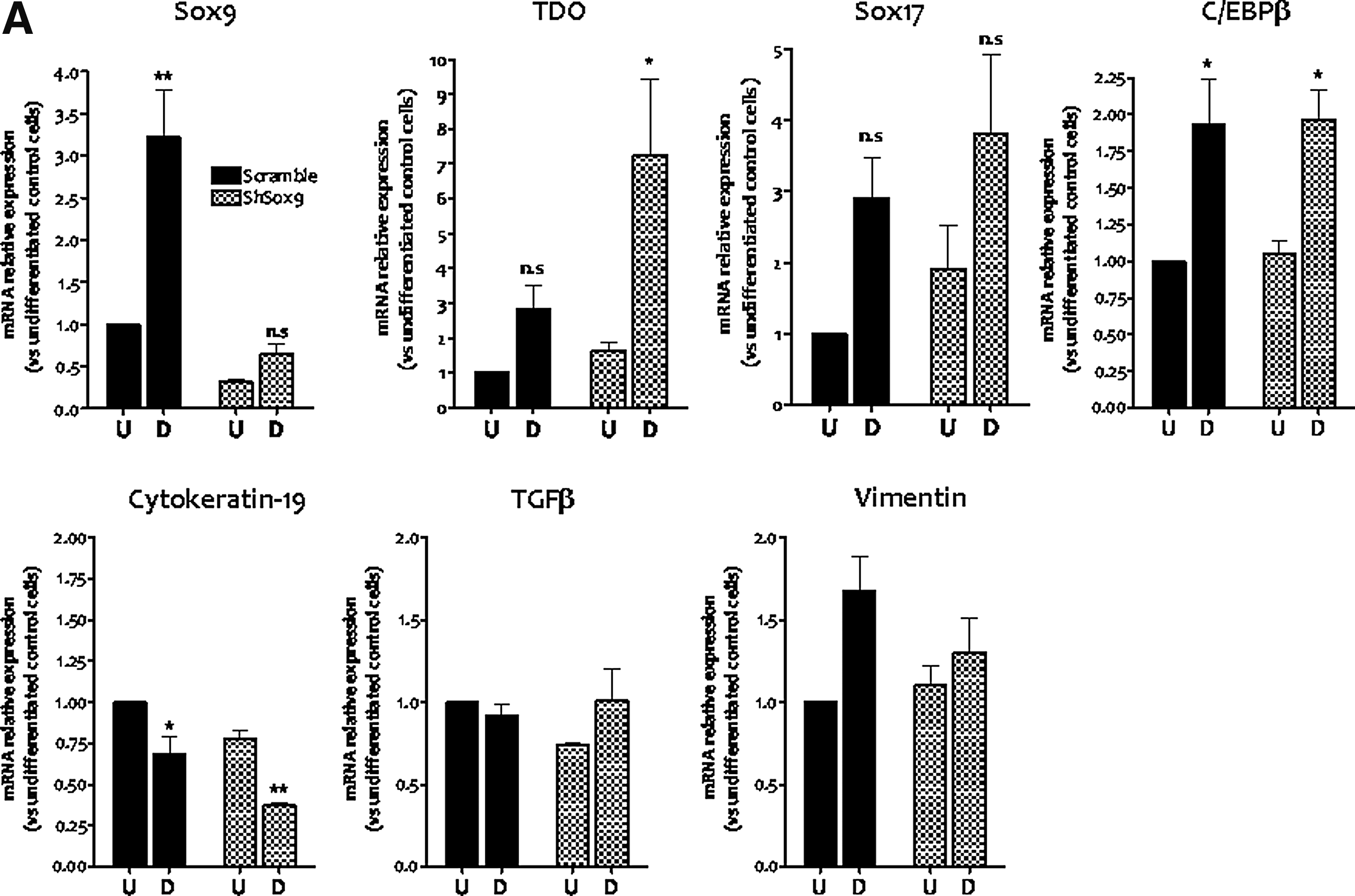
Stable silencing of Sox9 modulates the expression of hepatic markers in both early differentiated liver mesenchymal stem/progenitor and epithelial HepG2 cells.

In parallel, we investigated the impact of knocking down the expression of Sox9 on hepatocytic signature of epithelial HepG2 cells. The expression of shRNA Sox9 in tandem with dsRed allows the isolation of three subclones of transduced HepG2 cells, thanks to their fluorescence intensity, which is proportional to blunting of Sox9 expression efficiency (Supplementary Fig. S3). In the HepG2 cell subclone presenting the highest Sox9 silencing (Supplementary Fig. S4), no modulation of hepatocyte nuclear factor (HNF) transcription factors was seen (Fig. 6B). However, a significant downregulation of CYP1A1 and glucose 6-phosphatase as well as an upregulation of UGT1A, ornithine transcarbamylase (OTC), CYP2D6, and CYP3A4 were detected (Fig. 6C, D).
Sox9 expression is induced in hepatocytes of human diseased livers
Based on the data obtained in early-differentiated ShSox9-transduced ADHLSCs as well as ShSox9-transduced HepG2 cells, it could surmise that in vivo Sox9 expression can be perturbed in hepatocytes of diseased livers. Therefore, we investigated Sox9 expression in samples of liver disease patients using immunohistochemistry. Liver slices from both metabolic (glycogenosis type 1a and OTC deficiency) and structural (biliary atresia) diseases were used. In the healthy human liver, expression of Sox9 is exclusively limited to bile duct cells and no staining was observed in mature hepatocytes as reported above (Fig. 1C).
As shown in Fig. 7A, we demonstrated that Sox9 expression is upregulated in the nuclei of hepatocytes of diseased liver samples. Sox9 immunopositivity is mainly observed in the zone 1 close to periportal area. In biliary atresia (n=2) as well as OTC deficiency samples, Sox9 protein expression is induced in an increasing number of hepatocytes. To confirm that the alteration in Sox9-induced expression is not related to liver progenitor proliferation, we analyzed in serial slices the expression of KRT-19. Our results demonstrated that most Sox9 immunopositive hepatocytes are not stained with KRT-19.
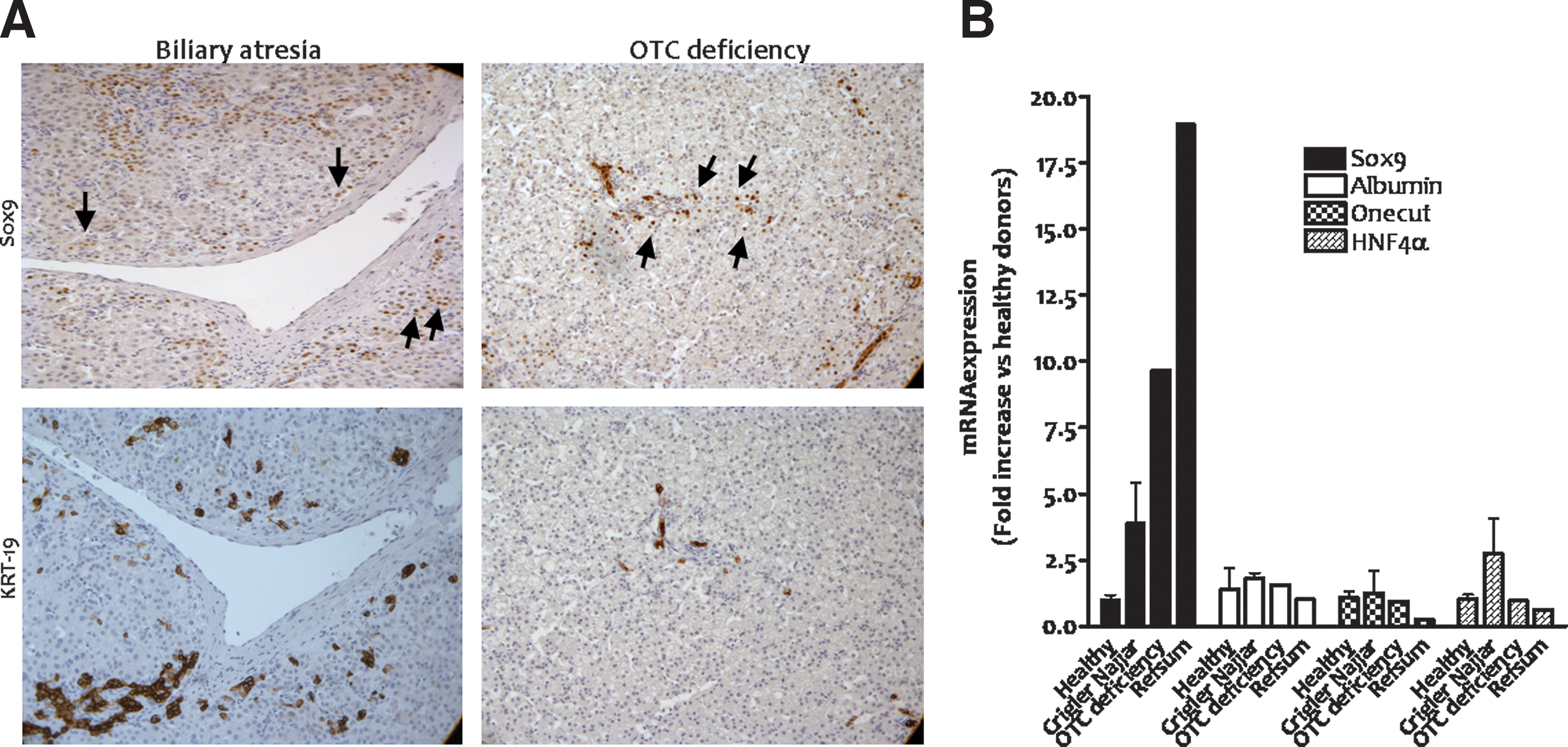
Sox9 expression is enhanced in hepatocytes of diseased human livers.
In parallel, we checked the expression of Sox9 mRNA levels in hepatocytes isolated from metabolic diseased livers [Crigler–Najjar disease (n=2), OTC deficiency (n=1), and Refsum disease (n=1)]. Data revealed that Sox9 expression is significantly induced in all diseased livers compared with healthy ones and other hepatic markers, like albumin and HNF4α (Fig. 7B).
Discussion
In the current study, we demonstrate that hepatogenic differentiation of liver mesenchymal stem/progenitor cells is accompanied with downregulation of Sox9 expression. The induction of Sox9 expression by TGFβ, a factor widely involved in liver development, alters the quality of hepatogenic differentiation of liver mesenchymal progenitor cells. Such data suggest the potential involvement of Sox9 transcription factor in hepatocyte lineage ontogeny.
Hepatoblasts being of endodermal origin give rise to hepatocytes and cholangiocytes, the principal cells of the adult liver [36 –38]. Understanding the molecular networks that control the differentiation of hepatoblasts toward a specific liver cell type is of great interest. Transcription factors have been shown to be significantly involved in all stages of hepatic development. All this knowledge has been investigated and translated to stem/progenitor cells since the clinical use of these cells has been proposed as an attractive alternative approach to restore liver mass and function. Sox9 is highly detected and plays a marked role in embryonic development of many tissues and organs, including chondrocytes, testis, heart, lung, pancreas, nervous system, retina, and hair follicles [15,18,39 –42]. At the hepatic level, Sox9 remained differentially expressed in liver cells. While absent in fetal and adult hepatocytes [25], Sox9 increased expression has been considered to be an earliest sign of biliary differentiation [30].
Basal Sox9 expression in the three studied MSC cell types suggests its implication in maintaining stem/progenitor cells in undifferentiated status as stated by Seymour et al. [18] and supports ADHLSC hepatic progenitor commitment. Sox9 mRNA levels detected in extra-hepatic MSCs are higher than ADHLSCs and mature hepatocytes. Western blotting analysis of Sox9 protein level reveals a great discrepancy with mRNA expression levels. Indeed, ADHLSCs exhibit higher protein level of Sox9 compared with all other MSCs and mature hepatocytes. Poor correlation between transcriptome and translatome is widely accepted in the literature. Such divergence is mainly due to complex and diversity of modulating mechanisms, including inconsistent mRNA and protein turnover rates and/or post-transcriptional regulation of the mRNA [43]. One could stipulate that the mechanism of Sox9 protein passes over the induction of its corresponding mRNA expression, but instead only translates a cellular pool of the mRNA into protein. An important discrepancy of Sox9 protein and mRNA expression has been reported in monolayer cultures of human articular chondrocytes [44]. In our study, ADHLSC monolayer cultures that derive from primary culture of liver parenchymal cell suspensions (in contrary to BM-MSCs and UCMSCs that are directly isolated from their corresponding tissues) may, as for chondrocytes, be responsible for this discrepancy and increased protein levels. Hence, Sox9 may not be transcribed de novo in ADHLSCs, whereas a residual pool of the corresponding mRNA could still be translated.
When ADHLSCs were submitted to an in vitro hepatogenic differentiation protocol, we demonstrated that both mRNA and protein expression of Sox9 was significantly diminished. The same modulation of Sox9 protein and mRNA expression was noticed in extra-hepatic MSCs, which may support Sox9 role in hepatocyte lineage development. This is clearly supported by the absence of detected Sox9 protein expression in mature hepatocytes of both primary cultures and liver slices.
The hepatogenic differentiation protocol we developed in our laboratory is based on sequential incubation with specific growth factors and cytokines [28,33]. Therefore, we investigated if the decrease of Sox9 expression occurred early or late in the hepatogenic differentiation process. Sox9 mRNA expression has been shown to be upregulated after the first step (combined treatment with EGF and FGF-2). If no corroborative data are available in the literature with respect to Sox9 regulation by EGF, FGF2 has been shown to upregulate the expression of this transcription factor as for instance in adipose tissue MSCs [45]. The same Sox9 regulation profile was observed in differentiated UCMSCs. At the end of the second step of differentiation (combined treatment with FGF-2 and HGF), Sox9 expression decreased in ADHLSCs suggesting that the induction effect of FGF-2 is reversed by a potent inhibition of HGF. Such inhibitory effect was not observed in UCMSCs as Sox9 decreased expression was only detected at the last step of the hepatogenic differentiation process. The absence of HGF effect in UCMSCs may be related to its documented role in promoting osteogenic differentiation of multipotent MSCs [46,47] and in modulating chondrocyte differentiation [48,49]. This is in accordance with the involvement of Sox9 factor in modulating mesodermal differentiation of MSCs.
Sox9 expression decreased in ADHLSCs once Oncostatin M was added in the culture medium (steps 3 and 4). Such an effect was in accordance with previous studies showing downregulation of Sox9 expression after interleukin (IL)-6 treatment [50]. To confirm the role of OSM in diminishing Sox9 expression, ADHLSCs were treated separately by each growth factor/cytokine for 3 or 7 days. Compared to the other growth factors tested, only OSM-treated ADHLSCs showed significant decrease of Sox9 expression.
The decrease in Sox9 mRNA and protein level demonstrated after in vitro hepatogenic differentiation of all studied MSCs is in line with previous studies reporting that high Sox9 level enforces proliferation, whereas silencing Sox9 reduces the rate of proliferation [51]. We also observed that Sox9 mRNA level is increased by more than three times in ADHLSCs from expansion culture conditions compared with cells cultured in serum-free conditions (unpublished data).
This suggests that the decrease of Sox9 we observed in differentiated cells may lead to an inhibition of the cell cycle progression from G1 to S phase as previously demonstrated in primary cells [52]. Indeed, Sox9 decreased/silenced expression has been shown to induce upregulation of negative regulators of cell cycle like p21. The expression of p21 has been shown to be upregulated by several growth factors and cytokines involved in liver regeneration and differentiation. It has been reported that p21 is critical mediator of the prodifferentiating and antiapoptotic effects of IL-6 type cytokines including OSM [53].
The recent description of Sox9 as a biliary marker [26,30] let us speculate that alteration of hepatogenic differentiation might be a biliary differentiation process. This latter may the result of an induced Sox9 expression consequently to TGFβ activation. More experiments are required to confirm this hypothesis, but they are beyond the scope of this study. However, we were interested in assessing whether re-establishing Sox9 expression would be associated with altered hepatogenic differentiation. Because TGFβ has been shown to induce Sox9 expression and modulate biliary differentiation, we added TGFβ during the whole differentiation process and noted that it significantly altered the morphological differentiation of ADHLSCs compared with non-TGFβ-treated differentiated cells. When TGFβ signaling is increased in the hepatic parenchyma, a biliary differentiation program is superimposed upon the hepatocytes leading to the development of hybrid hepatobiliary cells [54]. Therefore, we investigated whether TGFβ-induced morphological alteration observed after in vitro hepatogenic differentiation of ADHLSCs was correlated with a nonexhibition of hepatocyte metabolic features. Analysis of glucose-6-phosphatase activity as well as cytochrome P450 activity confirmed that TGFβ treatment altered the in vitro hepatogenic differentiation of ADHLSCs at both the morphological and functional levels. Such alterations remained correlated with an upregulation of Sox9 expression and confirmed the documented negative regulation of TGFβ on hepatocytic lineage [30,55]. Finally, we investigated whether the effect of TGFβ in lowering the quality of ADHLSC hepatogenic differentiation was more efficient in early or late steps of the hepatogenic differentiation protocol. Although the decrease of Sox9 expression was noticed from the second step of the differentiation process, its reversion using TGFβ (in that step) did not alter the functional quality of differentiated ADHLSCs. However, when added at the last maturation step, differentiated ADHLSCs were unable to exhibit CYP3A activity. Such data suggest that the decreased expression of Sox9 in ADHLSCs was more critical when occurring at the maturation step after OSM treatment.
The impact of stable-specific silencing of Sox9 was also evaluated on the quality of hepatogenic differentiation. Among the genes studied in early-differentiated ShSox9-transduced ADHLSCs, significant modulation has been noticed for TDO and KRT-19. This is in accordance with the literature data as genes involved in tryptophan metabolism have been documented to be downstream direct targets of Sox9 [56]. Furthermore, Sox9 and KRT-19 are concomitantly upregulated in the subpopulation of hepatoblasts initiating the expression of biliary-specific genes [1]. No modification was observed for the other investigated early markers. In parallel, we confirmed in the epithelial HepG2 cells the involvement of Sox9 in modulating hepatocytic markers mainly those involved in metabolic activity (OTC, G6Pase, and UGT1A) and xenobiotic detoxification (CYP2D6 and CYP3A4). No data related to Sox9 connections to pathways are available so far. In human liver disease samples, we reciprocally revealed that Sox9 expression is upregulated in hepatocytes at both the protein and mRNA levels as shown in OTC deficiency and biliary atresia. Stained hepatocytes were also seen in glycogenosis type 1a liver but at lesser extent (Data not shown). This suggests the participation of Sox9 in modulating the hepatocytic phenotype as a potential modulator and/or a target of these liver pathways.
In conclusion, this study demonstrates the downregulation of Sox9 expression after in vitro hepatogenic differentiation of liver mesenchymal stem/progenitor cells. Induced expression of this transcription factor using TGFβ is correlated with defective in vitro hepatogenic differentiation of these liver stem/progenitor cells, whereas stable silencing of Sox9 upregulated the expression of some hepatic markers more likely related to metabolic activity. The results from this study points out the potential original role of Sox9 as a marker of hepatogenic differentiation, including adult stem/progenitor cells. Further studies remain mandatory to gain insight into the molecular mechanisms modulated by and/or regulating such transcription factor to direct liver cell differentiation toward a hepatocyte or a cholangiocyte.
Footnotes
Acknowledgments
The current study was financially supported by Fonds Spéciaux de Recherche (FSR) from Université Catholique de Louvain. M.N. is a Principal Investigator of IREC (Institut de Recherche Expérimentale and Clinique).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.