
Abstract
Induced pluripotent stem (iPS) cells are considered as having the greatest potential for use in cell-based therapies. However, at least two hurdles remain: integrating viral transgenes and introducing the c-Myc and Klf4 oncogenes. In a previous study, fibroblasts were incapable of generating iPS cells in the absence of both oncogenes and viral infection. For the present study, we tested our hypothesis that iPS cells can be generated without oncogenes and viral infection under hypoxic conditions and used for cell therapies. By avoiding oncogenic factors and virus integration, this strategy would decrease the potential for cancer formation. According to our observations, the repeated transfection of two expression plasmids (Oct4 and Sox2) into mouse embryonic fibroblasts (MEFs) and combined hypoxic condition resulted in the generation of a novel iPS cell. At 6 h post-transfection, MEFs were subjected to hypoxic conditions (3% O2) for 24 h; this procedure was repeated four times. The resulting MEFs were seeded on feeder cells on day 9; iPS cell clones were observed 12 days post-seeding and designated as iPS-OSH. Data for cell morphology, stem cell marker staining, gene expression profiles, and embryonic body, teratoma, and chimeric mouse formation indicated iPS-OSH pluripotent capability. Neural precursor cells differentiated from iPS-OSH cells were used to treat an ischemic stroke mouse model; results from a behavior analysis indicate that the therapeutic group surpassed the control group. Further, iPS-OSH-derived neural precursor cells differentiated into neurons and astrocytes in mouse stroke brains. In conclusion, we generated a novel iPS-OSH in the absence of viral infection and oncogenic factors and could use it for ischemic stroke therapy.
Introduction
E
First generated in 2006 [5], induced pluripotent stem (iPS) cells represent a novel tool for stem cell research. iPS cells can be generated from human and mice fibroblasts after the introduction of Oct4, Sox2, c-Myc, and Klf4 genes [5,6]. They are similar to ES cells in terms of proliferation, morphology, gene expression, surface antigens, the epigenetic status of pluripotent cell-specific genes, and telomerase activity. The pluripotency of ES and iPS cells gives them exciting potential for tissue repair and replacement [1], but at least two challenges should be overcome: (a) the inefficiency of reprogramming primary cells, which makes it difficult to generate patient-specific iPS cells from a small initial cell population, and (b) the integration of viral and oncogenic transgenes such as c-Myc and Klf4 into the somatic genome [7,8]. c-Myc retrovirus reactivation has been shown to trigger tumor formation in chimeric mice derived from iPS cells [9,10].
Many novel strategies for generating iPS cells have been tried in response to these challenges. In 2008, Okita et al. described their successful generation of mouse iPS cells without viral vectors [11], thus reducing the potential of genomic change. By optimizing the reprogramming method, it is now possible to reprogram both mouse and human somatic cells using a non-virus system. To overcome the low efficiency challenge, Yoshida et al. have shown that hypoxic conditions can increase the efficiency of iPS cell generation [12]. Although Yoshida et al. avoided viral integration via plasmid transfection and increased reprogramming efficiency under hypoxic conditions, they still introduced four factors containing the oncogenes c-Myc and Klf4. For the present study, we used only two factors (Oct4 and Sox2) plus hypoxic conditions to generate novel iPS cells in the absence of oncogenic factors. Our results indicate that the proliferation, morphology, and gene expression of pluripotent cell-specific genes, as well as the ability of iPS cells to differentiate into three layers, were similar to the same characteristics in ES cells. We also used a different strategy than did Yoshida et al. for hypoxic treatment [12]. It is our hope that this method will eventually have utility for clinical applications.
To assess that potential, we differentiated novel iPS cells into neural precursor cells and transplanted them into mouse ischemic stroke brains to test their therapeutic potential, specifically focusing on neural precursor cells derived from the iPS cells. We also investigated whether a neuroplastic effect was achieved via the survival, migration, and differentiation of implanted iPS-OSH-derived neural precursor cells in our laboratory's mouse ischemic stroke model [13].
Materials and Methods
Mouse embryonic fibroblast cell cultures
We isolated primary mouse embryonic fibroblast (MEF) cells from 13.5-day-old C57BL/6 mice embryos. Embryos were retrieved by Cesarean section and internal organs, legs, and heads were removed. Tissues were minced with scissors and transferred to test tubes containing Trypsin for digestion. MEF cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS) (both Gibco BRL), penicillin (100 U/mL), streptomycin (100 μg/mL), non-essential amino acids (0.1 mM), and
Oxygen tension incubation
We used hypoxic conditions (3% O2) to determine the effects of oxygen tension on MEF cells. Cells were added to 10 cm dishes, placed in a hermetic chamber at 37°C and 3% O2, and held for 4, 8, or 24 h. Oxygen tension was maintained by regulating N2 concentration. Other gas concentrations were 5% CO2 and 92% N2 (ThermoForma Tri-Gas Incubator).
Western blot assays
Western blot procedures have been previously described [14]. Antibodies used in this study were mouse anti-Oct4 (Abcam), mouse anti-Sox2 (Chemicon), mouse anti-c-Myc (Chemicon), mouse anti-Klf4 (Abcam), and mouse anti-Actin (Millipore).
Construction of plasmids expressing Oct4 and Sox2
Oct4 and Sox2 cDNA were obtained from commercial clones (Thermo Scientific). Clones containing the Oct4 and Sox2 genes were digested with EcoRI to obtain cDNA fragments, and then ligased into pcDNA 3.1 vectors. The resulting pcDNA-Oct4 and pcDNA-Sox2 plasmids were used in our transfection experiments.
Transfection
pcDNA-Oct4 and pcDNA-Sox2 were purified using a Qiagen Megaprep Kit (Qiagen). MEF cells were co-transfected with DNA from both plasmids using FuGene HD transfection reagent (Roche). Cells transfected with pcDNA 3.1 were used as controls. Cells were analyzed 24 h post-transfection using real-time polymerase chain reaction (PCR) to detect Oct4, Sox2, c-Myc, and Klf4 expression levels.
Real-time PCR and reverse transcription PCR
Total RNA was extracted from MEF cells using TRIzol (Invitrogen) [14], with concentrations determined by spectrophotometry. Complementary DNA was produced from mRNA (5 μg) using a SuperScript III Reverse Transcriptase Kit (Invitrogen). Real-time PCR was used to determine Oct4, Sox2, c-Myc, and Klf4 gene expression levels as previously described [15]. PCR conditions were pre-denaturation at 94°C for 5 min followed by 28 cycles of amplification at 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, followed by a 10-min extension step at 72°C. PCR was performed with ExTaq (Takara) to detect the expression levels of the Ecat1, Eras Nanog, c-Myc, Esg1, Klf4, Oct4, Rex1, Sox2, and β-actin genes. PCR was also used to detect the genomic integration of plasmid DNA in iPS-OSH cells. Primer sequences used to amplify the Ampicillin resistance (Amp(r)), CMV promoter (pCMV), and Oct4 plasmid genes are listed in Table 1.
TaqMan6-mOct4 primers were used for real-time PCR. PCR-Oct3/4 primers were used for RT-PCR analysis, and G-Oct4 primers were used for detecting the integration of plasmid transfection.
PCR, polymerase chain reaction; RT-PCR, reverse transcription PCR.
iPS cells generation
As shown in Figure 2A, we co-transfected pcDNA-Oct4 and pcDNA-Sox2 into MEF cells once every 2 days (four times in total). Cells were transferred to hypoxic chambers 6 h post-transfection and held for 24 h. On day 9, the MEF cells were passaged on feeder cells and observed to detect ES cell-like clones. After seeding, the medium was changed to iPS cell culture medium (DMEM with 15% heat-inactivated FBS; both Gibco BRL), non-essential amino acids (0.1 mM),
Alkaline phosphatase staining and immunofluorescent antibody assays
Alkaline phosphatase staining was performed using a Vector Leukocyte Alkaline Phosphatase kit. For immunofluorescent antibody assays, cultured cells were placed on slides, treated with fixing solution I [4% paraformaldehyde plus 400 mM sucrose in phosphate-buffered saline (PBS)], and held at 37°C for 30 min. Slides were treated with fixing solution II (fixing solution I plus 0.5% Triton X-100) and held at room temperature for 15 min. After washing with PBS, slides were treated with blocking buffer (0.5% BSA in PBS) at room temperature for 1 h and washed thrice with PBS before reacting with different primary antibodies at 1:100 dilutions either overnight at 4°C or for 1 h at 37°C. The antibodies used were anti-Nanog (Novus), anti-Oct4 (Abcam), anti-Sox2 (Chemicon), anti-SSEA1 (Millipore), anti-GFAP (Millipore), anti-NeuN (Millipore), and anti-Tuj-1 (Sigma-Aldrich). Slides were washed five times with cold PBS before reacting with FITC-conjugated anti-mouse IgG or TRITC-conjugated anti-rabbit IgG (Sigma-Aldrich). After four more washes with cold PBS, slides were mounted and observed using a confocal fluorescence microscope (TCS-NT). DNA was stained with Hoechst 33258 fluorochrome (Sigma-Aldrich) to localize nuclei.
Spontaneous in vitro iPS cell differentiation
iPS-OSH cells were harvested by trypsinization and transferred to Ultra-Low attached culture dishes in ES medium without LIF. After 3 days, aggregated cells were plated onto gelatin-coated tissue culture dishes and incubated for another 3 days before staining with anti-α-fetoprotein (Cell Signaling), anti-alpha-smooth muscle actin (Chemicon), or anti-Tuj-1 monoclonal antibodies (GeneTex) plus DAPI.
Teratoma formation and histological analyses
iPS-OSH cells were suspended in iPS cell culture medium (5×107 cells/mL). Nude mice were anesthetized with Zoletil by an IP injection, with 100 μL of cell suspension (5×106 cells) injected subcutaneously into mice dorsal flanks. Tumors were surgically dissected from mice 4 weeks post-injection. Samples were weighed, fixed in PBS containing 4% formaldehyde, and embedded in paraffin. Sections were stained with hematoxylin and eosin to determine cell types.
Chimeric mouse generation
A chimeric mouse experiment was performed to demonstrate the pluripotency of iPS-OSH cells. Briefly, cells were microinjected into albino mouse blastocysts that were transplanted into the uteri of pseudo-pregnant mice to obtain chimeric mice; the chimeric percentages were determined by coat color.
DNA microarrays
Total RNA from ES, iPS-J (Riken Research) [5], iPS-OSH, and MEF cells were labeled with Cy3. Samples were hybridized to Mouse G3 Whole Genome Oligo 8×60K Microarrays (Agilent) according to the manufacturer's protocols. Arrays were scanned with a Microarray Scanner System, and data were analyzed using GeneSpring GX software (both from Agilent). All data are MIAME compliant; new raw data have been entered into the GEO system. Accession numbers are iPS-OSH (GSM789276), iPS-J (GSM789277), ESC (GSM789278), and MEF (GSM789279).
Neural precursor cell differentiation from iPS-OSH cells
All three-step differentiation procedures were performed according to standard protocols previously described [16,17]. Briefly, iPS-OSH cells were transferred to Ultra-Low attached culture dishes in ES medium without LIF to generate embryoid bodies. After 4 days, the embryoid bodies were transferred to normal dishes containing medium with an ITS media supplement for neural precursor cell selection. After 6 days, cells were transferred to poly-
Electrophysiology
Neural precursor cells were cultured in N2-containing medium without bFGF for 10 days to differentiate them into mature neurons, with individual neurons selected for recording based on morphological features. Whole-cell recording was performed in voltage-clamp mode using an Axopatch 200B (Molecular Devices). Whole-cell currents were recorded at a holding potential of −60 mV. Signals were filtered at 2 kHz and digitized at 10 kHz (Digidata 1440A). Recording pipettes (3–5 Mω) were filled with intracellular solution containing 140 mM CsCl, 10 mM HEPES, 4 mM Mg-ATP, and 0.5 mM BAPTA (pH 7.20, 290–295 mOsm). Cover slips were continuously superfused with extracellular solution containing 140 mM NaCl, 5.4 mM KCl, 10 mM HEPES, 1.0 mM MgCl2, 1.3 mM CaCl2, and 20 mM glucose (pH 7.4, 305–315 mOsm). To evoke glutamate receptor-mediated currents, we used fast perfusion of 1 mM glutamate with a computer-controlled multibarrel fast perfusion system (Warner Instruments). All experiments were performed at 23°C–25°C.
Mouse brain ischemia/reperfusion model
The adult male C57BL/6 mice used in this study were subjected to two-vessel ligation after being anesthetized by IP injections of Zoletil. Ligation of right middle cerebral arteries (MCAs) and right common carotids (CCAs) was performed using methods previously described [13]. Right CCAs were clamped with non-traumatic arterial clips, and right MCAs were ligated using 10-0 nylon sutures. Next, iPS-OSH-derived neural precursor cells (1×106) labeled with bisbenzimide were transplanted into three cortical areas that were adjacent to the right MCA, 3.0–3.5 mm below the dura. After 120 min of ischemia, the MCA sutures and CCA arterial clips were removed to enable reperfusion. Behavioral assessments were conducted at 7, 14, and 21 days post-cell transplantation. Mice were sacrificed on day 21, and brains were removed and prepared for immunofluorescent staining.
Neurological behavior measures
Our behavioral tests focused on locomotor activity, beam walking, and rotarod analyses. Behavioral assessments were performed 1 day before cerebral ischemia and at 7, 14, and 21 days post-cell transplantation. Locomotor activity was measured using a versamix animal activity monitor (Accuscan Instruments). We measured total distance (cm) and movement time (s) over 30 min to compare activity in the iPS-OSH-derived neural precursor cell-treated mice and a vehicle control group. For the beam walking analysis, we examined the ability of animals to remain upright and to walk along an elevated and narrow beam. Rotarod analysis consists of mice that are trained to walk on a rotarod moving at a speed of 5.0 rpm (Panlab/Harvard Apparatus). Training was considered complete when mice stayed on the rotarod for 180 s. Rotarod speed was then increased from 5 to 20 rpm over 2 min.
Statistical analyses
Student's t-tests were used to evaluate mean differences between control and treatment groups. Data lacking normal distribution were analyzed by one-way ANOVAs; statistical significance was established as P<0.05.
Results
Oct4, Sox2, Klf4, and c-Myc gene expression levels in MEF cells under hypoxic conditions
To determine whether hypoxic conditions increased Oct4, Sox2, c-Myc, or Klf4 expression, we performed western blot analyses of hypoxic condition-treated MEFs. As shown in Figure 1A, Oct4, Sox2, c-Myc, and Klf4 up-regulation was confirmed at the protein level after hypoxic treatment (3% O2). Gene expression levels were highest at 8 h post-treatment.
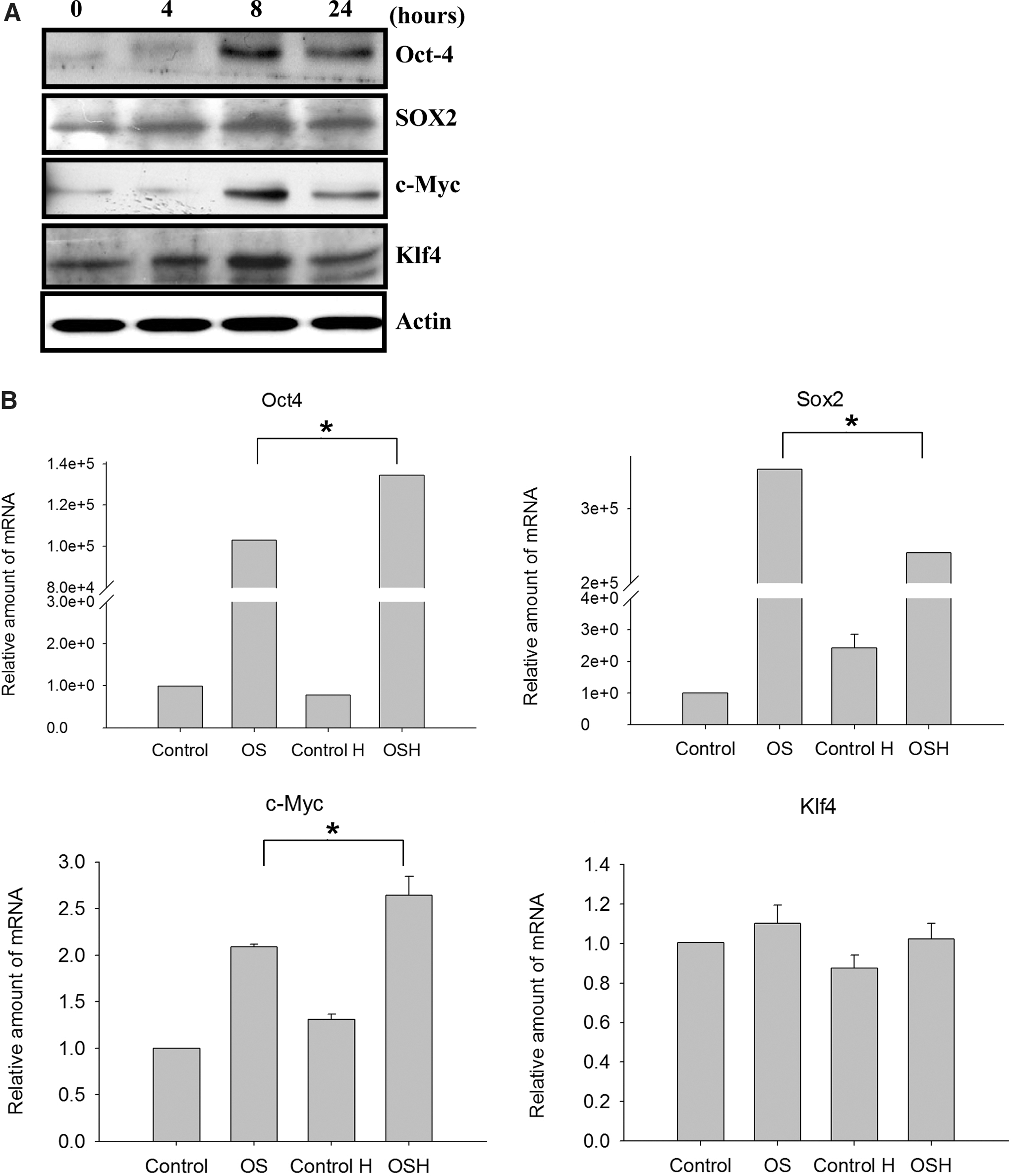
The expression levels of Oct4, Sox2, c-Myc, and Klf4 in mouse embryonic fibroblast (MEF) treated with 3% hypoxic condition.
Real-time PCR was used to detect the gene expression profiles of Oct4, Sox2, c-Myc, and Klf4 genes after pcDNA-Oct4 and pcDNA-Sox2 transfection under hypoxic conditions. The data indicate Oct4 and Sox2 over-expression in the transfected MEF cells (Fig. 1B). Oct4 and c-Myc gene expression levels in the hypoxic-treated transfected MEF cells were higher than in the oxygenic controls (Fig. 1B). However, Sox2 expression levels in the hypoxic-treated transfected cells were not higher than in the oxygenic controls. Finally, no differences in Klf4 gene expression were noted across the four cell groups (Fig. 1B).
iPS cell generation by Oct4 and Sox2 under hypoxic conditions
Our strategy is outlined in Figure 2A. We observed an average of 2.5 clones with ES cell-like morphologies at 12 days after seeding feeder cells from the original 1×105 MEF cells (21 days post-initial transfection) in three independent experiments. After passaging these clones and seeding the new feeder layers, we observed that the morphologies of the iPS-OSH cells were similar to those of the ES cells (Fig. 2B). iPS-OSH cells (including the original clones and passaged cells) tested positive for alkaline phosphatase staining (Fig. 2C).

The generation strategy, morphology, and stem cell marker staining of induced pluripotent stem (iPS)-OSH cells.
Stem cell marker staining
We used immunofluorescent staining to determine stem cell marker expression in iPS-OSH cells. iPS-OSH cells were stained with mouse antibodies against Nanog, Oct4, Sox2, and SSEA1. As shown in Figure 2D, the cells tested positive for all four markers. High-magnification images of immunofluorescence-stained Nanog, Oct4, and Sox2 showed that these three markers were expressed in a nuclear form (Fig. 2E). Nanog, Oct4, and Sox2 expression was co-localized with DAPI.
Stem cell-specific gene expression levels and karyotyping in iPS-OSH cells
Reverse transcription PCR was used to determine ES marker gene expression in iPS-OSH, iPS-J, MEF, and ES cells. Ecat1, Eras, Nanog, c-Myc, Esg1, Klf4, Oct4, Rex1, and Sox2 genes were detected. As shown in Figure 3A, the Ecat1, Eras, Nanog, Oct4, Rex1, and Sox2 gene expression profiles in iPS-OSH cells were equivalent to or higher than those in the ES and iPS-J cells. Chromosomal G-band analysis results indicate that the iPS-OSH cells had normal karyotypes (Fig. 3B).

The stem cell-related gene expression levels and karyotype in iPS-OSH cells.
In vitro and in vivo iPS-OSH cell pluripotency
We used embryoid body formation (in vitro) and teratoma formation (in vivo) to determine iPS-OSH cell pluripotency. In vitro experimental results indicated that iPS-OSH cells formed embryoid bodies in low-attached plates. We observed differentiation initiated by the iPS-OSH cells after placing the embryoid bodies in cell culture plates. Next, we used immunofluorescent staining to detect cells that were positive for α-fetoprotein (an endoderm marker), α-smooth muscle actin (a mesoderm marker), and Tuj-1 (an ectoderm marker). As shown in Figure 4A, we observed iPS-OSH cell differentiation in all three germ layer cells, including hepatocyte (α-fetoprotein), smooth muscle (α-smooth muscle actin), and neuronal cells (Tuj-1). For the in vivo experiments, we injected iPS-OSH cells into the dorsa of nude mice to observe teratoma formation. As shown in Figure 4B, the cells differentiated into all three types of germ layer cells, including respiratory epithelial, gut epithelial, muscle, adipose, epidermal, and neural tissues. Combined, these data indicate iPS-OSH cell pluripotency in vitro and in vivo.
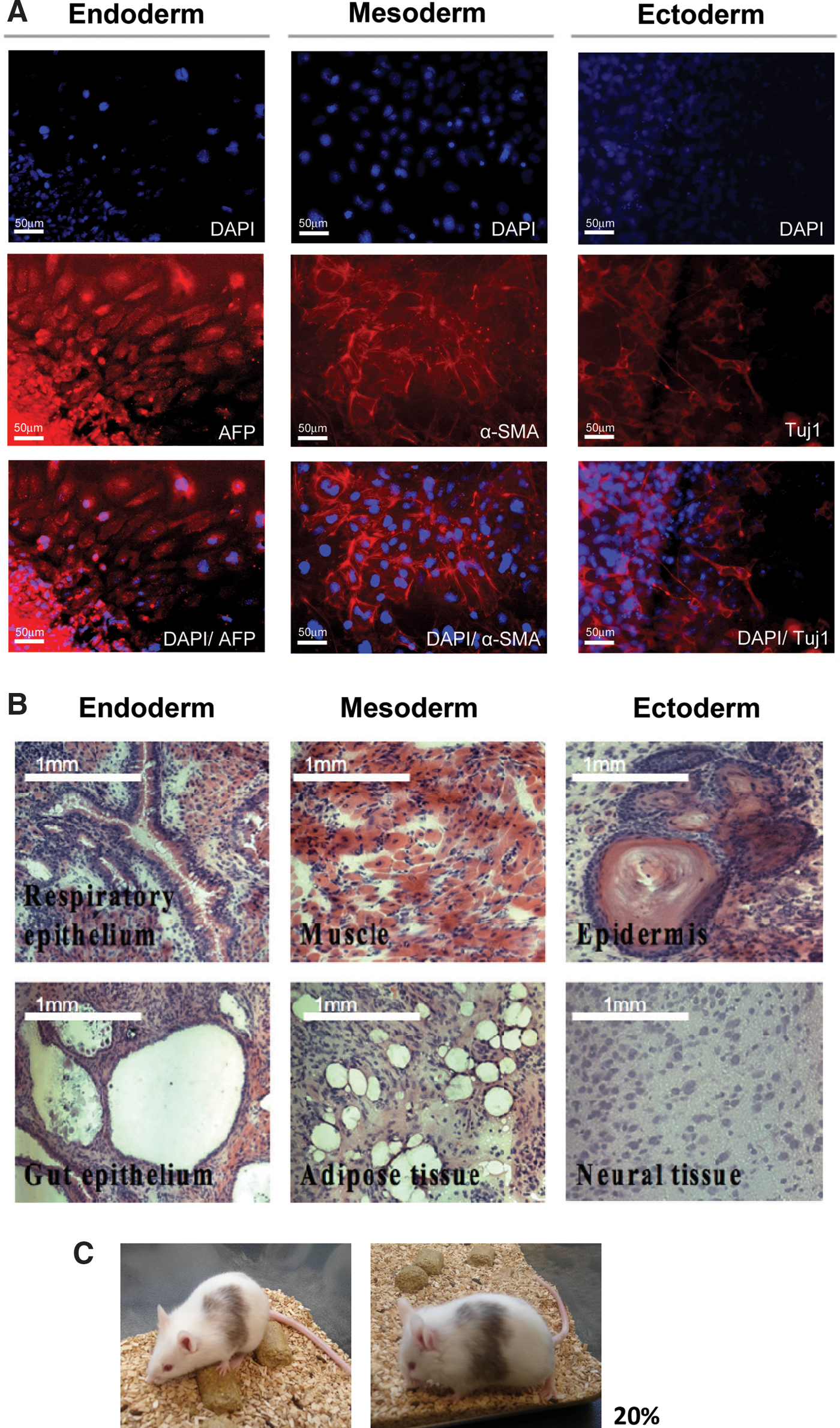
Detection of the pluripotency of iPS-OSH cells.
Chimeric mouse generation
As shown in Figure 4C, live-born chimeras were generated from iPS-OSH cells that were microinjected into albino mouse blastocysts. The chimeric percentage was ∼20%, indicating that the iPS-OSH cells had pluripotent capabilities.
Global gene expression profiles of iPS-OSH cells
Microarray analyses were performed to compare gene expression patterns in iPS-OSH, iPS-J, MEF, and ES cells. All data have been submitted to the GEO system (accession numbers GSM789276, iPS-OSH; GSM789277, iPS-J; GSM789278, ESC; and GSM789279, MEF). Results from a hierarchy clustering analysis using 40,777 genes indicate that the iPS-OSH cells clustered with iPS-J cells, then with ES, and, finally, with MEF cells (Fig. 5A). Results from a scatter plot analysis also indicate greater similarity in global gene expression levels between iPS-OSH cells and both iPS-J and ES cells (Fig. 5B). Oct4, Sox2, and Nanog gene expression levels in iPS-OSH cells were also similar to those in the iPS-J and ES cells, and considerably higher than in the MEF cells. Combined, these data confirm that iPS-OSH cells are similar to iPS-J and ES cells, but not to MEF cells.
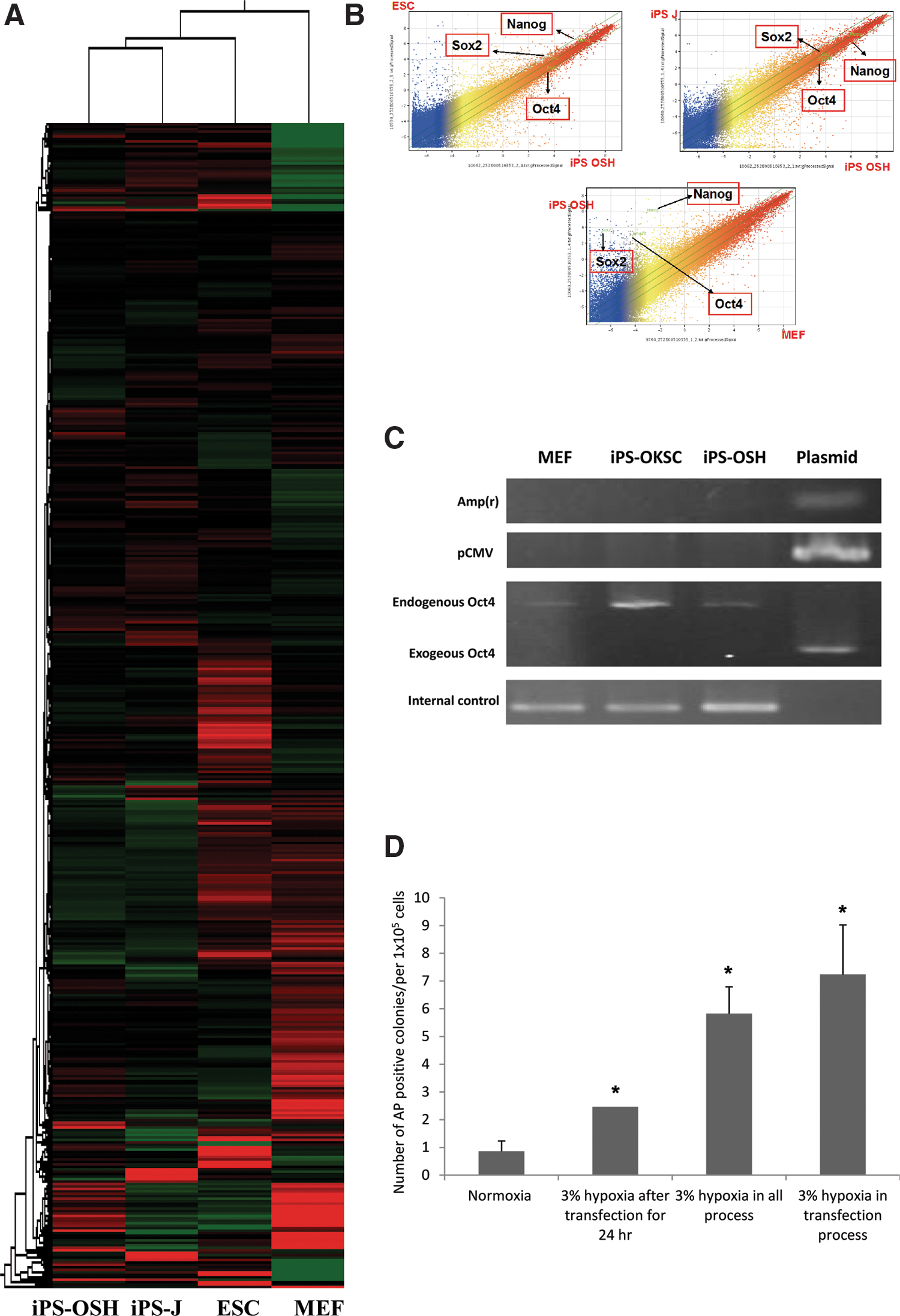
Comparison of the global gene expression levels in iPS-OSH, iPS-J, ES, and MEF cells by microarray analysis.
Detection of plasmid integration in iPS-OSH cells
To test for the genomic integration of plasmid DNA in iPS-OSH cells, we used PCR to detect three genes in plasmids: Amp(r), pCMV, and Oct4. iPS-OKSC cells were generated using plasmid transfection protocols previously described [11]. As shown in Figure 5C, the integration of any one of the three genes was not observed in the iPS-OSH cells, indicating a non-integration method for generating iPS cells.
Comparison of the four strategies for generating iPS cells
We established four groups for purposes of comparing iPS generation efficiencies among different hypoxic treatment strategies: (a) normoxia in all processes; (b) 3% hypoxia 24 h post-transfection (iPS-OSH generation strategy) (Fig. 2A); (c) 3% hypoxic treatment during all transfection processes followed by seeding in feeder layers; and (d) 3% hypoxic treatment during the transfection process and normoxia after seeding in feeder layers. Our data indicate significantly higher efficiencies in groups c and d than in group b, as well as significantly higher efficiencies in groups b, c, and d compared with group a (Fig. 5D). In short, the 3% hypoxic treatment increased iPS generation efficiency.
Differentiation of iPS-OSH cells into neural precursor cells
After generating and characterizing the novel iPS-OSH cells, we confirmed their ability to differentiate into neural precursor cells, and used those neural precursor cells with our cerebral ischemic stroke model. Procedures for differentiation entailed a modification of previously described protocols [16,17]. Neurotrophin receptor (p75), which is abundantly expressed in neural precursor cells, is considered a typical neural crest marker [18]. As shown in Figure 6A, data from flow cytometric analyses of p75 expression levels indicate that p75+ cells represented 63.2% of all cells after differentiation. Further, immunofluorescence staining results indicate that Nestin and Tuj-1 (two neural cell markers) were positively expressed in iPS-OSH-derived neural precursor cells (Fig. 6A). We also used electrophysiological tests for neuron function in mature iPS-OSH-derived neurons. As shown in Figure 6A, depolarization response to glutamate in the presence of Mg2+ indicates the expression of functional AMPA receptors.
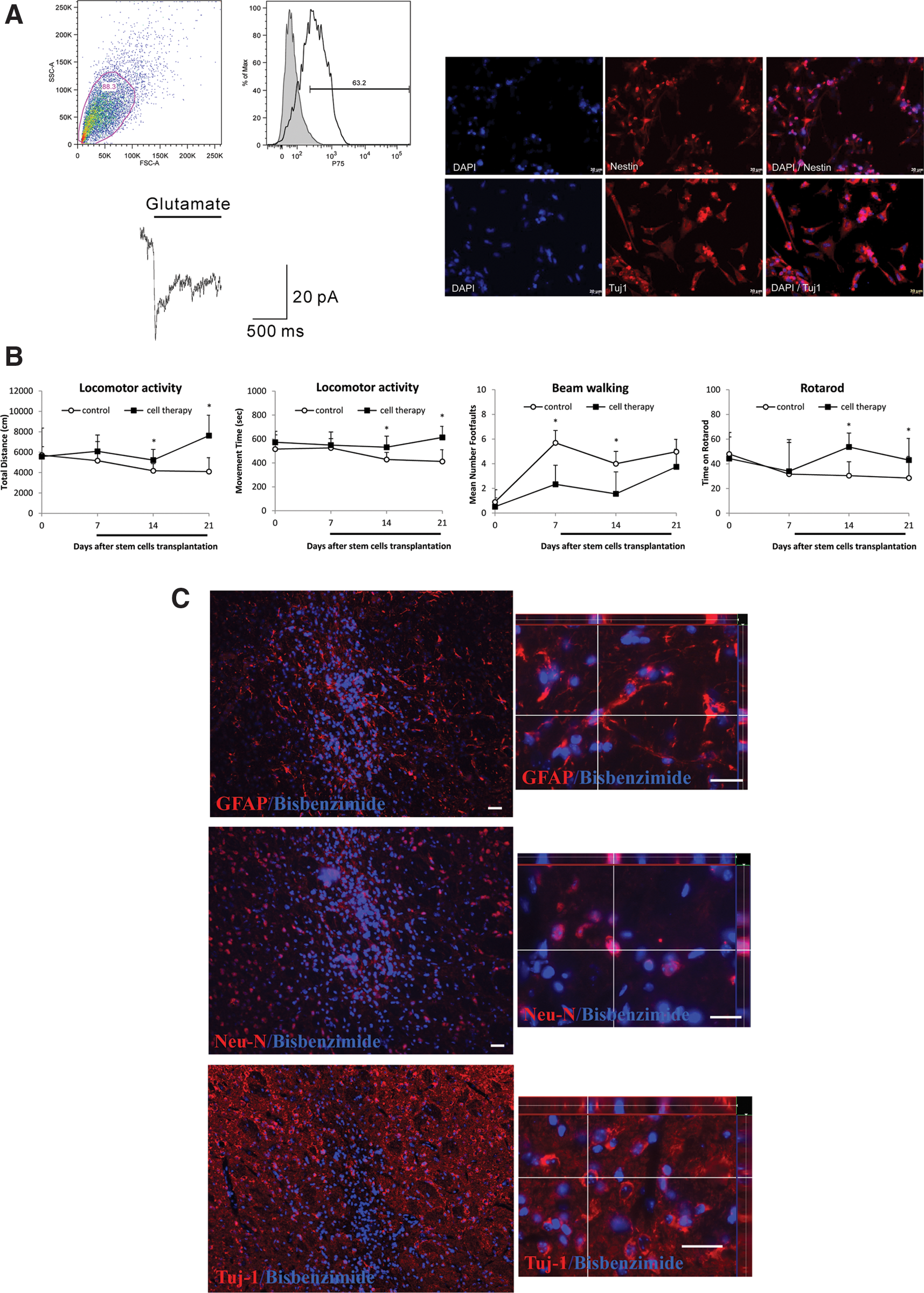
iPS-OSH was used to differentiate into the neural precursor cells and used for ischemic stroke therapy.
iPS-OSH-derived neural precursor cell transplantation improved neurological behavior in cerebral ischemic mice
iPS-OSH-derived neural precursor cells were transplanted into the cerebral ischemic stroke model to evaluate their therapeutic effects. Treated and vehicle control animals were tested for locomotor activity, beam walking, and rotarod movement to evaluate the effects of transplanted cells on ischemic stroke injury recovery. As shown in Figure 6B, locomotor activity results indicate that mice treated with iPS-OSH-derived neural precursor cells exhibited significant improvements in total distance (cm) and movement time (s) compared with the vehicle controls at 14 and 21 days post-transplantation; beam walking data indicated significant decreases in the number of foot faults in the treatment group compared with vehicle control mice at 7 and 14 days post-transplantation; and rotarod observations show that treated mice stayed on the rod significantly longer than vehicle control mice at 14 and 21 days post-transplantation. In short, the data indicate faster recovery in the iPS-OSH-derived neural precursor cell-treated group compared with the vehicle control group.
Neural differentiation after intracerebral transplantation of iPS-OSH-derived neural precursor cells
Mice were sacrificed, and brains were harvested at 21 days post-cell transplantation. To determine whether transplanted iPS-OSH-derived neural precursor cells differentiated into neurons or astrocytes at ischemic sites, we used double-immunofluorescent staining to analyze the colocalization of specific markers and bisbenzimide-labeled cell nuclei. As shown in Figure 6C, the bisbenzimide-labeled cells colocalized with antibodies for the glial fibrillary acidic protein (GFAP), neuronal nucleus (NeuN), and neuron-specific class III β-tubulin (Tuj-1) neural markers in the brain tissue of mice treated with the iPS-OSH-derived neural precursor cells. According to these data, the cells differentiated into neurons after intracerebral transplantation.
Discussion
Although ES cells are capable of differentiating into three germ layer cell types and are applied to therapeutic approaches in animal models [19,20], there are at least two significant limitations to their use for human transplantation: immune rejection and ethical concerns. iPS cell technology was developed to overcome ES cell limitations. The first iPS cells were generated in 2006 by introducing four transcription factors (Oct4, Sox2, c-Myc, and Klf4) into somatic cells, and then reprogramming the somatic cells to become pluripotent stem cells [5]. Nowadays, there are many ways to establish iPS cells, including the use of retroviruses [5,6], lentiviruses [21], adenoviruses [22], reprogrammed proteins [23], transposons [24], microRNA [25], and plasmid transfection [11,26]. Although plasmid transfection avoids viral integration into iPS cell genomes, oncogenic factors make therapeutic applications unfeasible. The major purpose behind using iPS cells in clinical applications is to establish a safe strategy for cell generation. In this study, we attempted to reprogram somatic cells without oncogenic factors or viral integration. It is our hope that this strategy will have applications for therapeutic research and development.
The c-Myc oncogene has been shown to trigger tumorigenesis in iPS cells containing chimeric mice [10]. These data indicate a risk factor for oncogenes in iPS cells and the iPS cell-derived differentiation cells. In 2009, Hong et al. reported that 10% of transduced MEFs lacked p53 after they were reprogrammed into iPS cells [27]. This finding suggests that oncogene over-expression during iPS cell generation was more dangerous and resulted in iPS cells for tumor-like cells. In a previous study, the transfection of an oncogene into a normal fibroblast via calcium ions had the potential to transform normal fibroblasts into cancerous cells [28]. Accordingly, it is important to prevent oncogene transfection when generating iPS cells, even when using a plasmid transfection method.
At least two researchers have reported that hypoxic treatment increases iPS cell generation efficiency. According to Yoshida et al., hypoxia enhances iPS cell generation [12], and according to Foja et al., hypoxia supports the reprogramming of mesenchymal stromal cells via the induction of Oct4, Nanog, and ES cell-specific microRNA-302 clusters [29]. We believe there are two important differences between the present study and Yoshida's work: (a) We used only two factors (neither one oncogenic), and (b) we waited until 6 h post-transfection to subject our MEF cells to hypoxic treatment and to hold on for only 24 h. In addition, Yoshida et al. kept their somatic cells under hypoxic conditions for a minimum of 12 days after seeding into feeder cells, whereas we held ours for only 4 days (total time) during transfection, and did not keep cells under hypoxic conditions after seeding. Lee et al. reported that continuous hypoxia for 60 h increased apoptosis and reduced cell viability and proliferation in mouse ES cells [30]. Other researchers have described hypoxia-induced mitochondrial apoptosis in mouse ES cells via reactive oxygen species generation [31]. Combined, these studies indicate that long-term hypoxic conditions induce mouse ES cell apoptosis. For this reason, we used a shorter hypoxic (24 h) process to generate iPS-OSH cells.
The primary motivation behind the use of iPS cells in clinical applications is to create a safer strategy for iPS cell generation. In 2011, Okita et al. found a more efficient method using episomal plasmids for generating integration-free human iPS cells [32], and Yoshioka et al. used synthetic self-replicating RNA to generate iPS cells [33]. We believe that our method represents a novel approach with the potential for therapeutic development. Our future plans are to focus on cell differentiation and therapeutic applications.
While focusing on ischemic stroke therapy, Jiang et al. have described transplanted iPS cell migration to injured brain areas and subsequent differentiation into neuron-like cells [34]. They claim that iPS cells can be directly transplanted into an animal model without differentiation. In the present study, we used iPS-OSH-derived neural precursor cells as a therapeutic approach, a method that we consider safer than direct iPS cell transplantation into mouse brains. In a previous study, we used several different cell types for stroke therapy, including human umbilical cord-derived mesenchymal stem cells [13], intracerebral peripheral blood stem cells (CD34+) [35], human bone marrow stromal cells [36], and murine olfactory ensheathing cells [37]. Here, we used novel iPS-OSH cells to test our hypothesis that neural precursor cells are capable of promoting neuroplasticity in mouse stroke models. Similar to our previous study, the data indicate that iPS-OSH-derived neural precursor cells are capable of differentiating into neural-like cells in stroke brains, resulting in improved responses to ischemic stroke injuries.
In summary, we generated a novel iPS-OSH cell line in the absence of viral infection and oncogenic factors that are used for stroke therapy. This procedure of this study has the potential for clinical application in the future.
Footnotes
Acknowledgments
This research was supported in part by the Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH102-TD-B-111-004), China Medical University (CMU99-N1-05), and the Republic of China National Science Council (NSC 98-2314-B-039-028-MY2). The authors wish to thank Shinya Yamanaka and the RIKEN Bioresource Center for providing the iPS-J cells. They also thank the Transgenic Mouse Model Core facility of NRPGM for generating the chimeric mice used in this study, and Jon Lindemann for his editing assistance.
Author Disclosure Statement
The authors declare no conflicts of interest.