
Abstract
Besides the presence of somatic stem cells in hair follicles and dermis, the epidermis also contains a subpopulation of stem cells, reflecting its high regenerative capacity. However, only limited information concerning epidermis-derived epithelial-like stem/progenitor cells (EpSCs) is available to date. Nonetheless, this stem cell type could prove itself useful in skin reconstitution after injury. After harvesting from equine epidermis, the purified cells were characterized as EpSCs by means of positive expression for CD29, CD44, CD49f, CD90, Casein Kinase 2β, p63, and Ki67, low expression for cytokeratin (CK)14 and negative expression for CD105, CK18, Wide CK, and Pan CK. Furthermore, their self-renewal capacity was assessed in adhesion as well as in suspension. Moreover, the isolated cells were differentiated toward keratinocytes and adipocytes. To assess the regenerative capacities of EpSCs, six full-thickness skin wounds were made: three were treated with EpSCs and platelet-rich-plasma (EpSC/PRP-treated), while the remaining three were administered carrier fluid alone (PRP-treated). The dermis of EpSC/PRP-treated wounds was significantly thinner and exhibited more restricted granulation tissue than did the PRP-treated wounds. The EpSC/PRP-treated wounds also exhibited increases in EpSCs, vascularization, elastin content, and follicle-like structures. In addition, combining EpSCs with a PRP treatment enhanced tissue repair after clinical application.
Introduction
I
Currently, epidermal-like stem cells have been obtained by either dedifferentiating adult epidermal cells [11] or inducing pluripotent stem cells [12]. The isolation of skin-derived precursor cells with neurogenic properties has been previously reported from human dermis [13 –15] and hair follicles [16] as have the isolation and partial purification of adult multipotent stem cells from human and murine epidermis [17,18]. For instance, Fujimori et al. reported the isolation of human epidermal stem/progenitor cells by means of gravity-assisted cell sorting [17]. However, the practice of selecting for stem cells based on size (diameter) is problematic given that there exists a significant proportion of somatic cells with equally small diameters, ultimately resulting in a heterogeneous yield of cell types. On the other hand, Nowak and Fuchs utilized fluorescence-activated cell sorting (FACS) to isolate epidermal stem cells from mice [18]. However, due to the lack of EpSC-specific markers, cell sorting with a single marker was not sufficient to isolate a pure population of stem cells. Ergo, EpSC purification was the initial goal of the present study and was based on two principles: (i) Since EpSCs are located in the epidermal basal layer, the separation of this skin layer from the underlying dermis would avoid contamination by a large number of unwanted non-epithelial cells; and (ii) by seeding the isolated cells at a clonal density in non-coated flasks, EpSCs might be purified, as only those cells that have the ability to clonally expand in suspension (a stem cell-specific property) would survive [19], thereby substantially reducing the survival of differentiated cell clusters.
The existence of undifferentiated cells that are capable of surviving in suspension was first reported in 1986 [20]. Subsequently, these cells were designated as stem cells because of their bipotent properties [21,22]. To date, different stem cell types have been isolated, such as mammary stem/progenitor cells [23], corneal stem/progenitor cells [24], and neural stem cells [25]. The use of non-coated flasks was later employed to isolate spherical cell clusters derived from human hair follicles [16]. Using embryonic stem cell culture conditions, Yu et al. [16] succeeded in forming cell clusters containing stem cells from skin follicles. The isolated stem cells were not strictly EpSCs, as they exhibited neurogenic properties. To our knowledge, there are no reports of the harvesting of a homogenous population of cells from mammalian epidermis that fulfil all the criteria to merit classification as EpSCs. Therefore, the present study is likely the first to describe the isolation, purification, and immunophenotypical and functional characterization of equine EpSCs.
Clinical applications of non-EpSCs for skin wound healing have been described [26,27]. In this regard, the improved healing capacity of persistent leg wounds in human diabetic patients in response to bone marrow-derived mesenchymal stem cell (MSC) treatment was reported [28]. Moreover, it has been demonstrated that peripheral blood-derived stem cells moderate the development of granulation tissue and crust formation of naturally occurring wounds in horses, which were unresponsive to more conservative therapies [29]. However, in both studies, there was no evidence of a decrease in scar tissue formation or an effect on other functional parameters, such as elastin or collagen production. In order to achieve restitutio ad integrum, it might prove beneficial to use stem cells already committed toward the keratinocyte fate. To our knowledge, there are no reports of an experimentally controlled double-blinded study of mammalian skin wound treatment with EpSCs. Therefore, the second goal of this study was to compare induced full-thickness skin wounds (positive control), skin wounds treated with EpSCs plus platelet-rich plasma (EpSC/PRP-treated), and skin wounds treated with PRP alone. Since it has been previously indicated that PRP might have synergistic effects on stem cell regeneration [30,31] and that a fibrin-based matrix could support selective adhesion, proliferation, and differentiation of keratinocyte progenitors [32], we elected to use endogenous PRP as a carrier for the EpSCs. Macroscopic and histological examinations revealed significant differences between wound-healing parameters of EpSC/PRP-treated and PRP-treated samples.
Materials and Methods
Sampling, epidermal cell harvesting, and epidermosphere formation
Samples were collected immediately after the sacrifice of three horses in a government-certified slaughter house. Skin tissue was obtained by excising 1 cm2 of full-thickness skin in the neck region after clipping and surgical preparation (scrubbing with iodide and disinfecting with 70% ethanol). The samples did not show any macroscopic abnormalities and were transported to the laboratory within 6 h in phosphate-buffered saline (PBS) 1× (without calcium and magnesium) with 2% of penicillin-streptomycin-amfotericine B (P/S/A; Sigma-Aldrich). The tissue samples were treated with 0.25% trypsin-ethylenediamintetraaceticacid (EDTA) (Gibco) solution at a temperature equal to or lower than room temperature, for 12–18 h. Afterward, the epidermis was further mechanically detached from the dermis (skin peeling). Subsequently, the epidermis was cut into small pieces, and a second enzymatic dissociation step with 2% collagenase III (Worthington Biochemical Corporation) at 37°C for 60 min was performed. The collagenase III was inactivated by adding EpSC medium, consisting of Dulbecco's modified Eagle's medium (DMEM)/F12 (Gibco), 20% fetal bovine serum (FBS; Gibco), 2% P/S/F, 20 ng/mL human recombinant-basic fibroblast growth factor (FGF), and 20 ng/mL epidermal growth factor (all from Sigma-Aldrich). The suspension was then filtered (40 μm; BD Falcon) and washed with EpSC medium by centrifugation at 300g for 8 min at room temperature. Approximately 5×103 cells/cm2 were planted per well of an ultralow attachment six-well plate (Corning; Elscolab). The EpSC medium was refreshed thrice a week by means of centrifugation of the epidermospheres at 300g for 8 min. After 1 week, the epidermospheres were seeded on adhesive tissue culture dishes in EpSC medium.
Epithelial cycle assay, colony-forming unit assays, and population doubling time
After the first epidermosphere cycle, which developed from skin tissue-derived cells on ultralow attachment plates (as described earlier), all epidermospheres were collected at day 7 post-seeding and plated onto adhesive tissue culture dishes in EpSC medium. On reaching 80% confluency, the adherent cells were trypsinized with 0.25% trypsin-EDTA and seeded at a clonal density of 1 and 100 cells/cm2 on ultralow attachment plates to initiate a second epidermosphere cycle and of 1 cell/cm2 on adherent plates for 1 week for colony-forming unit (CFU) assays for the three skin samples in triplicate. The adherent CFUs were counted after fixation with 90% ethanol for 10 min at −20°C. Crystal violet staining (Sigma-Aldrich) was performed to visualize CFUs macroscopically, and the total number of CFUs per six-well plate was counted. In addition, the second-cycle epidermospheres were counted and related to the number of planted cells.
Population doubling time (PDT) in days was calculated from P 0 to P 10 as previously described [4], using the following formula: T/[log(N f/N i)/log2] with T the time in days, N f the final number of cells, and N i the initial number of cells.
Flow cytometry
To characterize the epidermosphere-derived cells immunophenotypically, the expression of several stem cell markers was evaluated by flow cytometry. Per series, 200,000 cells were used and labeled with the following primary antibodies: mouse anti-human CD29-FITC IgG2a (Abcam; clone B-D15, 1:10), rat anti-mouse CD44-FITC IgG2b (Abcam; clone IM7, 1:20), rat anti-mouse CD49f IgG2a (Abcam; clone GoH3, 1:10), mouse anti-dog CD90 IgM (VMRD; clone DH24A, 1:100), mouse anti-human CD105-PE IgG1 (AbD Serotec; clone SN6, 1:50), mouse anti-horse major histocompatibility complex (MHC) class I IgG2a (Washington State University; 1:50), mouse anti-horse MHC class II IgG1 (Abd Serotec; 1:50), and rabbit polyclonal anti-human Ki67 IgG (Abcam; 1:200). For Ki67, cells were fixed with 4% paraformaldehyde for 10 min and, subsequently, permeabilized with 0.1% Triton X for 2 min at room temperature. Cells were incubated with the primary antibodies for 15 min on ice in the dark and washed twice in washing buffer, consisting of DMEM with 1% bovine serum albumin. For CD49f, CD90, MHCs, and Ki67, secondary goat anti-rat-FITC (Abcam, 1:100), goat anti-mouse-Cy5 (Abcam; 1:100), rabbit anti-mouse-FITC (Abcam, 1:100), and goat anti-rabbit-Cy5 (Abcam; 1:100) antibodies were used, respectively, to identify positive cells after 15 minutes of incubation on ice in the dark. Finally, all cells were washed thrice in washing buffer, and at least 10,000 cells were evaluated using a FACS. All analyses were based on (i) autofluorescence and (ii) control cells incubated with isotype-specific IgGs, in order to establish the background signal. All isotypes were matched to the immunoglobulin subtype and used at the same protein concentration as the corresponding antibodies.
Differentiation experiments
Differentiation of putative EpSCs toward two major cell types present in the skin, namely adult epithelial cells (or keratinocytes) and adipocytes, was induced using distinct differentiation protocols. The first protocol consisted of culturing 5×103 EpSC/cm2 in keratinocyte differentiation medium, containing DMEM/F12 (Gibco), 20% FBS (Gibco), 5 μg/mL insulin, 1 μg/mL hydrocortisone, and 0.1 mM beta-mercaptoethanol (all from Sigma-Aldrich) for 10 days. Media were refreshed every 3–4 days, and immunohistochemistry (IHC) was performed to evaluate the expression of markers present in differentiated epithelial cells (see next). Keratinocyte differentiation was repeated in suspension cultures using the differentiation medium mentioned earlier for 10 days as well. The second protocol was needed for adipogenic differentiation, and here, 4×104 cells/cm2 were planted in adipogenic inducing medium (adapted from Spaas et al. [5]) consisting of DMEM (Gibco) that was supplemented with 10 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, 10 μg/mL recombinant human-insulin, 0.5 mM indomethacin, and 15% rabbit serum (all from Sigma-Aldrich). Differentiation was evaluated after 3 days of cultivation using Oil Red O (Sigma-Aldrich) staining. As a control for the bilineage differentiation, putative EpSCs were cultivated for the same time in EpSC medium, at the same density, and in identical culture vessels.
IHC on cells
Adhesive cell cultures were stained in their culture dishes, whereas epidermospheres cultured in suspension were seeded onto normal tissue culture plates for 1 h to enable adherence, or cytospins were prepared. After that, cells were fixed for 10 min with 4% paraformaldehyde and permeabilized for 2 min with 0.1% Triton X, both of which were at room temperature. Cells were then incubated with hydrogen peroxide for 5 min followed by washing with PBS before incubation for 2 h at room temperature with the following primary mouse IgG1 monoclonal antibodies: anti-human cytokeratin (CK)14 (Abcam; clone LL002, 1:50), anti-human CK18 (Abcam; clone C-04, 1:30), anti-human Pan CK (Dako; clone, AE1/AE3, 1:50), and anti-porcine vimentin (Abcam; clone V9, 1:100); the mouse anti-human smooth muscle actin (SMA) IgG2a Ab (Dako; clone 1A4, 1:200); and the following rabbit antibodies: anti-human Wide CK (Abcam; 1:50), anti-human Ki67 (Abcam; 1:200), anti-human casein kinase 2β (Abcam; clone EP1995Y, 1:50), and anti-human p63 (Abcam; 1:50). After washing with PBS, secondary ready-to-use goat anti-mouse and anti-rabbit peroxidase (PO)-linked antibodies (Dako) were added and incubated for 30 min at room temperature. Finally, 3,3′-diaminobenzidine (DAB) was added for 2–10 min, and a counter staining with hematoxylin was performed to visualize the surrounding cells. As controls, identical staining was performed on undifferentiated EpSCs, and background staining was assessed by using the proper isotype-specific IgGs. All isotypes were matched to the immunoglobulin subtype and used at the same protein concentration as the corresponding antibodies.
In vivo study
After local anesthesia and surgical preparation, six full-thickness skin wounds of ∼4 cm2 were induced in a 5-year-old gelded horse dorsally from the musculus gluteus medius (three on the left side and three on the right side). Three randomly assigned wounds received allogenic putative EpSCs (derived from a slaughter horse skin sample) in combination with autologous PRP (EpSC/PRP-treatment) as a carrier, and the three remaining wounds were treated identically, but without putative EpSCs (PRP injection only). The EpSC/PRP-treatment consisted of two approaches: First, an intradermal injection (0.5 mL in each of the four wound edges) of 1 mL of DMEM containing 8×106 freshly prepared putative EpSCs at passage 3 in combination with 1 mL of PRP (prepared as previously described [30,33]) was administered. Immediately afterward, a topical application of 0.5 mL DMEM containing 4×106 putative EpSCs and 0.5 mL of PRP was performed. The topical application was repeated after absorption of the liquid, which was after 24 h. The PRP treatment was performed in exactly the same manner; however, the DMEM did not contain EpSCs in this group. Wounds were not sutured, but covered with an adhesive, non-absorbable plastic in which micropores were made in order to enable oxygen access. The horse was bound in his stable for 24 h in order to prevent rolling, and photographs were taken every 3–4 days. Wounds were measured and surfaces were calculated immediately before and 30 days after treatment by a veterinarian under double-blinded conditions. The experimental protocol was approved by the ethics committee (EC_2012_002) of Global Stem cell Technology (LA1700607).
Histology of tissue sections
On day 30, an 8 mm punch biopsy was taken from the center of the wound (covering the whole wound area) for tissue sampling. The specimens were fixed in neutral-buffered 10% formalin, embedded in paraffin, sectioned at 4 μm thickness, and stained with Hematoxylin and Eosin, Van Gieson (VG), and Elastin stains according to standard protocols.
All samples were blindly analyzed by an ECVP-certified pathologist (S.M.) using a modified scoring system, adapted from Abramov et al. [34] and Babaeijandaghi et al. [35] (Table 1). Briefly, the epidermis was scored for thickness (0–3), crust formation (0–1), dermo-epidermal separation (0–2), and completeness of re-epithelization (0–3). Sections of positive control skin originating from the same horse were made from the samples at the time of wound induction (establishing a maximal score). The dermis was evaluated using six main parameters: (i) edema (0–3); (ii) amount and morphology of the stroma (thickness measured and scored 0–3, amount of collagen, and the morphology of collagen fibers (thin/immature, intermediate, or thick/mature) using VG stain and scored 0–2, amount of elastin using elastica Van Gieson stain and scored 0–1); (iii) thickness and morphology of the granulation tissue (thickness measured and scored 0–3, maturation of fibroblasts scored 0–3); (iv) neovascularization (scored 0–3 and average number of capillaries counted per 200× magnification field) on immunohistochemical stain for von Willebrand factor (vWF); (v) acute inflammation/neutrophilic infiltrate (0–3); and (vi) chronic inflammation/lymphoplasmacytic infiltrate (0–3). Again, positive control sample values set a maximal score. The average of three fields was used to evaluate different histopathological parameters, and all measurements were performed with a computer-based program (LAS V4.1; Leica Microsystems).
VG, Van Gieson; vWF, Von Willebrand Factor; IHC, immunohistochemistry.
IHC on tissue sections
In order to localize epithelial cells within the epidermis, skin glands, and hair follicles, Pan CK and Wide CK were used. Casein Kinase 2β and Ki67 indicated where proliferating [36] epithelial [37] cells were located within the sample. To examine the role of EpSCs in elastin production and neovascularization, tissue sections were stained with mouse anti-bovine elastin IgG1 (Leica; clone BA-4, 1:100) and rabbit anti-human vWF (Dako; 1:6,400), respectively. Furthermore, the expression of the mesenchymal marker vimentin [38] and the myogenic marker SMA [39] was assessed. Immunolabeling was achieved with a high-sensitive horseradish PO mouse or rabbit diaminobenzidine kit with blocking of endogenous PO (Envision DAB+kit; Dako) in an autoimmunostainer (Cytomation S/N S38-7410-01; Dako). A commercially available antibody diluent (Dako) with background-reducing components was used to block hydrophobic interactions. All skin samples (including positive and negative controls) were submitted to the same immunohistochemical staining procedure.
Statistical analysis
Data were analyzed using the two-tailed Student's t-test for group comparisons of normally distributed variables. Values were given as means±standard deviation (bars). P-values were calculated using an Excel spread sheet (2007; Microsoft Corp.), and P<0.05 was considered a probabilistically significant difference between the compared groups of samples.
Results
Epidermis-derived cells are classified as EpSCs
Epidermis-derived cells are capable of epidermosphere formation, proliferate clonally in suspension, and exhibit similar expansion capacities on substrate adhesion as well as in suspension
After performing the skin peeling protocol, the epidermis could be removed without the inclusion of hair follicles or other dermal structures (Supplementary Fig. S1; Supplementary Data are available online at

The epithelial-like stem/progenitor cell (EpSC) cycle. After epidermosphere formation on ultralow attachment plates
Moreover, after dissociating the epidermospheres into single cells and seeding 100 EpSCs/cm2 (1,000 EpSCs/six-well), approximately the same amount of spheres were formed after 1 day and the average number of spheres (1,203 vs. 1,240 vs. 1,157) remained relatively constant during the 1 week cultivation period (Fig. 2A), indicating that most epidermosphere-derived EpSCs maintained their phenotype and were capable of giving rise to subsequent generations of spheres. Indeed, since sphere formation was initiated within 1 day and it takes at least 24 h for EpSCs to divide (data not shown), we may conclude that all the EpSCs were at the origin of the newly formed epidermospheres. In addition, the number of EpSCs significantly increased at day 4 (P=0.0005) and 7 (P<0.0001) from the first day of culture, indicating that all EpSCs were capable of clonal expansion in suspension (Fig. 2B).

Epidermosphere formation assay. After planting 100 EpSCs/cm2 (1,000 EpSCs/six-well), the average number of spheres (white histograms) of the three skin samples remained approximately the same at day 1, 4, and 7 (Y-axis) after seeding
To compare the self-renewal capacity and clonogenic expansion of EpSCs in adherent cultures as well as in suspension, a critically low number of cells (1/cm2) was seeded at a clonal density on large surfaces and cultured for 1 week. Although the CFU assay for the first skin sample demonstrated significantly (P=0.013) more CFUs in adhesion than epidermospheres in suspension, the second skin sample exhibited the opposite trend (P=0.0059). Summing the three samples together, an average of one CFU (one epidermosphere per cm2) or 11 CFUs (spheres) per six-well plate were formed within the first week (Fig. 2C). This implied that every epidermosphere-derived cell had the capacity to clonally multiply both in adhesion as well as in suspension. Another indication of the self-renewal capacity of the EpSCs was their PDT (in days) over multiple passages (from P 0 up to P 10). The EpSCs from all three skin samples divided rapidly as shown by a positive PDT varying between 0.78 and 1.16 for all passages tested.
Marker expression of epidermis-derived EpSCs
The epidermis-derived EpSCs were immunophenotypically characterized by flow cytometry, and approximately all cells were positive for the stem cell surface markers CD29, CD44, CD49f, and CD90 (Fig. 2D). Moreover, EpSCs were positive for the proliferation marker Ki67. Cross-reactivity of all the markers was evaluated with the appropriate positive controls using flow cytometry or immunofluorescence (data not shown). No fluorescence signal was obtained with the MSC marker CD105 (Fig. 2C), with MHC II (Supplementary Fig. S2), or with the appropriate isotype controls. Moreover, a low expression of MHC I (Supplementary Fig. S2) was observed. Peripheral blood mononuclear cells were used as positive controls to confirm MHC cross-reactivity (Supplementary Fig. S2).
Equine EpSCs can differentiate into different keratinocyte subtypes and adipocytes
After culturing EpSCs in the keratinocyte differentiation medium for 10 days, two different adult cell types appeared in the culture. Stellate-shaped cells (form several cobblestone-like cells after differentiation for 15 days, Supplementary Fig. S3) became evident that were positive for specific keratinocyte [40] and follicular cell markers (cfr. positive control data), including CK14, CK18, Pan CK, and Wide CK in adhesion as well as in suspension (Fig. 3; Supplementary Figs. S4 and S5). Within the differentiated cell culture, cylindrical-shaped cells arose that were positive for vimentin (Fig. 3G) and SMA (Fig. 3H). EpSCs and the differentiated cultures were negative for relevant isotype controls. Undifferentiated EpSCs stained negative in adhesion (Fig. 3A, C, E) as well as in suspension (Supplementary Fig. S4) for CK18, Pan CK, and Wide CK. Moreover, undifferentiated EpSCs expressed low cytoplasmic levels of CK14 (Supplementary Fig. S5B, C), but were clearly positive for the proliferation marker Ki67 [36] (Supplementary Fig. S6) and the epithelial marker Casein Kinase 2β [37] (Supplementary Fig. S6), both of which sharply decreased on differentiation (Supplementary Fig. S6). Finally, most of the EpSCs as well as several of their more differentiated progeny were positive (nuclear and weaker cytoplasmic signal) for the epithelial stem cell marker p63 [41,42] in adhesion as well as in suspension (Supplementary Fig. S7). Cross-reactivity of all the markers was evaluated with the appropriate positive controls via dot blots (data not shown) and/or via IHC (data not shown).
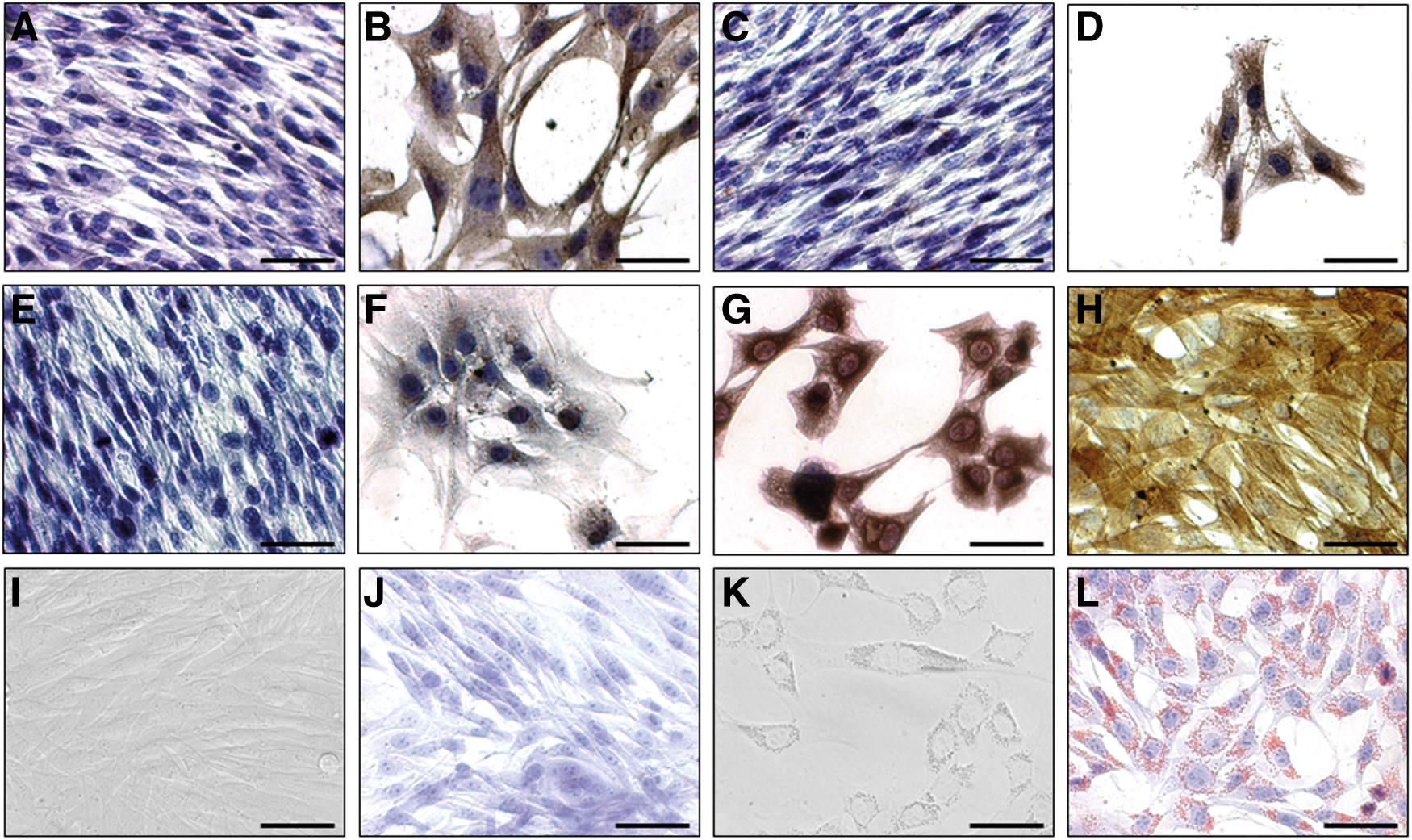
Differentiation of EpSCs into keratinocytes and adipocytes. Immunohistochemistry on the EpSCs confirmed that cytokeratin (CK) markers, CK18
Interestingly, within 3 days of culturing in adipogenic-inducing medium, EpSCs changed from a spindle-shaped to a round morphology that was concomitant with the production of small intracellular granules, microscopically detected in all cultures (Fig. 3K). Subsequently, Oil Red O staining confirmed the presence of lipid droplets in these cells (Fig. 3L), indicating that EpSCs are capable of differentiating into adipocytes. Non-differentiated control EpSCs maintained their spindle-shaped morphology (Fig. 3I) in monolayers after cultivation in EpSC medium for 3 days and stained negatively for lipid droplets (Fig. 3J).
EpSC/PRP-treated skin wounds display a higher regenerative profile than PRP-treated wounds
EpSC/PRP-treated wounds display macroscopic-enhanced healing capacities
Wounds were measured at the day of induction and again 30 days later (Fig. 4). Despite the examining veterinarian perceiving no difference, surface calculations were performed by dividing the remaining repair tissue in mm2 by the initial wound area in mm2×100. This revealed an average of 79% (100%−surface calculation) of the wound surface that was filled (granulation and epithelialization) in the EpSC/PRP-treated group, which was significantly different (P=0.0083) from 72% for the PRP-treated group (Fig. 4).

Macroscopic wound evaluation. Macroscopic images of the wound site at different time points (d=day) after platelet-rich-plasma (PRP) treatment with (B, D, F) and without (A, C, E) skin-derived EpSCs. At each time point, the left picture represents wounds (A–C) at the left side of the horse and wounds (D–F) at the right side. The table represents the percentage of wound filling, calculated after measuring the areas before treatment (after induction at d0) and 30 days later (d30). In the EpSC/PRP-treated wounds, a significantly larger area was filled at d30.
EpSC/PRP-treated wounds display microscopic-enhanced healing capacities
In all skin samples, a normal to mild acanthotic, mature epidermis with crust formation (Fig. 5B, C, E, F) was constituted at day 30. Image-based length measurements revealed a considerably higher amount of collagen and a significantly (P=0.017) thicker dermis in the PRP-treated samples (7.4±0.6 mm, Fig. 5B, E) compared with the EpSC/PRP-treated (5.6±0.9 mm, Fig. 5C, F) and positive control samples (5.7±0.4 mm, Fig. 6A, D). PRP-treated wounds also contained a considerably higher amount of granulation tissue (6.4±0.4 mm), compared with the EpSC/PRP-treated wounds (5.1±0.8 mm). In all skin samples, a mild acute inflammation (polymorphonuclear cells) was present and in five out of six cases, a mild (Fig. 5G) to moderate (Fig. 5H) chronic inflammation (lymphocytes and plasma cells) was observed that was mainly restricted to perivascular regions, yet in some instances extended to the interstitial regions. In one sample from the PRP-treated group, an abundant number of inflammatory cells could be noticed in a localized area (Fig. 5I). There were no significant differences in histopathology scoring with regard to the degree of edema and the morphology of the dermal stroma or granulation tissue.
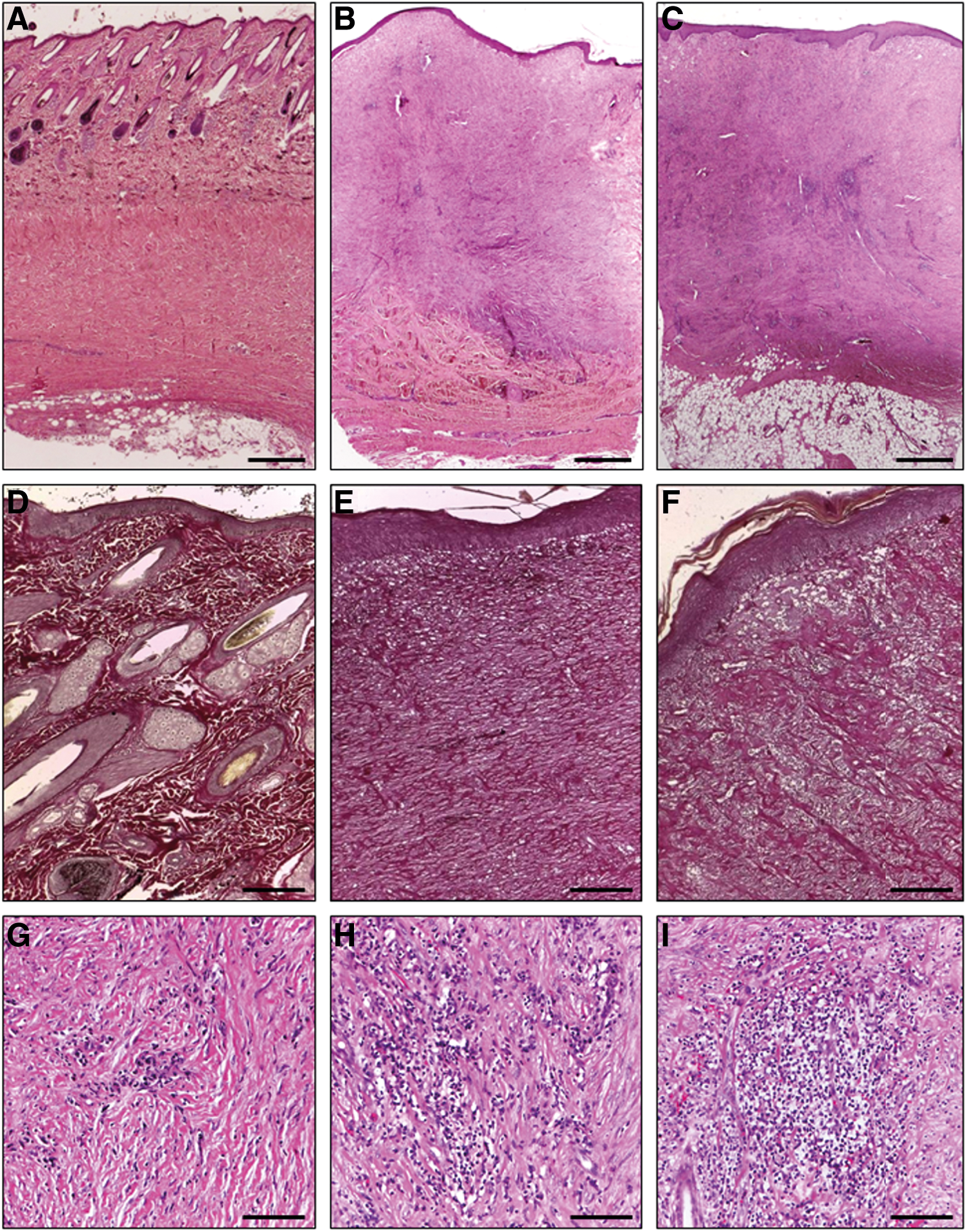
Hematoxylin-eosin
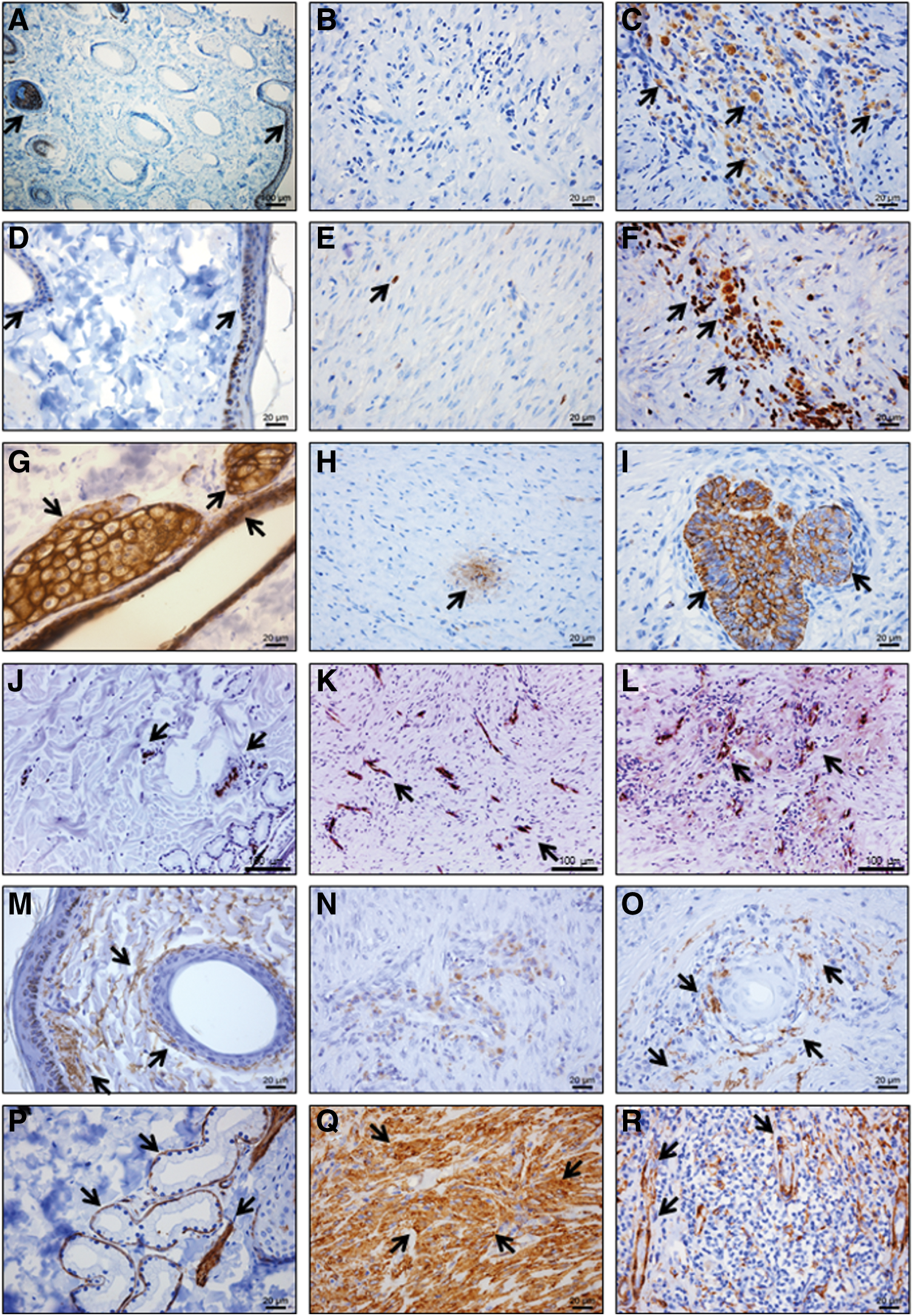
Immunohistochemistry staining on the intact skin samples [left column:
When evaluating Casein Kinase 2β and Ki67—which were present in the EpSCs and decreased during differentiation—a few cells in the epidermal basal cell layer and the hair follicle stained positively in the nucleus (Fig. 6A, D). Within the newly formed dermis, the EpSC/PRP-treated wounds revealed areas with many positive cells for Casein Kinase 2β (Fig. 6C) and Ki67 (Fig. 6F), whereas PRP-treated wounds contained, at most, a single positive cell per 400× magnification field (Fig. 6B, E). When evaluating epithelial markers (Wide CK and Pan CK) in the intact skin sections, epidermal cells, adnexal glands, and follicular epithelial cells stained strongly positive (Fig. 6G). When evaluating these markers in follicle-like structures in the dermis, these structures seemed more developed in the EpSC/PRP-treated group (Fig. 6I) than in the PRP-treated group (Fig. 6H). In both treatment groups, the newly formed epidermis stained diffusely positive for CK markers (data not shown). Neovascularization was evaluated by means of vWF staining and a considerably, yet non-significant (P=0.26), higher average of dermal blood vessels (31±4) per 200× magnification field was noticed in EpSC/PRP-treated wounds (Fig. 6L) compared with the PRP-treated wounds (23±4) (Fig. 6K).
Histochemical elastin staining revealed no considerable differences between the EpSC/PRP- and PRP-treated skin samples (data not shown), whereas IHC revealed a remarkably higher elastin production around follicle-like structures in the dermis of EpSC/PRP-treated wounds (Fig. 6O), resembling intact skin structures (Fig. 6M). On the contrary, there was only scarce elastin staining in the PRP-treated group (Fig. 6N). SMA cytoplasmic immunolabeling revealed a thin line of positive cells surrounding hair follicles and sweat glands (Fig. 6P). Moreover, the arrector pili muscles and vascular tunica media stained strongly positive for SMA (Fig. 6P), as well as a subset of follicular cells (mainly in lower follicular outer root sheath and higher inner root sheath layer). Both PRP- and EpSC/PRP-treated wounds contained dense areas of SMA (Fig. 6Q)-positive cells in the dermis. However, EpSC/PRP-treated wounds also contained large zones with minimal SMA staining, a feature that was absent in PRP-treated wounds. In addition, a thin layer of SMA-positive cells encircling dermal follicle-like structures was present in EpSC/PRP-treated wounds (Fig. 6R), a pattern very similar to SMA staining in intact skin.
Discussion
Although the isolation of sphere-forming progenitor cells was described in 1986 [20], the main problem using suspension cultures for stem cell isolation was potential contamination with other cell types which are capable of surviving this selection process by insinuating themselves to surviving stem cells in the clusters. It should be noted that the present study describes a peeling technique using mechanical and enzymatic digestions to separate epidermal cells from their surrounding tissue as well as a method to enrich for EpSCs by seeding at a low density, followed by epidermosphere formation. Finally, the isolated EpSCs were characterized and clinically evaluated.
Briefly, the equine EpSCs were immunophenotypically characterized by using the universal stem cell markers CD29, CD44, CD49f, and CD90. The nuclear marker Ki67, which is expressed during the (G)1, synthesis (S), and G2/mitosis (M) phases of the cell cycle [36], was used to give an indication of the self-renewal capacity of the EpSCs. The markers CK18, CD105, and p63 were used to make a clear distinction from equine MSCs. MSCs are positive for both CK18 (data not shown) and CD105 [5], which is not the case for EpSCs; whereas p63 is absent in MSCs [43,44] and was present in the nucleus and cytoplasm of EpSCs. These results are in agreement with previous reports examining p63 expression in corneal stem cells [41,42] and circulating keratinocyte progenitor cells [32]. Moreover, the EpSCs in the present study were positive for Casein Kinase 2β, which is reputed to inhibit the development of the mesenchymal morphology and phenotype [37,45], and plays an important role in the functional regulation of epithelial cells [46 –48].
To date, different markers for stem cell localization in mammalian skin have been identified; however, EpSC-specific markers are lacking due to lack of tissue specificity. For instance, stem cell factor has been reported as an indicator for skin stem cells, despite its expression by mature melanocytes [49]. Other markers, such as CK19 [12] and β-catenin, are expressed not only in skin stem cells [50], but also by differentiated epidermal cells [11]; whereas β-catenin is also present in lymphocytes [51]. In this study, Casein Kinase 2β and Ki67 were used to quantify the number of proliferating [36] epithelial cells [37]. These markers were present in EpSCs in vitro and became further up-regulated in the EpSC/PRP-treated group in vivo. Blanpain and Fuchs found that stem cells in the epidermis divide asymmetrically to enhance the EpSC pool as well as to provide a more differentiated progeny pool [52], helping explain the increased incidence of EpSCs 30 days after EpSC/PRP treatment. Moreover, in the intact skin samples, both markers only coincided in areas where EpSCs were located: the epidermal basal cell layer, transition of dermal papilla, and bulge of the hair follicle. Indeed, the presence of stem cells has been previously reported in these areas [8,9].
Sphere formation is a frequently used method to enrich for stem/progenitor cells by culturing enzymatically digested cells on ultralow attachment plates to prevent adhesion [53]. These spheres commonly consist of a mixture of stem/progenitor cells, their progeny, and non-stem progenitor cells, as previously described by Stingl in 2009 [54]. In contrast to the study of Toma where a crude dermal cell suspension was cultured and sphere formation was obtained [14], our study describes the cultivation of epidermis-derived cells at a clonal density under ultralow attachment conditions. All the epidermosphere-derived cells had the capacity to self-renew in a clonal manner with, or without, the necessity for a surface to attach on. Moreover, the number of planted cells (1 or 100 per cm2) corresponded with the average number of CFUs or epidermospheres (1 or 100 per cm2) and the number of EpSCs per epidermosphere significantly increased during the cultivation period, indicating that the sphere-forming efficiency of these EpSCs approached 100%. By contrast, Dontu reported sphere formation in only 0.4% of planted mammary cells (4 per 1,000) [53]. This would also imply that the epidermospheres are 100% homogenous.
In addition, a successful bilineage differentiation was achieved toward two major skin cell types: keratinocytes within 10 days and adipocytes within 3 days. EpSC differentiation was confirmed in adhered cells as well as in suspension by CK14, CK18, Pan CK, and Wide CK expression. Since it has been previously reported that human embryonic stem cells strongly up-regulate CK18 expression during epithelial differentiation [55] and our positive control samples show the presence of CK18 in sweat gland epithelial cells (data not shown), up-regulation of this marker can be considered a reliable indication for differentiation toward epithelial lineages. Moreover, the low level of CK14 expression in the EpSCs indicated that these cells were early enough in differentiation, similar to the stage II cells during keratinocyte differentiation of human embryonic stem cells [56]. The increase in CK14 expression we observed, on the other hand, might correspond to the stage III cells detected in the study mentioned earlier, which represented more differentiated keratinocytes.
Furthermore, after EpSC differentiation, a cell population arose that stained positive for vimentin and SMA. In this regard, it has been reported that human basaloid keratinocytes [57] and equine lamellar cells [58] were positive for vimentin. In addition, up-regulation of SMA has been reported in human epidermal keratinocytes under certain conditions (hydrogen peroxide treatment) as well [59]. In our hands, the intact skin samples that were used as positive controls also contained vimentin and SMA positive epidermal and follicular cells (data not shown). Whether the positive cells in our keratinocyte differentiation culture were a keratinocyte subpopulation or displayed a myofibroblast phenotype remains to be determined.
In the present study, most of the EpSCs and several differentiated cells expressed p63, which is a transcription factor that is essential for epithelial development and proliferation as well as for epidermal differentiation and stratification [60,61]. It has been reported that more than 60% of isolated human corneal and limbal epithelial cells were positive for this marker [62]. Unlike previous findings in horse skin [63], it should be mentioned that in this study, using a rabbit polyclonal antibody directed to a mix of p63 isoforms, the expression of p63 was not restricted to the nucleus. Immunoblotting analyses later confirmed cross-reactivity of the antibody with our equine EpSCs. Dilution optimization, as well as isotype and positive controls further validated these results, revealing cytoplasmic localization (data not shown). By contrast, Carter et al. described strictly nuclear signals using mouse monoclonal antibodies directed to the delta N region of the peptide in equine skin samples [63]. It is likely that the noted discrepancies in cellular localization associated with the different antibodies arise from the differences in epitope specificity. In several studies [32,64], other markers for adult keratinocytes were used, such as CK5 and E-Cadherin; however, we were unable to evaluate the expression of these markers in or on our equine EpSCs or their differentiated progeny, due to a lack of cross-reactivity of several commercially available antibodies tested (data not shown). In this regard, it has been reported that only about 4% of human antibodies reacts with the equivalent equine epitopes [65]. Finding suitable antibodies is, therefore, a challenge in equine studies.
Adipogenic differentiation is usually described after 21 days for MSCs [5]; however, recently, adipogenesis of mammary stem/progenitor cells has been reported as soon as 5 days [66] and even 3 days in the present study. If MSCs would be stained after 3 days of adipogenic differentiation under the same circumstances, adipocytes might also be detected. In that case, the percentage of stained cells should provide more insights into this matter. It should be mentioned that adipogenic differentiation of cells derived from the epidermal basal cell layer was somehow unexpected. However, adipogenic differentiation capacities of (human amnion and chicken oviduct) epithelial cells have been previously reported by other groups as well [67,68]. Further study would be essential to demonstrate the potency of these cells and their capacity for complete tissue reconstitution in vitro or in vivo.
For a second intention of wound healing, skin grafts from other parts of the body are the treatment of choice [69]. However, accessory skin loss, infection, and difficult attachment to the underlying and surrounding tissues are major complications, reducing the chance of success of skin transplantation in horses [70 –73]. Since skin allograft rejection is a well-known risk in humans [74] as well as horses [75], skin tissue from other horses is mainly being used as a biologic dressing [76]. With regard to regenerative medicine, phase II clinical trials have been reported using allogenic MSCs to treat graft-versus-host disease in humans [77,78]. In the present study, the EpSCs were low in MHC I and negative for MHC II, reflecting low immunogenicity. In this regard, it has been reported that allogenic MSCs, which were also negative for MHC II, did not induce an immune response after intravenous and intradermal injections in horses [79,80]. Whether or not other allogenic non-EpSC cell types would have the ability to cause similar clinical improvements remains to be proved.
We observed statistically significant (P=0.0083) improvements in macroscopic wound filling after EpSC/PRP treatment. Other studies have also observed enhancement in wound healing with other stem cell classes using visual assessment [28,29]. In order to assess whether or not restitutio ad integrum (full restitution of tissue integrity and functionality) was achieved, a double-blinded histological study was performed in this study. Via such an approach, a significantly thinner granulation tissue and a more normalized diameter were apparent in all EpSC/PRP-treated wounds when compared with PRP-treated wounds. Both the more moderate dermis and granulation tissue formation in the EpSC/PRP-treated wounds indicate the formation of a tissue that more closely resembles the original intact skin (positive controls). Considerably more mature follicle-like structures could also be noticed in the EpSC/PRP-treated wounds, further demonstrating a more advanced regenerative state and tissue integrity. The follicles mentioned earlier were commonly surrounded by SMA-positive cells in all treated wounds, a feature that was absent in PRP-treated wounds.
In addition, the EpSC/PRP-treated wounds contained significantly more elastin fibers and closely resembled positive controls at 30 days post injection, whereas the PRP-treated wounds revealed only a minor elastin-like substance at the same point in time. In this regard, it has been recently reported that the amount of elastin correlates with scarless healing [81]. Indeed, elastin provides skin resistance, matrix synthesis, and supports a range of cell activities, including cell migration and proliferation [81]. Therefore, the presence and organization of elastin is a desirable trait in wound-healing evaluation. Neovascularization was evaluated by means of vWF staining, and a considerably higher average amount of blood vessels was noticed in the EpSC/PRP-treated group (31±4 vs. 23±4 per 200×). Indeed, stem cell therapy has been earlier associated with an increase in vascularization [26,27,82], and there is the possibility that an enhanced nutrition supply to the wound site also has beneficial effects on wound healing. To date, different angiogenic regulating factors have been identified: interleukin-8 [82], fibroblast growth factors [83], vascular endothelial growth factors (VEGFs) [84], angiopoietins [85], platelet-derived growth factors (PDGFs) [83], and matrix metalloproteinases [86]. In this regard, it has been described that adult stem cells (MSCs) promote angiogenesis through cytokine and protease expression [87]. In addition, it has been hypothesized that MSCs can function as pericytes, surrounding the blood vessels [88,89]. Although no such data are available for EpSCs, one might postulate that these cells exert a trophic influence on blood vessel formation. Alternatively, platelets in the PRP-treated wounds could secrete different growth factors, such as PDGF and VEGF, which also have an angiogenic effect [90]. Nevertheless, it has been reported that platelet degranulation induces pro-angiogenic signaling in epithelial cancer cell lines [91]. This might explain how the combination of EpSCs and PRP increased the vascularization in comparison to PRP only
Conclusion
In conclusion, this study is the first that reports the isolation and purification of equine EpSCs. Moreover, we describe an immunophenotypical and functional characterization of the isolated cells. In addition, a double-blinded clinical study revealed significant macroscopic as well as microscopic improvements in different wound-healing parameters after EpSC/PRP treatment.
Footnotes
Acknowledgments
This study was supported by Pell Cell Medicals. The authors wish to thank Sarah Loomans from the Department of Pathology, Bacteriology, and Poultry Diseases for her technical assistance.
Author Disclosure Statement
S.Y.B. and J.H.S. declare competing financial interests, and Pell Cell Medicals declares a patent application.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.