
Abstract
Adenosine-to-inosine (A-to-I) RNA editing is a post-transcriptional, site-specific modification process that is catalyzed by Adenosine Deaminase Acting on RNA (ADAR) gene family members. Since ADARs act on double-stranded RNA, most A-to-I editing occurs within repetitive elements, particularly Alu elements, as the result of the inherent property of these sequences to fold and form double strands. ADAR1-mediated A-to-I RNA editing was recently implicated in the regulation of human embryonic stem cells (hESCs). Spontaneous and neuronal differentiation of hESC was shown to result in a decrease in A-to-I editing levels. Knockdown of ADAR1 in hESCs results in an elevation of the expression of differentiation-related genes. In addition, we found that hESCs over-expressing ADAR1 could not be generated. The current study shows that the editing levels of induced pluripotent stem cells (iPSCs) change throughout reprogramming, from a source cell level to a level similar to that of hESCs. Up- or down-regulation of the ADAR1 level in human foreskin fibroblast (HFF) cells before induction of reprogramming results in varied reprogramming efficiencies. Furthermore, HFF-iPSC early clones derived from source cells in which the ADAR1 level was down-regulated lose their iPSC properties shortly after iPSC colony formation and instead exhibit characteristics of cancer cells. Taken together, our results imply a role for ADAR1 in the regulation of pluripotency induction as well as in the maintenance of early iPSC properties.
Introduction
R
Human embryonic stem cells (hESCs) are derived from in vitro fertilized oocytes that are grown in culture to the blastocyst stage [24]. ESCs are characterized by maintenance of the undifferentiated state during prolonged propagation in vitro, retaining a stable normal karyotype, and, most importantly, by pluripotency, the ability to differentiate, in vitro and in vivo, into potentially all cell lineages [25].
Several recent publications have reported the involvement of ADAR1 and A-to-I RNA editing in the regulation of hESCs. Chen and Carmichael showed that edited RNA escape nuclear retention in undifferentiated hESCs [26], suggesting a specific role for non-coding edited RNA in hESCs. The level of editing of Alu sequences in hESCs was reported to decrease during spontaneous differentiation toward embryoid bodies (EBs) as well as during neuronal differentiation [27]. The role of editing in the regulation of hESCs was further studied by manipulation of ADAR1 expression. ADAR1 knockdown (KD) in hESCs resulted in an increased expression of genes related to differentiation, particularly neural lineage genes [27]. We recently found that hESCs as well as human embryonal carcinoma do not over-express the p110 isoform on p110 transduction or transfection [28]. Taken together, these reports suggest a role for ADAR1 and A-to-I RNA editing in the regulation of the pluripotent state of hESCs.
The introduction of induced pluripotent stem cells (iPSCs) [29] holds great potential in personalized and regenerative medicine. iPSCs can also serve as an excellent model for studying the mechanisms underlying the stem cell pluripotent state. iPSCs are generated by ectopic expression of defined transcription factors, most commonly by Yamanaka's four factor set: OCT4, SOX2, KLF4, and c-Myc (OSKM) [29]. To further investigate the role of ADAR1 and A-to-I RNA editing in the regulation of the pluripotent state, we focused our current work on the means by which ADAR1 and RNA editing affects the induction of pluripotency. We report here that A-to-I editing within several transcripts changes during the course of reprogramming to a level similar to that of hESCs, and that ADAR1 expression level affects the frequency of iPSC colony formation. Furthermore, newly formed iPSC colonies derived from cells in which ADAR1 was down-regulated lost their pluripotent properties and instead exhibited oncogenic characteristics, suggesting a role for ADAR1 in the maintenance of the early induced pluripotent state.
Materials and Methods
Cell culture
hESC clones H9.2, I6, and I3.2, and iPSCs were cultured on a mouse embryonic fibroblast (MEF) feeder layer using standard protocols [24,30]. EBs were formed as follows: Undifferentiated hESC or human-induced pluripotent stem cell (hiPSC) colonies were detached from the feeder layer by collagenase, moved to a non-adherent petri dish, and grown in serum containing medium for 14–27 days. Additional cells and culture conditions are described in the Supplementary Materials and Methods section (Supplementary Data are available online at
Viral infection
The pTK-p110 OE construct was previously described [28]. Lentiviral virus infection was performed by transfection of constructs harboring the target transgene, packaging (Gag-Pol) and VSVG (envelope) (at a ratio of 10:9:1; 20 ng overall) by jetPEI (Polyplus transfection™) into 293T. Two days post-transfection, supernatant from 293T cells was collected, filtered through a 0.45 μ filter, and transferred to target cells. The retroviral expression construct pSUPER (PS), which expresses synthetic short interfering RNA (siRNA) [31], was designed to suppress ADAR1. siADAR1 pSUPER forward sequence:
GATCCCCCGCAGAGTTCCTCACCTGTTTCAAGAGAACAGGTGAGGAACTCTGCGTTTTTGGAA
siADAR1 pSUPER reverse sequence:
AGCTTTTCCAAAAACGCAGAGTTCCTCACCTGTTCTCTTGAAACAGGTGAGGAACTCTGCGGGG.
Retroviral viral particles were created by transfection of target plasmid into the Phoenix-Eco virus assembly line followed by infection as described earlier for lentivirus. Infected cells were subjected to 3 days of 1 μg/ml puromycin selection. An empty PS vector was used as a control.
Reprogramming
The reprogramming of human foreskin fibroblast (HFF) procedure is described in the Supplementary Materials and Methods section. The derivation of human hair follicle keratinocytes (HFKT) iPSC and human dermal fibroblast (HDF) iPSC was previously described [32]. HDF and HFKT cells were obtained from healthy individuals who signed a consent form. Procedures were performed according to approval No. 3611 by the Helsinki Committee for Experiments on Human Subjects at the Rambam Health Care Campus.
Alkaline phosphatase staining was performed using a detection kit (Millipore), according to the manufacturer's instructions. Teratoma assays were performed by an injection of iPSCs into the hind limb of NOD/SCID mice. After 10 weeks, the injected mice developed large tumors. Teratomas were dissected, fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) overnight, and embedded in paraffin wax for sub-sequential sectioning.
Immunostaining is described in the Supplementary Materials and Methods section.
RNA extraction and cDNA synthesis
Cells were harvested and total RNA were extracted using the Aurum total RNA mini kit (BIO-RAD, Hercules, CA), according to the manufacturer's instructions. Briefly, 1 μg of total RNA was reverse transcribed by iScript (BIO-RAD) according to the manufacturer's instructions, using random primers to create cDNA. All RNA samples were subjected to DNase treatment to protect against genomic DNA contamination, using the DNA-free kit (Ambion) according to the manufacturer's instructions.
Polymerase chain reaction and quantitative real-time polymerase chain reaction
Genomic DNA was extracted with the Wizard Genomic DNA purification kit (Promega, Madison, WI) according to the manufacturer's instructions. Polymerase chain reaction (PCR) was performed using DreamTaq green master mix (Fermentas). Primers are specified in Supplementary Table S1. Quantitative real-time (QRT) PCR analysis was performed in triplicate and normalized by the internal endogene GAPDH expression. The reaction was performed with either Platinum SYBR Green or TaqMan reaction mix (Applied Biosystems), and analyzed using the Relative Quantification study method. Primers for SYBR Green reaction are specified in Supplementary Table S1. TaqMan reaction primers (Applied Biosystems) were as follows: GAPDH-Hs99999905; ADAR1-p110- Hs01017596; ADAR1-p150-Hs01020780; and ADAR2 Hs 00953731.
Western blot
Western blot was performed according to standard protocols. ADAR1 primary antibody (No. K188) was a generous gift of Prof. C. Samuel, UCSB.
Editing quantification by primer extension combined with Sequenom analysis
Global RNA editing quantification was carried out using MALDI-TOF mass spectrometer using the primer extension method [33] (Sequenom; described in detail under Supplementary Materials and Methods section). Primer sequence and genomic localization are listed in Supplementary Table S2. MDM4, RBBP9, CARD11, and BRCA1 undergo editing within Alu elements that are embedded in non-coding 3′UTR or introns; and BLCAP undergoes editing within the coding sequence (editing within the site tested results in the substitution of the second amino acid of BLCAP from Y to C).
Editing detection by direct sequencing
cDNA was amplified with gene-specific primers flanking editing clusters followed by gel electrophoresis separation. The PCR fragment was excised from the gel using an extraction kit (Geneaid). The extracted PCR product was subjected to a sequencing reaction using the 3100 Genetic Analyzer (Applied Biosystems). Since Inosine is reverse transcribed as Guanosine, the sequence histogram analyzed by Sequencher 4.7 (Gene Codes Corporation) shows both A and G peaks in an edited site. When the reverse direction is sequenced, Thymine and Cytosine peaks are received. Editing frequency is determined as the % ratio of the G peak over the sum of G and A peaks in the sequencing chromatogram. Peak areas were quantified by the DS Gene program (Accelrys). Primers are listed in Supplementary Table S1.
Affimetrix array processing and analysis
Expression data microarray analysis was performed with the Affymetrix Hu Gene 1.0st oligonucleotide arrays (url1). Gene level RMA sketch algorithm (Affymetrix Expression Console and Partek Genomics Suite 6.2) was used for crude data generation. Detailed array processing is described in the Supplementary Materials and Methods section. Genes that changed by at least twofold were filtered. Further processing included functional analysis by Ingenuity Pathways Analysis (
Matrigel implants assay
The formation of tumors in vivo was evaluated using a xenograft model of Matrigel implants. 1×106 HFF-AKD cells were re-suspended in 200 μL phenol-red free matrigel (BD Biosciences; 1:1) on ice. Matrigel mixture was then injected subcutaneously into immune-deficient NOD/SCID 8–12 week-old mice (two injection sites for each mouse). Tumors were removed 8–10 weeks post-injection, fixed in 4% PFA, and sectioned.
Soft agar colony formation assay
A bottom layer of agar containing 1.2 mL of 0.6% low melting agar (Sigma) dissolved in hESC media was poured into wells of a six-well cell culture dish and allowed to set at 4°C for 20 min. HFF-AKD colonies were trypsinized to a single-cell suspension, counted, and resuspended in 0.3% of low melting agar dissolved in hESC media. A top layer (1 mL) containing 5×103 cells was poured over the first layer and allowed to set at 4°C for 20 min. hESC medium (2 mL) was added and replaced every 3 days. After 21 days, colonies were fixed, stained for 1 h with 0.005% crystalviolet, and incubated with PBS overnight to remove excess crystal violet. RL95-2 endometrium carcinoma cell line (ATCC;
Results
A-to-I editing levels of Alu elements in iPSC differ from those of their source cells
Reprogramming of somatic cells into PSCs requires substantial changes in their intrinsic properties. These include changes in the gene expression program and major epigenetic changes, including ablation of the DNA methylation pattern, chromatin remodeling, and telomerase elongation (reviewed in [34]). We, therefore, set out to examine whether A-to-I RNA editing levels also change throughout the reprogramming process, a question that has not yet been addressed. By comparing the editing levels of iPSC clones with those of their respective somatic sources, and with those of hESCs, we monitored changes in editing occurring throughout the reprogramming of somatic cells to iPSCs.
For this comparison, we used iPSCs that had been previously created in our lab [32,35], which were reprogrammed by ectopic expression (mediated by both retro and lentiviral infection) of the Yamanka's OSKM factors from three types of source cells: HFF, HFKT and HDF. We analyzed 5–6 iPSC clones from each group; two of the HFKT-iPSC clones were free of viral segments after excision of the integrated viral cassette.
To assess changes in editing levels that occurred during reprogramming, we tested seven genes previously confirmed to undergo editing in multiple clustered sites within Alu elements in hESCs and other cell types [27,28].
By means of Sequenom MassArray, we tested single sites of nucleotide substitution in five of the genes (Fig. 1A). The editing levels of three genes RBB9, CARD11, and MDM4 were low in all cell types examined, ranging from 4% to 9%. However, in all three comparison groups, the editing levels were higher in the iPSCs compared with their parental source (Fig. 1A). BRCA1 editing varied between source cells exhibiting high levels in HFF and HFKT (58% and 92% respectively), and lack of editing in HDF. Levels of editing of HDF-iPSCs and HFF-iPSCs were ∼30% higher than those of their parental sources, while HFKT-iPSCs exhibited lower levels of editing than did HFKT parental cells (∼85%; Fig. 1A). Editing levels of HDF-iPSCs and HFF-iPSCs were ∼10% (from ∼12% to ∼22%) higher in BLCAP than in their parental source cells, while the HFKT-iPSC BLCAP editing level did not differ from that of its source cells.
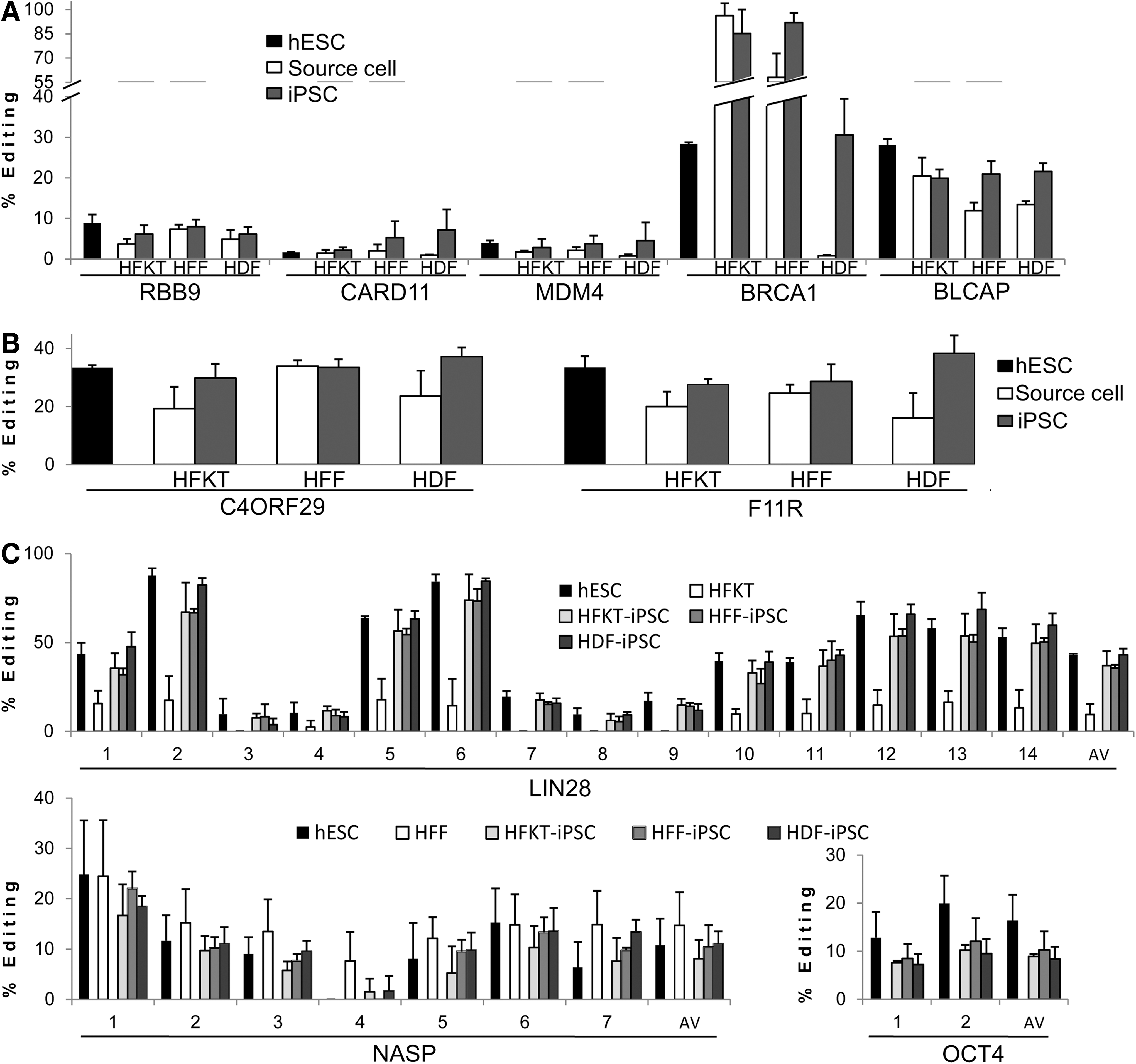
Adenosine-to-Inosine RNA editing levels of Alu elements in induced pluripotent stem cells (iPSCs) differ from those of their source cells, and resemble those of human embryonic stem cells (hESCs). RNA editing level analysis was performed on three groups of reprogramming source cells [human hair follicle keratinocytes (HFKT), human foreskin fibroblast (HFF), and human dermal fibroblast (HDF)] and three respective iPSCs (HFKT-iPSC, HFF-iPSC, and HDF-iPSC). Mean levels and standard deviations of RNA editing for the three samples of source cells, hESCs, and 4–6 clones of each iPSC group are presented. Editing levels of hESC lines H9.2 and I6, as presented in
In two additional representative genes, F11R and C4ORF29, we performed by direct sequencing, a more detailed analysis of multiple edited sites within an edited cluster (Fig. 1B). For the C4ORF29 editing cluster, the combined mean editing levels of ten sites in HFKT-iPSC and HDF-iPSC were higher than in their respective source cells by 10% and 14%, respectively (Fig. 1B). In contrast, HFF-iPSC C4ORF29 editing levels were similar to those of the HFF parental cells (Fig. 1B). Importantly, consistently higher editing levels in HFKT-iPSCs and HDF-iPSCs, compared with their respective source cells, were measured for all ten sites of the cluster that was analyzed (Supplementary Fig. S1). Similar consistency was observed in F11R; the editing levels of eight sites in all three iPSC groups were higher than in their respective source cells (Fig. 1B and Supplementary Fig. S1). Exceptions were observed at a few sites, at which the editing levels of source cells and iPSCs were similar (HFF to HFF-iPSC comparison, sites 2 and 4, and HFKT-iPSC site 7; Supplementary Fig. S1).
Overall, for the three comparison groups analyzed, the mean editing levels were statistically different (P<0.001 according to Wilcoxon signed-rank test) for iPSCs compared with source cells in 19 out of 21 comparison pairs [the two exceptions being BLCAP in HFKT-iPSCs (Fig. 1A) and C4ORF29 in HFF-iPSCs (Fig. 1B)]. Furthermore, for six of the seven genes tested, the mean editing levels of iPSCs were higher than for their respective source cells. The only exception was the editing level of BRCA1 in HFKT-iPSCs. However, the observation that this gene was almost completely edited in the HFKT source cells may explain the opposite trend. The observed consistency in the elevation of editing levels in iPSCs compared with their source cells suggests that this is a general phenomenon occurring during reprogramming rather than gene or site specific.
The A-to-I editing level of hiPSCs resembles that of hESCs regardless of their origins
The editing levels of hiPSCs, in all three iPSC groups of different origins, approached those of hESCs in five of the seven genes analyzed (with the exception of HFKT-iPSCs BLCAP editing; Fig. 1A, B). In CARD11, editing in source cells was similar to that of hESCs, and the observed editing levels for the three iPSC cell types were higher than those of hESCs (Fig. 1A). A more complex trend was observed in the BRCA1 gene. In HDF-iPSCs, the editing level was similar to that of hESCs; while that of HFF-iPSCs was higher than that of the respective source cell, and, thus, more distinct from the hESC editing level (Fig. 1A). In contrast, the decrease in the editing level in HFKT-iPSCs was toward the hESC editing level. Nevertheless, for BRCA1, the HFKT-iPSC editing level remained very high compared with that of hESCs (Fig. 1A).
For a further comparison of A-to-I editing levels of iPSCs and hESCs, we examined genes that are related to hESC pluripotent state regulation. Edited clusters within three pluripotent genes were selected. The reprogramming factor LIN28, a key regulatory factor in the maintenance of pluripotency [36], was previously reported to undergo A-to-I editing in hESC [26]. We identified editing targets in an Alu repeat within an intron of OCT4, the master regulator of pluripotency [37], and in nuclear autoantigenic sperm protein (NASP), an H1 histone-binding protein, which was reported to exhibit highly conserved expression in ESC across species [38]. We confirmed that the level of editing for these three transcripts does, in fact, change during hESC spontaneous differentiation (Supplementary Fig. S2), as was previously demonstrated for other genes in hESC [27].
The mean editing levels of the three pluripotency-related genes in all iPSC groups were similar, regardless of their origin, and lower than the hESC editing level (Fig. 1C). Interestingly, LIN28 was also expressed in HFKT, and editing was observed in some of the 14 sites analyzed. However, the editing levels of these sites were lower than those of the iPSCs (Fig. 1C). For NASP editing, no significant differences were observed between hESCs and iPSCs. NASP was also expressed in HFF, and editing levels were higher in the HFF source cells than in hESCs and HFF-iPSCs (Fig. 1C). For OCT4, the average editing level was lower in iPSCs than in hESCs (Fig. 1C). As expected, the expression of OCT4 was not observed in any of the source cells.
Taken together, for 7 of the 10 genes analyzed (RBB9, MDM4, CARD11, F11R, C4ORF29, LIN28, and NASP) and for one of the two edited sites in OCT4, no statistically significant differences between hiPSC and hESC editing levels were found (Mann–Whitney test U>0.05). For BRCA1 and BLCAP, differences between hESC and iPSC editing levels were statistically significant (Mann–Whitney test U<0.05).
We suggest that, on the whole, A-to-I RNA editing levels are altered during reprogramming, and that editing levels in iPSCs resemble respective editing levels in hESCs, regardless of their parental source.
ADAR1 expression in hiPSCs is elevated compared with that of source cells and hESCs
Recent reports suggest that the editing of Alu sequences in hESCs is highly dependent on ADAR1 [27]. We found elevated expression levels of ADAR1-p110 transcript and protein in each of the HFKT, HFF, and HDF sourced iPSC clones analyzed compared with their source cells (Fig. 2). Interestingly, ADAR1-p110 protein expression was higher in most iPSC clones than in the H9.2 hESC clone. The elevated expression of ADAR-p110 in iPSCs correlates with the elevation in the Alu sequence editing level observed in most of the genes tested in iPSCs (Fig. 1).

The adenosine deaminase acting on RNA (ADAR)1-p110 expression in human-induced pluripotent stem cells (hiPSCs) is elevated compared with that of source cells and hESCs. The expression level of ADAR1-p110 in hiPSCs was compared with that in source cells and in hESCs.
ADAR1-p110 expression level affects reprogramming efficiency
The elevated expression of ADAR1-p110 protein in iPSCs, as well as the changes observed in A-to-I editing levels in iPSCs compared with their source cells, may result indirectly from the global changes that the source somatic cells undergo during reprogramming. Our goal, therefore, was to examine whether ADAR1 and A-to-I RNA editing have a functional role in the induction of pluripotency. We designed a system in which the level of the ADAR1 enzyme was down- or up-regulated in HFF cells; these cells were then induced to pluripotency by infection with retroviruses carrying the OSKM factors. A schematic illustration of the experiment design is presented in Fig. 3A.
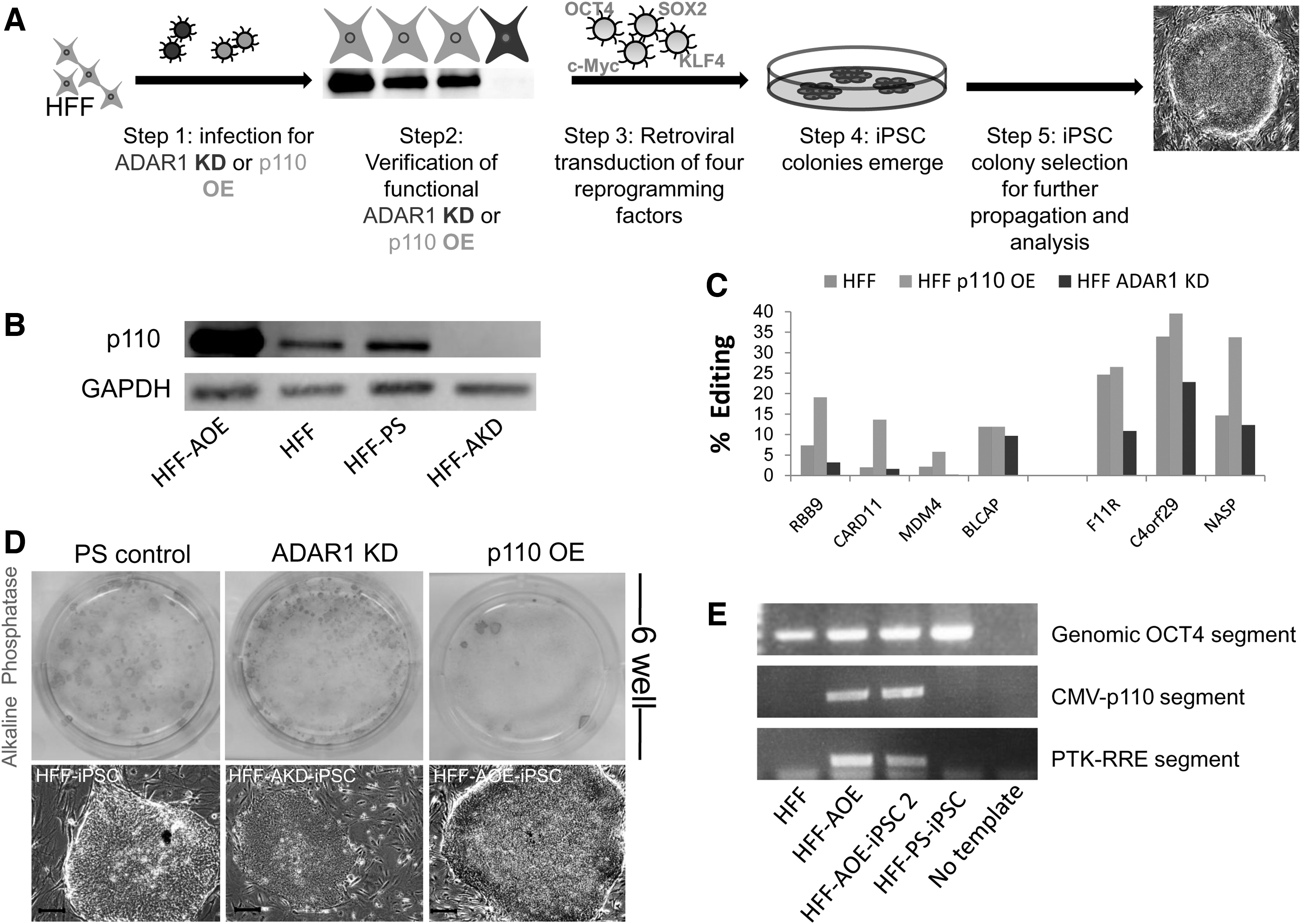
iPSC colonies emerged from source cells in which the ADAR1 level was manipulated. The ADAR1 level in HFF source cells was down- or up-regulated, followed by induction of pluripotency. iPSC colonies emerged about 21 days later. Different ADAR1 treatments generated variable colony formation efficiency.
For down-regulation of ADAR1, HFF cells were infected with a retrovirus expressing siRNA sequence targeted against ADAR1 (an empty vector PS was used as a control). To up-regulate ADAR1, we infected HFF cells with a lentivirus harboring the ADAR1-p110 isoform under the CMV promoter [green fluorescent protein (GFP) was used as a control]. Successful OE and KD of the ADAR1-p110 isoform in HFF cells was confirmed by western blot (Fig. 3B). To ensure a functional effect of ADAR1 OE or KD (increased or decreased editing levels, respectively), we examined the A-to-I RNA editing levels of seven genes. Five of these genes exhibited decreased editing levels after ADAR1 KD (Fig. 3C). BLCAP and CARD11 exhibited minor to no decrease (less than 2%). After ADAR1-p110 OE, more than 10% elevation in editing levels was observed in RBB9, CARD11, and NASP. Between 4% and 6% elevation was detected in MDM4 and C4ORF29, and no alterations in editing levels were detected in F11R and BLCAP.
Next, the four types of source HFF cells were simultaneously infected with the OSKM reprogramming factors. As we and many others reported, small, tight non-iPSC colonies (which seem to comprise of incomplete reprogrammed, or transformed cells) first appeared at 10–14 days post-infection. Colonies resembling hESC morphology emerged 16–21 days post-infection (Fig. 3D). Interestingly, in the OE treatment culture, almost no non-iPSC colonies emerged and ESC-like colonies appeared later then they did with control treatments.
The efficiency of ES-like colony formation was measured by counting colonies with hESC-like morphology that were also positive for live TRA-1-60 staining [39]. The average efficiency, measured in three separate experiments (two repeats in each experiment), is presented in Table 1. The highest efficiency was observed for the KD treatment (Fig. 3D; Table 1), and the lowest (by 10-fold) efficiency was observed for the OE treatment (Fig. 3D; Table 1). The control PS and GFP/untreated efficiencies, as well as the KD treatment (Fig. 3D; Table 1), resembled our previous results with this reprogramming system [32], and concur with other reports [34]. Four to six clones (for each treatment) of the TRA-1-60-positive ES-like colonies were mechanically picked and clonally expanded. Other TRA-1-60-positive ES-like colonies from each treatment were mechanically picked and grown non-clonally in a pool. HFF-PS-iPSC control clones and HFF-AOE-iPSC clones were grown under hESC culture conditions; their ES-like morphology was preserved for more than 20 passages. Since each iPSC clone originates from a single HFF cell, we verified that the HFF-AOE-iPSC-obtained clones were derived from HFF-AOE cells and not from a possible rare cell which was not infected by the ADAR1-p110 OE virion, by ensuring insertion of the CMV-p110 segment to the HFF-AOE-iPSC genome (Fig. 3E). Some clones from each treatment were tested for their pluripotent properties by analyzing pluripotent marker expression (Supplementary Fig. S3), and for their ability to differentiate into derivatives of the three germ layers, both in vitro by spontaneously differentiating into EBs (Supplementary Fig. S4) and in vivo by an injection into NOD/SCID mice and teratoma formation (Supplementary Fig. S4). This ensured that for each treatment, bonafide iPSCs were obtained.
Colonies that exhibit hESC-like morphology and which are TRA-1-60 positively stained are considered true iPSCs and are counted from the total number of infected source cells. Results are reported as the mean±SD of at least three experiments.
P<0.05 versus PS control (two-tailed student t-test).
ADAR, adenosine deaminase acting on RNA; SD, standard deviation; GFP, green fluorescent protein; PS, pSUPER; OE, over-expression; KD, knockdown; hESC, human embryonic stem cells; iPSCs, induced pluripotent stem cells.
Most iPSC clones originating from HFF-AKD did not maintain their iPSC properties
Surprisingly, in 8 of the 11 HFF-AKD-iPSC clones (clones from three separate experiments, which were successfully propagated from a single mechanically picked colony to a full populated well) and in all HFF-AKD-iPSC pools (six pools), small colonies that comprised loosely packed cells appeared between passages 4 and 8 (3–6 weeks; Fig. 4A). Two to three passages after their appearance, these new cells took over the culture, with no reminisce of the hESC-like iPSC colonies.
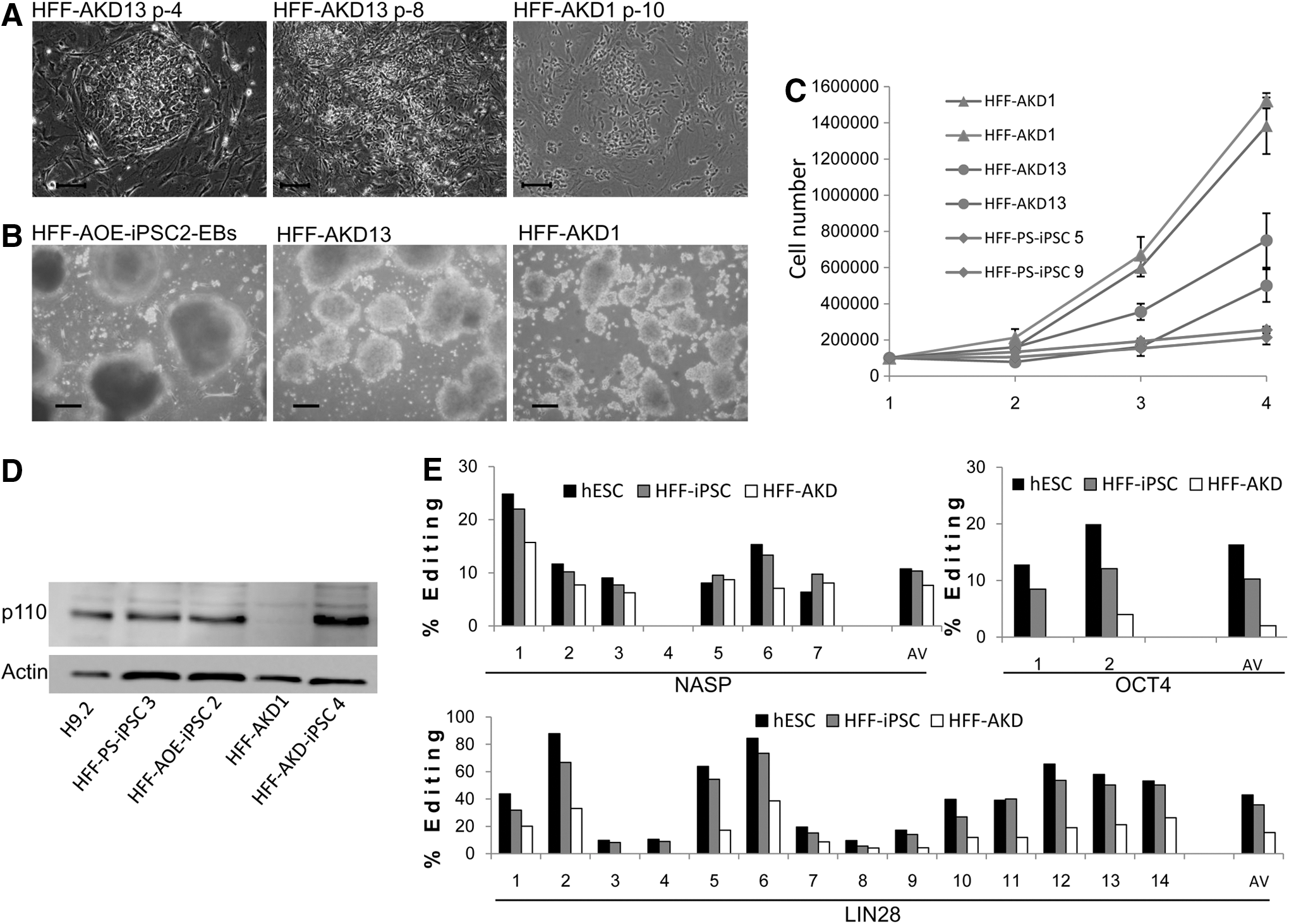
iPSC clones originating from HFF-AKD do not maintain their iPSC properties. HFF-AKD-derived iPSC clones lost their ES-like morphology, did not differentiate into typical EBs, and proliferated very aggressively.
The transformed cells grew in hESC conditions, retaining small spread colony formation and proliferating very rapidly both on MEF feeder and in feeder-free conditions. The HFF-AKD transformed population grew by ∼2.5- to 3-fold every 24 h until vessel size limited its expansion (Fig. 4C). In contrast, for HFF-PS-iPSC clones, population doubling time was about 48 h. This measured HFF-PS-iPSC proliferation rate resembles the population doubling time of 36–48 h reported in the literature for hESCs and iPSCs [29,30]. The only pluripotent marker expressed by the transformed cells was OCT4 (Supplementary Fig. S3). After enzymatic detachment, the transformed HFF-AKD colonies formed small tight floating bodies that did not exhibit typical EB morphology (Fig. 4B), and did not develop, form cysts, or differentiate (Fig. 4B and Supplementary Fig. S4). In addition, an injection of the HFF-AKD cells into the hind limb of NOD/SCID mice did not result in teratoma formation. Such morphology transformation and loss of iPSC features was not observed in any of the control cells and not in many of our previous reprogramming experiments, either with HFF source cells or with any other source cells. To exclude the possibility that the observed transformation of the HFF-AKD cells occurred due to genomic instability originating from the reprogramming processes, we performed G-banding karyotype analysis of HFF-AKD representative clones and of one of the pools. We found no chromosomal abnormalities (Supplementary Fig. S5).
Some HFF-AKD-iPSC clones did not undergo the transformation described earlier. These clones were grown for more than 20 passages and exhibited standard iPSC properties such as pluripotent marker expression (Supplementary Fig. S3) and in vitro and in vivo differentiation into the three germ layers.
ADAR1-p110 isoform protein levels in iPSCs derived from different ADAR1 level HFF sources were similar in all iPSC clones measured by western blot compared with H9.2, including the HFF-AOE-iPSC clones and the HFF-KD-iPSC clone 4 that retained its iPSC morphology (Fig. 4D). In contrast, ADAR1-p110 was almost undetected in the transformed clones. Accordingly, the editing level of three hESC related genes (discussed in Fig. 1C) showed a consistent decrease in multiple sites (Fig. 4E). The fact that ADAR1 protein expression was not silenced in the HFF-AKD-iPSC clones which did not undergo transformation strongly supports the notion that the down-regulation of ADAR1 was involved in the transformation of HFF-AKD-iPSC clones.
HFF-AKD-transformed clones exhibit similar expression patterns, which are distinct from those of iPSCs
As a first step in the investigation of the transformations of the early HFF-AKD-IPSCs, we compared the expression profile of two HFF-AKD clones, 1 and 13 (clones from separate experiments), with an early passage (p-5) HFF-PS-iPSC control clone (which represents a pre-transformation state), by means of Affymertix microarray analysis (Supplementary Fig. S6A). The expression profile of the two HFF-AKD clones differed considerably from that of the control clone. The HFF-AKD1 clone expressed 3,408 genes and the HFF-AKD13 clone expressed 1,191 genes, for which there were more than twofold differences in expression level compared with the control iPSC clone (Supplementary Fig. S6A). Of the genes that were differentially expressed more than twofold, 938 were common to both HFF-AKD clones. This shows a significant overlap of genes that were affected in the HFF-AKD clones (P-value<10−16, Chi-squared test) (Supplementary Fig. S6B; genes are listed in Supplementary Table S3). Of these overlapping genes, 559 genes were up-regulated in both HFF-AKD clones compared with the HFF-PS-iPSCs, and 365 genes were down-regulated. These results suggest that a common transformation process occurred in both clones. Verifying the microarray results for several genes by QRT (Supplementary Fig. S6C), we found a similar trend for expression levels to that observed by the microarray. Of note, expression levels of the five genes examined were similar for the HFF-PS-iPSC clone and the hESC clone H9.2 (Supplementary Fig. S6C).
To functionally characterize the 938 genes that were differentially expressed by more than twofold in both HFF-AKD tested clones compared with an HFF-PS-iPSC control clone, we performed functional analysis using the Ingenuity Pathway Analysis software. The genes were found to be associated with several functional classes and diseases. Of the 10 most over-represented biological functional groups (presented in Supplementary Fig. S6D), cancer-related genes comprised the largest group. Notably, many pluripotency-associated genes were down-regulated in HFF-AKD clones compared with the control clone (genes are listed in Table 2).
HFF-AKD exhibit cancer-related properties both in vivo and in vitro
Some cancer cell lines have been reported to express very low levels of ADAR1 [22]. Based on their morphology, high proliferation rate, low ADAR1 levels, their inability to differentiate, and their expression of cancer-related genes, we speculated that HFF-AKD-iPSC clones underwent a kind of oncogenic transformation. To further test this hypothesis, we investigated the ability of HFF-AKD clones to form colonies in an anchorage-independent manner by the soft agar colony-formation assay, which is a common method for the detection of malignant transformation. The RL95-2 endometrium carcinoma cell line was used as a positive control. Both HFF-AKD and the RL95-2 clones formed colonies in similar efficiencies 21 days post-embedding in the soft agar (Fig. 5A). In contrast, the control HFF-iPSC cells did not proliferate in the soft agar (Fig. 5A), concurring with reports for other human iPSC clones [40].
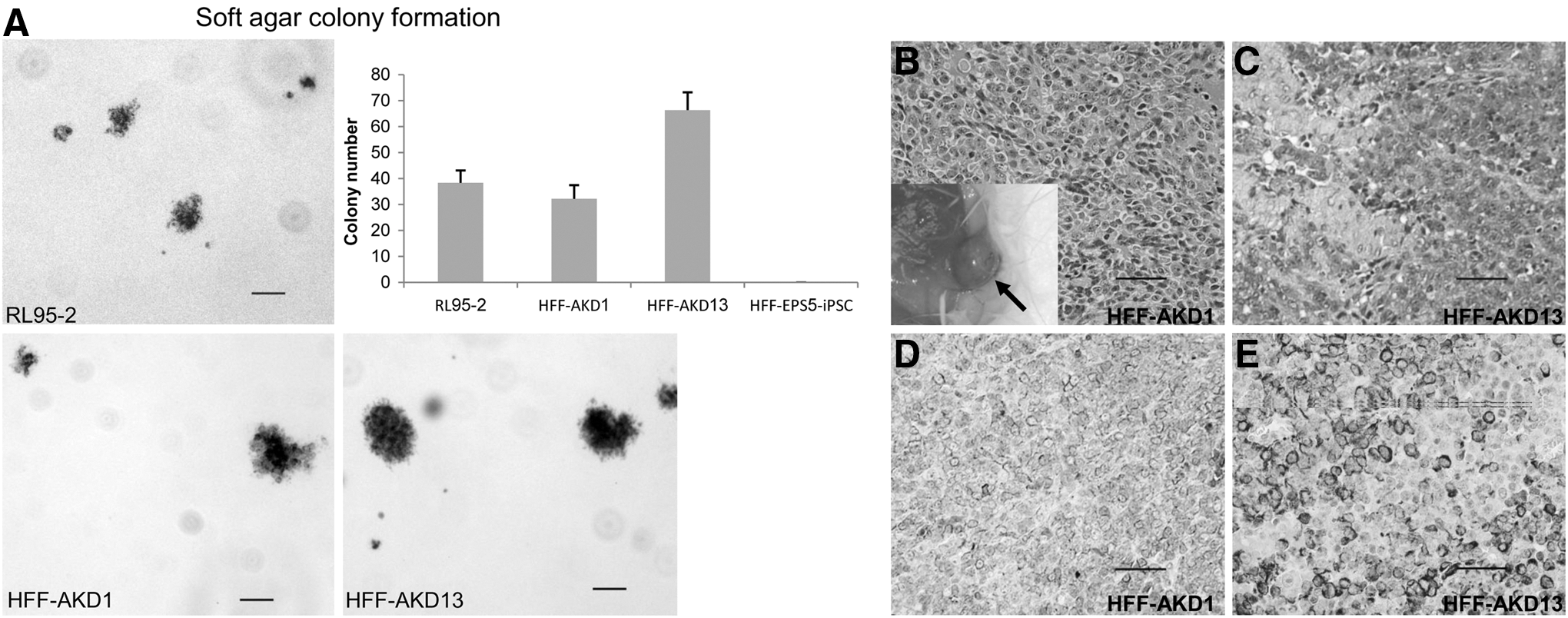
HFF-AKD-transformed clones exhibit cancer-like characteristics in vitro and in vivo. The transformed HFF-AKD clones 1 and 13 were further tested for cancer characteristics both in vitro by the soft agar colony formation assay and in vivo by a subcutaneous injection into NOD/SCID mice
A subcutaneous injection of matrigel-embedded hESC cells was reported to enhance the frequency of tumor formation [41]. At 8–10 weeks after a subcutaneous injection of 1×106 HFF-AKD cells embedded in 200 μL matrigel into both sides of the back of NOD/SCID mice, large vascularized tumors appeared at both injection sites in five out of seven mice (Fig. 5B; three out of four for HFF-AKD1 and two out of three for HFF-AKD13). The two other injected mice died a few weeks earlier and were excluded from the experiment. Histological analysis revealed that the tumors were mostly undifferentiated with necrotic areas (Fig. 5C, D). We further characterized the tumors and found that they stained positive for vimentin (Fig. 5D) but not for epithelial carcinoma markers such as p63 and cytokeratin (not shown). Taken together, these data suggest that the early HFF-AKD-iPSC clones could not maintain the pluripotent state and underwent oncogenic transformation.
Discussion
Recently, accumulating evidence attests to widespread differences in RNA and DNA sequences across the human transcriptome [42,43]. Though the exact extent of RNA editing is still a matter of debate, adenosine to inosine (represented as guanosine by sequencing) appears to be the major editing change occurring in RNA sequences, distinguishing them from the genomic sequences from which they were transcribed.
We and others recently reported the involvement of ADAR1 and A-to-I RNA editing in the regulation of PSCs [26 –28]. Since we were unable to generate any ADAR1-p110 over-expressing hESC or embryonal carcinoma clones [28], we suggested that the ADAR1 level is tightly regulated in PSCs.
The emergence of the field of iPSCs offers a new, very useful, and robust model for studying the mechanisms regulating to pluripotency, self-renewal, and differentiation. To further test our hypothesis of the involvement of ADAR1 and A-to-I editing in the regulation of pluripotency, we examined whether the induction of pluripotency by reprogramming entails a change in A-to-I RNA editing levels. The finding that the general level of A-to-I RNA editing of Alu sequences in hiPSCs changes throughout the reprogramming process (Fig. 1A, B) is consistent with previous reports that showed A-to-I editing levels to be tissue or cell-type specific, rather than identical across different tissues of a certain individual [27,28,44]. Thus, changes from one type of cell to another (i.e., reprogramming) result in changed editing levels. For most of the genes analyzed in the current study, editing levels in hiPSC clones were higher than those of their respective source cells, and resembled hESC editing levels, regardless of their parental source (Fig. 1). In contrast, most iPSC clones expressed higher levels of ADAR1-p110 than did hESCs (Fig. 2). This supports the notion that the level of ADAR1 expression does not necessarily correlate with the level of editing (e.g. higher NASP editing levels in HFF than in HFF-iPSC while ADAR1-p110 protein expression was higher in HFF-iPSC). Taken together, these findings suggests that gene-specific editing levels are important characteristics of PSCs.
This study showed differences between the A-to-I editing levels of hiPSCs and hESCs. One example is the higher editing levels of BRCA1 in HFF-iPSCs and in HFKT-iPSCs compared with hESCs. Though iPSCs were initially thought to be very similar to ESCs [45,46], differences in expression profile [47,48] and in epigenetic status [49,50] have been observed between them. Epigenetic memory [49,50] retained from source cells has been suggested as a possible explanation for these differences. Similarly, differences found in the current study in the editing levels of some iPSC and ESC targets could be due to the retaining of gene-specific editing levels from source cells. It has been suggested, however, that iPSCs become more similar to ESCs with prolonged passaging [47,48,50]. Accordingly, in the current study, the editing levels of most targets examined in post-excision iPSC clones and in iPSC clones cultivated for longer periods (later passages) appeared more similar to the editing levels of hESCs than did pre-excised and iPSC clones of earlier passages (data not shown).
Since A-to-I editing level alteration may be an indirect result of massive changes occurring during reprogramming, we examined whether ADAR1 has a functional role in the process of pluripotency induction. We, therefore, knocked down ADAR1 or alternately over-expressed the ADAR1 isoform p110 in HFF cells that were subsequently used as source cells for reprogramming. We achieved an inverse ratio of ESC-like colony formation at the ADAR1 level. OE of ADAR1-p110 resulted in significantly lower reprogramming efficiency. In contrast, KD of ADAR1 resulted in slightly higher efficiency of iPSC colony formation than in control cells. A number of studies have suggested that proliferation rate is a major roadblock during the onset of the reprogramming process: Induction of higher proliferation rates in source cells increases the chances for a successful reprogramming fate [51,52]. In addition, OE of ADAR1 in glioma cell lines [22] and of ADAR2 in astrocytoma cell lines [23] was shown to inhibit proliferation rates, suggesting that A-to-I editing may be involved in the regulation of proliferation. Thus, the lower reprogramming efficiency observed for HFF-AOE, and in contrast, the slightly higher efficiency of reprogramming of HFF-AKD cells, could be due to changes in proliferation rate.
Analyzing the ADAR1-p110 protein level revealed that the iPSC-AOE clones did not retain elevated expression of p110 (Fig. 4C). This concurs with our previously unsuccessful attempts to achieve ADAR1-p110 OE in hESCs, and further supports our hypothesis that the ADAR1 level is tightly regulated in PSCs [28]. The non-elevated p110 level in the iPSC-AOE clones strongly suggests that high levels of ADAR1 may interfere with the reprogramming process or the maintenance of early generated iPSCs. Therefore, only a small number of cell clones that have low expression levels of the p110 transgene can undergo reprogramming and survive (which may also explain the lower efficiency of HFF-AOE reprogramming). Another possible explanation for the non-elevated p110 level in the iPSC-AOE clones is that an unknown mechanism down-regulates expression levels of p110 in early iPSCs.
We cannot rule out the possibility that inserted CMV promoter-derived p110 expression was silenced in the newly formed iPSC clones, as the silencing of exogenous CMV promoters is associated with prolonged passaging of PSCs. However, we believe that this is the less likely explanation, as in an earlier study we were not able to establish ADAR1-p110 OE in PSCs by short period assays (28).
Most of the newly formed HFF-AKD-iPSC clones began losing their ESC-like morphology after 4–8 passages (Fig. 4A). These cells exhibited very low levels of ADAR1-p110 expression (Fig. 4D) and decreased editing levels (Fig. 4E). Thus, it is tempting to speculate that the low levels of ADAR1 expression are responsible for the iPSC transformation. Some of the HFF-AKD-iPSC clones retained iPSC properties for a long period, which concurs with them retaining a high level of ADAR1-p110 expression. This correlation between the efficiency of ADAR1 KD and the process of transformation further supports the notion that ADAR1 activity is essential for the maintenance of reprogrammed iPSCs. The exact stage at which the depletion of ADAR1 caused instability of the HFF-AKD cells is not clear. The fact that hESC resembling colonies (that also stained for TRA-1-60) were formed from HFF-AKD cells and with an efficiency which is only slightly different from that of control cells suggests that ADAR1 depletion affected the cells in the HFF-iPSC (early) stage rather than during the reprogramming from HFF. Nevertheless, further experiments such as ADAR1 KD at different time points during and after reprogramming should be conducted to further elucidate the ADAR1 role in the induction of pluripotency and the maintenance of iPSC.
The heterogeneity of ADAR1 silencing between different clones can be explained by epigenetic mechanisms. Each clone represents a different integration site of the shRNA transgene, which might be prone to different epigenetic effects. For example, if the shRNA transgene is integrated near or within a condensed hetrochromatin region, its expression level will be low and the KD efficiency of the clone will also be low. Alternatively, if the transgene is integrated within an “open,” transcription permissive chromatin region, high expression of the shRNA transgene can be expected [53].
In previous studies in which ADAR1 was down-regulated, inducible knockout of ADAR1 in mouse hematopoietic stem cells resulted in apoptosis, demonstrating an essential role for ADAR1 in stem cell maintenance [54]. In contrast, ADAR1 KD in hESCs [27] and knockout in mouse ESCs [8,9] did not have a dramatic effect on ESC survival or on the loss of pluripotent properties. These data suggest that ADAR1 and/or editing in itself may have different roles in different types of stem cells.
A-to-I RNA editing is much more abundant in humans than in other mammals [55]. Thus, it is possible that in contrast to mESCs, human pluripotent cells require “correct” levels of ADAR1 protein expression to sustain normal growth. Reprogramming to a pluripotent state is a prolonged process that does not end with iPSC colony formation [47,48]. It is possible that appropriate high levels of ADAR1 expression are essential in early iPSCs (but not in hESCs), as iPSCs are not yet fully reprogrammed to an ESC state. Further characterization of HFF-AKD properties versus those of control cells, such as apoptosis rate, epigenetic state, and microRNA expression, can contribute to our understanding of stem cell regulation by ADAR1 and RNA editing. In addition, ADAR1 KD in other types of source cells and rescue of ADAR1 depletion by OE, at different time points during and after reprogramming, will be also important for a more comprehensive understanding of the editing role of ADAR1 and RNA in iPSCs.
Evidence is growing that malignant tumors are initiated and maintained by a population of tumor cells which share biological properties similar to those of normal stem cells [56]. This “cancer stem cell” model is based on the observation that tumors arise from cells which share many of the particular properties of stem cells, such as self-renewal and transcriptional programming [57]. A number of studies have shown additional links of iPSCs to cancer [58,59], by showing that the p53 tumor suppressor pathway is a barrier to reprogramming and by supporting a functional link between tumor suppression and reprogramming.
The HFF-AKD transformed clones exhibit some characteristics of cancer: We found large differences in RNA transcription between two transformed HFF-AKD clones and an early HFF-PS-iPSC control clone. A function analysis of the common genes that were differentially expressed by more than twofold in both clones suggests a connection to cancer and metastasis (Supplementary Fig. S6D). The HFF-AKD cells proliferated very rapidly and did not differentiate in vitro. A subcutaneous injection of HFF-AKD cells into NOD/SCID mice resulted in the formation of undifferentiated tumors. These tumors also stained positive for vimentin, which is known to be expressed in most types of sarcoma. This is not surprising, considering the fibroblastic origin of the tumors. HFF-AKD clones exhibited additional cancer-like properties such as formation of colonies in an anchorage-independent manner in soft agar (Fig. 5A).
A recent work comparing reprogramming and formation of oncogenic foci (a method for in vitro production of tumor cells) suggests that these are related processes [60]. The authors proposed a model in which reprogrammed fibroblasts first acquire changes that lead to down-regulation of the differentiated state, followed by subsequent divergence of pathways into either an oncogenic or a pluripotent phenotype, depending on factors such as the expression of pluripotency genes. Based on our results, we suggest that low ADAR1 expression level may divert early iPSCs in the oncogenic direction rather than along a path of “normal” reprogramming to pluripotency.
Our findings support the involvement of ADAR1 in pluripotent state regulation. The mechanism for such involvement is yet to be revealed. ADAR1 can affect PSCs by site-selective editing of a specific gene (or a limited number of genes), while normal activity requires a certain editing state. Another possibility is that a general level of promiscuous non-selective editing is needed for normal pluripotent cell survival. ADAR1 may also affect reprogramming and regulate pluripotency in an editing-independent manner; for example, ADAR1 and ADAR2 mutated in their catalytic domain were also shown to regulate expression and small RNA biogenesis through double-stranded RNA binding [61,62]. Another recent study showed that ADAR1 facilitates loading of microRNA onto RNA-induced silencing complexes by forming heterodimers with DICER [63]. This suggests an additional, editing-independent mechanism, by which ADAR1 can regulate hPSCs. In all scenarios, the further study of the involvement of ADAR1 and A-to-I RNA editing in the regulation of pluripotent cells is of great interest.
Conclusion
We found that A-to-I RNA editing levels of hiPSCs resemble those of hESCs regardless of the hiPSC parental source cell. In addition, down- or up-regulation of the editing enzyme ADAR1 in HFF cells, which were subsequently reprogrammed, resulted in varied reprogramming efficiencies. Furthermore, newly formed hiPSC colonies derived from cells with down-regulated expression of ADAR1 exhibit oncogenic characteristics. This suggests an important role for ADAR1 in the regulation of PSCs. Future studies should elucidate the mechanisms by which ADAR1 regulates PSCs.
Footnotes
Acknowledgments
This work was conducted in the Berlin Family Laboratory for Stem Cell and Tissue Regeneration Research at the Sohnis and Forman Families Center for Stem Cell and Tissue Regeneration Research, Ruth and Bruce Rappaport Faculty of Medicine, Technion—Israel Institute of Technology (Haifa, Israel). The authors wish to thank Dr. Ayelet Dar for her help with the in vivo experiments and for her comments on this article. This work was supported in part by a grant from the Flight Attendant Medical Research Institute (FAMRI) and by Technion Research and Development Foundation (TRDF). J.I.-E. holds the Sylvia and Stanley Shirvan Chair in Cell and Tissue Regeneration Research at the Technion - Israel Institute of Technology. G.R. holds the Djerassi Chair in Oncology at the Sackler Faculty of Medicine, Tel-Aviv University. This work was performed in partial fulfillment of the requirements for a PhD degree (to S.O.), Sackler Faculty of Medicine, Tel-Aviv University.
Author Disclosure Statement
The authors indicate that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.