
Abstract
Mesenchymal stem cells (MSC) are multipotent and possess high proliferative activity, and thus are thought to be a reliable cell source for cell therapies. Here, we isolated MSC from adult tissues—bone marrow (BM-MSC), dental tissue (DT-MSC), and adipose tissue (AT-MSC)—to compare how autotransplantation of these MSC effectively supports the repair of bone fracture and ischemic tissue. An analysis by in vitro differentiation assays showed no significant difference among these MSC. The degree of calcification at the joint region of bone fracture was higher in mice transplanted with AT-MSC than in mice transplanted with BM-MSC or DT-MSC. To compare the abilities of MSC, characterize how those MSC affect the repair of ischemic tissue, vascular occlusion was performed by ligation of the femoral artery and vein. Of note, the blood flow in the ischemic region rapidly increased in mice injected with AT-MSC, as contrasted with mice injected with BM- or DT-MSC. The number of CD45- and F4/80-positive cells at the femoral region was higher in AT-MSC recipients than in recipients of BM-MSC or DT-MSC. We evaluated the mRNA expression of angiogenic and migration factors in MSC and found the expression of CCL5 mRNA was higher in AT-MSC than in BM-MSC or DT-MSC. Transplantation of AT-MSC with impaired expression of CCL5 clearly showed a significant delay in the recovery of blood flow compared with the control. These findings have fundamental implications for the modulation of AT-MSC in the repair of vasculature and bone fracture.
Introduction
M
Since no single MSC-specific cell surface marker is known, MSC are isolated from various tissues by their capacity to adhere to cell culture dishes and by a combination of several surface markers [5]. Recently, various cell surface markers have been used in attempts to identify putative MSC. A profile of the more consistently reported markers is CD44, human leukocyte antigen (HLA)-ABC, STRO-1, CD73, CD90, CD105, CD120a, and CD146 positive and CD14, CD34, CD45, and HLA-DR negative [6]. It was reported that bone marrow-derived MSC (BM-MSC) can be isolated using the anti-CD271 antibody [7]. In addition, analysis of CD271-positive cells revealed CD140b, CD340, and CD349 expression [7]. Other studies report analysis of BM-MSC found in SSEA-1 and SSEA-4 expression [8,9].
MSC have also shown a tendency to improve tissue damage following injury or disease. The transplant of BM cells can improve serum albumin levels in the peripheral blood of patients with liver cirrhosis [10]. Furthermore, it has been reported that MSC have the potential for improving ischemia. MSC have been used for cell therapy to treat cardiac tissue damaged by myocardial infarction [11]. MSC engraft into the injured myocardium and conceivably differentiate into cardiac myocytes [12]. Transplanted MSC improve ischemia in animal models [13,14]. Enhancing angiogenesis and peripheral blood flow, MSC are incorporated into the site of angiogenesis after tissue ischemia in a limb [15].
MSC can be isolated not only from BM, but also from dental tissue (DT), adipose tissue (AT), umbilical cord blood, peripheral blood, tendon, and placenta [16 –18]. DT-MSC were identified as odontogenic precursor cells, which are similar to BM-MSC, have high proliferative potential, and are able to regenerate dentin [19]. AT-MSC exist in white fat, such as subcutaneous fat and omental fat.
Procedures to isolate MSC from AT and DT are less invasive than those required to obtain samples from BM [20]. Ease of isolation is an important point to consider for clinical application of cell therapy. In addition, MSC from adipose or dental tissue are more useful for autotransplantation than MSC from BM. There are few reports comparing MSC derived from different adult human tissues [21]. Our results contribute a more detailed characterization of the three MSC subtypes BM-MSC, DT-MSC, and AT-MSC, contributing to the usefulness of therapy using these cells.
It has been reported that MSC secrete angiogenic factors, including vascular endothelial growth factor (VEGF), hepatocyte growth factor, placental growth factor, fibroblast growth factor-2 (FGF-2), transforming growth factor beta (TGF-β), and angiopoietin (Ang)-1. Through these factors, MSC can support neovascularization in ischemic regions [22,23]. The chemokine system is also involved in the regulation of angiogenic responses, including the control of migration, activation, proliferation, and apoptosis of leukocytes and EC [24,25]. Chemokines influence wound healing, inflammatory diseases, and tumor growth. CXC and CC chemokine families play roles in the modulation of angiogenesis [26 –28]. The CC chemokine CCL5/RANTES has been shown to be involved in several chronic inflammatory conditions [29,30]. It is poorly understood whether CCL5 acts directly in angiogenesis.
In this study, we demonstrate that AT-MSC express the highest level of CCL5 compared with BM- and DT-MSC, and furthermore, knockdown expression of CCL5 in AT-MSC show impaired recovery form bone fracture and vascular occlusion. Thus, we propose that chemokine/chomokine receptor pathway is highly involved in MSC-based therapy and the characteristics of MSC derived from various tissues should be investigated carefully before use for clinical application.
Materials and Methods
Isolation of primary cell cultures from human tissue
With permission from the ethics authorities at the University of Tsukuba, human samples were collected. Human AT was rinsed with phosphate-buffered saline (PBS) and minced with scissors and scalpels into less than 3 mm pieces. These pieces were treated with 0.1% collagenase (Nitta Gelatin, Osaka, Japan) in PBS, 20% fetal bovine serum (FBS; Hyclon, South Logan, UT) for 45 min at 37°C, and filtered through a 100-μm nylon mesh (BD Biosciences, San Jose, CA). After incubation, samples were centrifuged to remove the collagenase solution. The isolated cells were cultured in the Iscove's modified Dulbecco's medium (IMDM) (Invitrogen, Carlsbad, CA)/10% FBS/2 mg/mL
The BM samples were treated with HetaSep (StemCell Technologies, Vancouver, BC) and RosetteSep (StemCell Technologies) solutions as described previously [17]. Treated samples were put onto a density gradient buffer (Histopaque 1,083 g/cm3; Sigma-Aldrich, St Louis, MO) and centrifuged at 1,500 rpm for 20 min at room temperature. Cells were collected from the middle layer and then cultured by the same method as used for AT-derived samples, described above.
The dental samples were harvested from the dental root. The root tissue was gently separated from the crown. The tissues were digested in a solution of 0.1% collagenase for 45 min at 37°C. Single-cell suspensions were obtained by passing the cells through a 100-μm nylon mesh. Cells were isolated and cultured by the same method as used for AT-derived samples, described above.
These cells were maintained in a 25-cm2 culture flask (Sumitomo Bakelite, Osaka, Japan) at 37°C in 5% CO2 and in a humidified atmosphere. The culture medium was replaced with a fresh medium twice a week. After isolated cells reached a subconfluent state, they were harvested with 0.05% trypsin–EDTA (Invitrogen) and purified for CD31−/CD45− cells by FACS (MoFlo; Beckman Coulter, Brea, CA) to remove ECs and hematopoietic cells. Frozen cell stocks were prepared using the Cell Banker (Zenoaq, Koriyama, Japan) solution and stored in liquid nitrogen for further experiments. All experiments were performed using at least three different donors.
Flow cytometry
After BM-, AT-, and DT-MSC were expanded several passages, the surface markers were analyzed using flow cytometry (FACSCalibur; BD Biosciences). MSC were stained with various combinations of monoclonal antibodies conjugated with fluorescein isothiocyanate (FITC), phycoerythrin (PE), and allophycocyanine (APC): FITC-labeled HLA-ABC (W6/32), FITC-labeled CD90 (5E10), PE-labeled anti-CD31 (WM59), and APC-labeled anti-CD45 (HI30) (BioLegend, San Diego, CA); FITC-labeled anti-CD105 (SN6) (AbD Serotec, Oxford, United Kingdom); PE-labeled anti-CD166 (3A6), anti-CD73 (AD2), anti-CD14 (M5E2), and APC-labeled anti-CD34 (581) and anti-mouse IgG (BD Biosciences).
In vitro differentiation assay of MSC
For the osteogenesis experiment, 2×104 cells were plated in four-well plates (Nalge Nunc; Nunc, Rochester, NY) and treated with the osteogenic differentiation medium for 3 weeks. The osteogenic differentiation medium consisted of the IMDM supplemented with 1% FBS, 0.1 mM dexamethasone (Sigma-Aldrich), 10 mM β-glycerol-2-phosphate (Sigma-Aldrich), 0.2 mM ascorbic acid (Sigma-Aldrich), and 50 ng/mL human EGF (Wako, Osaka, Japan) [17]. The culture medium was replaced with a fresh medium once a week. The formation of mineralized matrix was revealed by staining with Alizarin Red S (Kodak, Rochester, NY) solution for 5 min. Then, cells were dissolved with 0.2 N HCl (Wako) and 5% sodium dodecyl sulfate, and the absorbance at 480 nm was measured.
For adipogenesis experiments, 2×104 cells were plated in four-well plates (Nalge Nunc) and treated with the adipogenic differentiation medium for 3 weeks The adipogenic differentiation medium consisted of the IMDM supplemented with 10% FBS, 0.1 mM dexamethasone (Sigma-Aldrich), 0.5 mM 3-isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich), 2 mg/mL insulin (Wako), and 0.1 mM indomethacine (Sigma-Aldrich). The culture medium was replaced with a fresh medium once a week. The adipogenic cultures stained with the Oil Red O solution (Muto Pure Chemicals, Tokyo, Japan) for 30 min at 42°C. After the staining, cells were dissolved with 4% IGPAL CA630 (Sigma-Aldrich) in isopropanol, and the absorbance at 492 nm was measured.
For chondrogenic experiments, 2.5×105 cells were plated in 96-well spheroid plates (Sumitomo Bakelite) and treated with the chondrogenic differentiation medium for 4 weeks. The chondrogenic differentiation medium consisted of the IMDM supplemented with 1% FBS, 0.1 mM dexamethasone (Sigma-Aldrich), 1 mM sodium pyruvate (Invitrogen), 0.25 nM ascorbic acid, 50 mg/mL ITS premix (BD Bioscience), 40 mg/mL proline (Sigma-Aldrich), 10 ng/mL TGF-β1 (Wako), and 10 ng/mL BMP-2 (Wako). The culture medium was replaced with a fresh medium once a week. For microscopy, the spheroids were embedded in OCT-compound (Tissue Tek, Tokyo Japan), cut into 5 μm sections, and stained with Toluidine Blue (Muto Pure Chemicals).
Analysis of MSC in bone fracture mouse model
Animal experiments were carried out in a humane manner with approval from the Institutional Animal Experiment Committee of the University of Tsukuba and in accordance with the Regulations for Animal Experiments of our university and Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under justification of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
A mouse model to investigate bone healing was produced as previously described [17]. Briefly, adult C57/BL6 mice were anesthetized (tribromoethanol; 125 mg/kg) and fractures were made at the middle of the femur. The fractured femurs were connected with pins. Using a 27G needle, an incision was made at the site where bones were reconnected.
MSC (5×105) were seeded on 2×2 mm Gelfoam scaffolds (Pfizer, New York, NY) and incubated at 37°C for 90 min before transplantation to mice. The cell-seeded Gelfoam construct was fixed at the site, where bones were fractured and reconnected. Immunosuppression was performed by intraperitoneal injection of cyclosporin-A (Wako) at 20 mg/kg body weight 2 days before the operation [17]. The injection of cyclosporin-A was continued every second day for the entire period of the assay. X-ray examination of mice was performed at 28 days after transplantation. The density of bone formation in the bone gap area shown in X-ray images was measured with NIH-Image software as described previously [17].
For histological staining, bones were fixed with 4% paraformaldehyde (Wako) for 3 days and decalcified with the Plank-Rychlo solution (Muto Pure Chemicals) for 20 days. Paraffin-embedded samples were cut into 7-μm-thick sections and stained with the hematoxylin–eosin solution (Muto Pure Chemicals).
Mouse vascular occlusion model
Adult C57/BL6 mice underwent unilateral femoral artery and vein ligation. These arteries and veins were ligated with 6-0 silk sutures from the proximal end of the femoral vessels to the popliteal vessels [18], while nerves were carefully kept unchanged. After 24 h, injection of MSC was performed intramuscularly by injecting cells, resuspended in 100 μL PBS, at four different sites of the ischemic leg. Instead of the injection of MSC, recombinant CCL5 (100 pg/mouse; R&D Systems, McKinley Place NE, MN) was injected intramuscularly every day for 2 weeks. Blood flow was measured using a laser Doppler blood flow meter (FLO-C1; Omegawave, Tokyo, Japan) at an appropriate day. Data obtained was the ratio of blood flow in the ischemic limb site divided by that in the nonischemic site.
Mice were analyzed for angiogenesis 2 weeks after transplantation of MSC to the region of hindlimb ischemia. The capillary density at the thigh muscle was assessed by immunofluorescence using Banderiraea simplicifolia lectin I-TRITC (0.1 mg/mL; Sigma-Aldrich) as reported previously [31]. The frozen sections were mounted and observed under a microscope equipped with the appropriate filters (Olympus, Tokyo, Japan). The number of capillaries in the muscle was measured in 10 different randomized fields for each mouse. Frozen sectioned samples were obtained 7 days post-transplantation and fixed with 4% paraformaldehyde (Wako). The sections were stained with anti-mouse CD45 antibody (MEC13.3) (BD Bioscience) and F4/80 (A3-1) (AbD Serotec). The number of CD45-positive cells and F4/80-positive cells was scored in distinct sections derived from different mice.
Localization of transplanted MSC
Localization of MSC into the region of hindlimb ischemia was analyzed 1 week after transplantation. MSC were conjugated with PKH67 (Sigma-Aldrich) and the capillaries were detected using lectin I-TRITC (Sigma-Aldrich). Frozen section samples were obtained 7 days post-transplantation and were fixed with 4% paraformaldehyde. Transplanted MSC were analyzed using confocal microscopy (TCS SP5; Leica, Solms, Germany).
Quantitative reverse transcription (RT)-PCR
Total RNA (1 μg) was reverse transcribed using an RT-PCR kit (Toyobo, Osaka, Japan) as described previously [17]. Resulting cDNA was amplified by GeneAmp PCR System 9100 (Applied Biosystems, Foster City, CA) for 23–35 cycles using 95°C for 5 s, 60°C for 30 s, and 72°C for 30 s. The reaction mixtures for quantitative polymerase chain reaction (PCR) were prepared using POWER SYBR® Green PCR master mix (Applied Biosystems) and analyzed by a 7700 Sequence Detector (Applied Biosystems). The data DDCt method was used to calculate relative quantification. Experiments were performed in triplicate. The sequence of primer sets used for the PCR reactions are shown in Table 1.
FGF, fibroblast growth factor; TGF-β, transforming growth factor beta; VEGF, vascular endothelial growth factor; IGF, insulin-like growth factor; MMP, matrix metalloproteinase; CXCR4, C-X-C chemokine receptor type 4.
shRNA system
We used the shRNA MISSION lentiviral transduction system (Sigma-Aldrich). Infected cells were selected using 2 μg/mL puromysin (Wako). After puromysin selection, expression levels of CCL5 were analyzed by quantitative PCR.
Microscopy analysis
Cell samples were viewed with an Olympus IX71 microscope system (Olympus) using UPlanF objectives lenses at 4x/0.13PhL and 10x/0.30Ph1. Sample slides were viewed with an Olympus BX51 microscope system (Olympus) using UPlanSApo objectives lenses at 4x/0.16PH, 10x/0.40PH, and 20x/0.75PH (Olympus) and mounting reagent (Muto Pure Chemicals). Data acquisition was performed using a DP70 digital camera attached to the microscope and DP controller software (Olympus). Images were processed using Adobe Photoshop version 8.0 software (Adobe System, San Jose, CA).
Statistical analysis
Statistical evaluations of data were conducted using the Student's t-test for per-comparison analysis. Data are presented as mean±SD.
Results
Characteristics of MSC from BM, DT, and AT
After adherent cells were collected from each tissue, CD45-negative and CD31-negative cell fraction was selected to remove hematopoietic cells and EC. Cells derived from each tissue had fibroblastic morphology (Fig. 1A). Subsequently, we used an Alizarin Red staining assay to examine the ability of these cells to differentiate toward the osteogenic lineage. Each type of MSC was able to differentiate toward the osteogenic lineage; there was no significant difference among the cell types (Fig. 1B). We also examined the capacity of the cells for adipocyte and chondrocyte differentiation by Oil Red O staining (Fig. 1C) and Toluidine Blue staining (Fig. 1D), respectively. These results showed that the differentiation ability of cells derived from BM, DT, and AT are at nearly the same level.
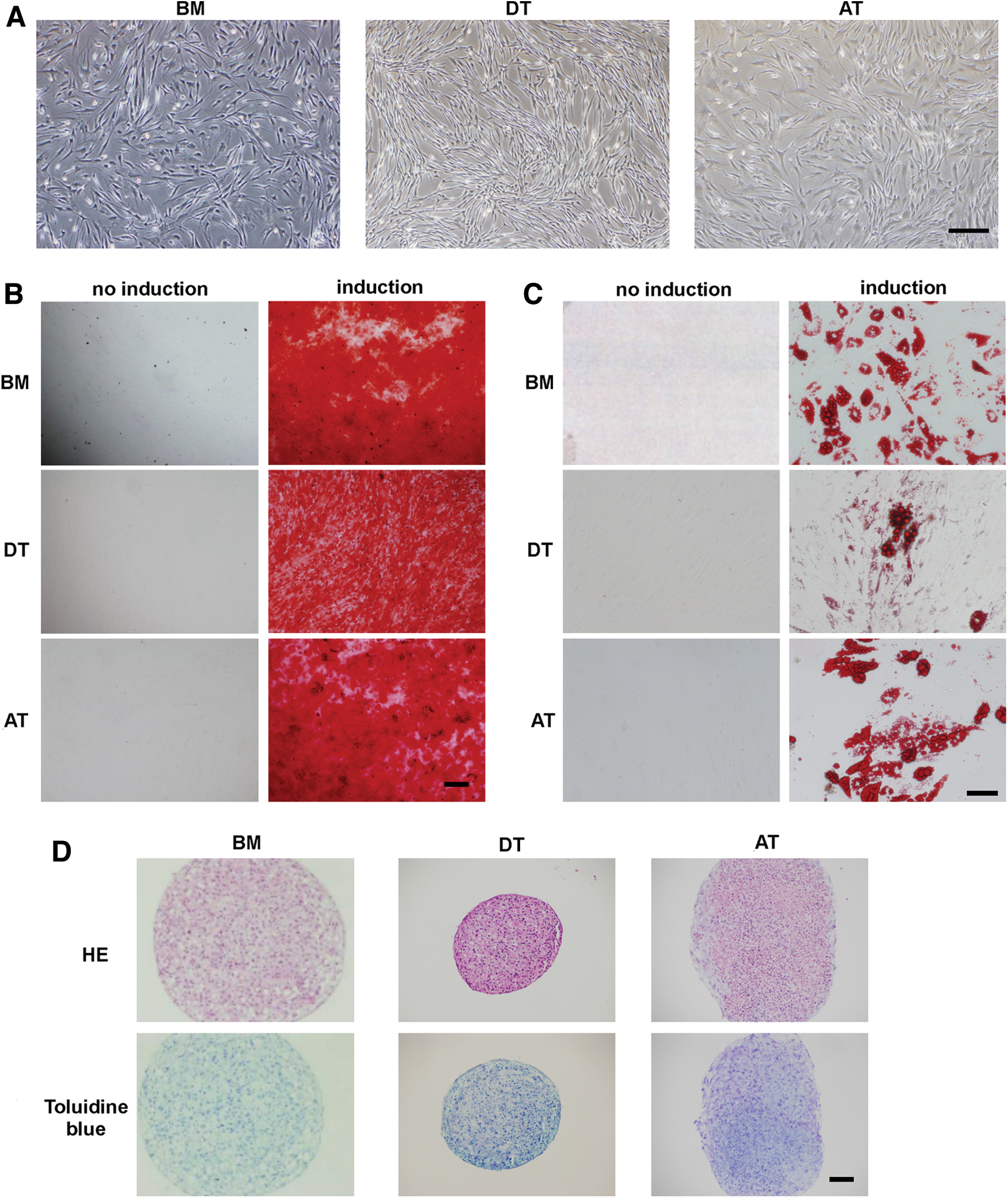
Isolation of mesenchymal stem cells (MSCs) from human bone marrow (BM), dental tissue (DT), and adipose tissue (AT).
Then, we examined cell surface markers by flow cytometry (Fig. 2). We found that cells selected were negative for CD10, CD14, CD24, CD31, CD34, CD36, CD38, CD45, CD49d, CD117, CD133, and HLA-DR and positive for CD13, CD29, CD44, CD73, CD90, CD105, CD166, and HLA-ABC, as described previously for MSC [18]. We could not discern any distinct characteristics exhibited by the MSC derived from different tissues.

Analysis of cell surface marker expression. FACS analyses of cell surface markers on these MSC. BM-
The effects of each MSC engraftment on bone repair
We transplanted BM-, DT-, and AT-MSC into immunosuppressed mice with surgically fractured femurs and examined the processes of bone repair (Fig. 3) [17]. At 4 weeks post-transplantation, new bone formation was evaluated by the degree of calcification revealed by X-ray. The defect without implantation of cells, which was injected with PBS as a control, was defective in bone formation. On the other hand, MSC engraftment resulted in the infiltration of mononuclear cells, including inflammatory blood cells and osteocytic cells at the implantation site. These results showed that implanted MSC can stimulate host-derived mesenchymal cells and inflammatory cells into the region of implantation.
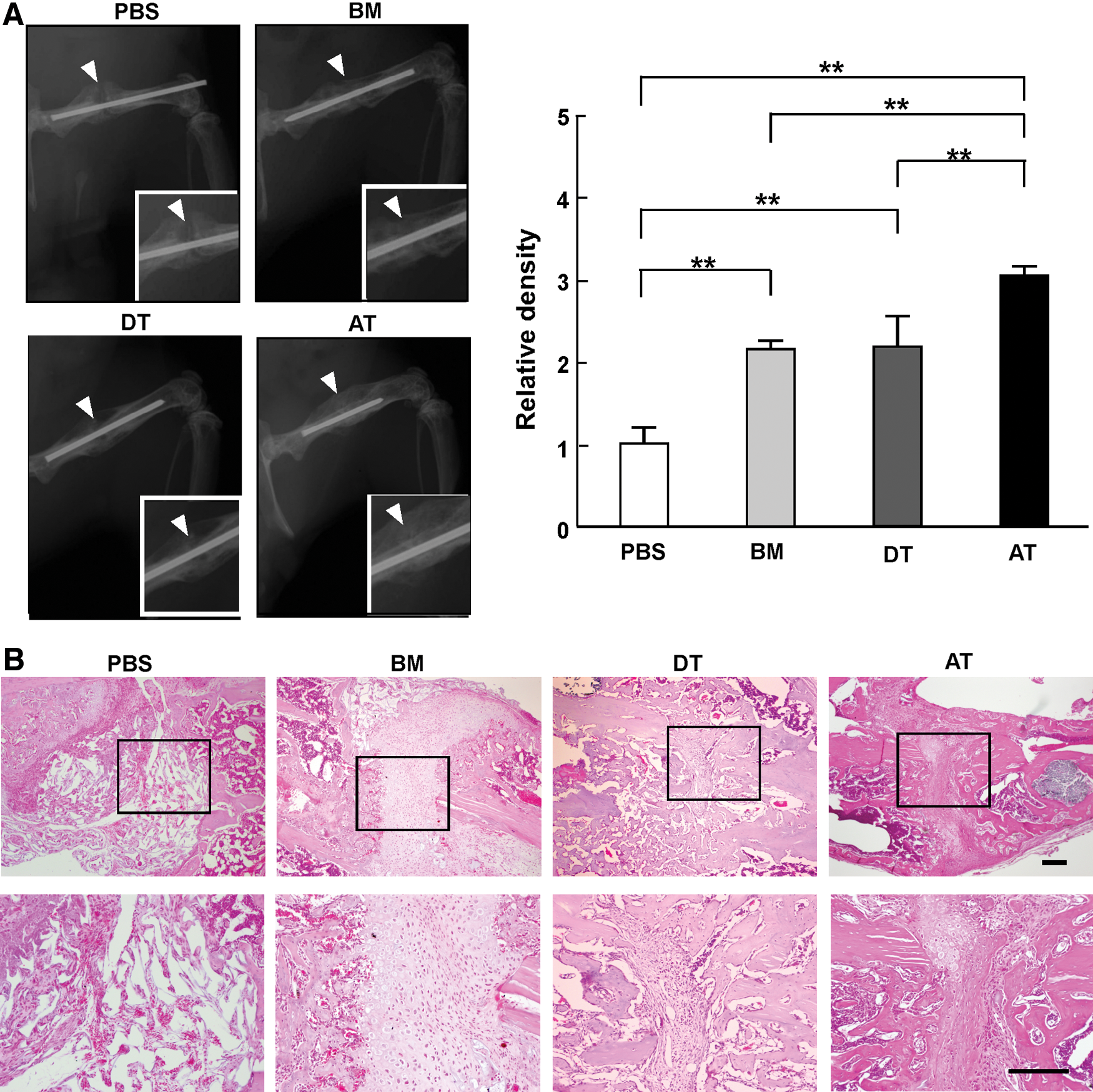
Repair of bone fracture by transplantation of each MSC.
Bone calcification was evaluated using X-ray radiographic images (Fig. 3A). X-ray examination revealed that the degree of calcification at the joint of each fractured femur was significantly higher in mice that received MSC transplantation compared with mice injected with PBS (PBS: 1.0±0.2; BM-MSC: 2.2±0.1; DT-MSC: 2.2±0.4; AT-MSC: 3.1±0.1; **P<0.01). Of note, transplantation of AT-MSC promoted bone calcification at the defect site more highly than did BM-MSC and DT-MSC.
Furthermore, histological analysis showed prominent lamellar formation in the joint regions when MSC were transplanted (Fig. 3B). The formation of lamellar bone on pre-existing hyaline cartilage represents bone substitution during the repair process. In mice that received a transplantation of AT-MSC, lamellar bone abounded in the central region of the joint, indicating that transplantation of AT-MSC accelerated bone substitution and repair in this bone fracture model.
Transplantation of AT-MSC showed significant arterio/angiogenic effects following vascular occlusion
We made a vascular occlusion model in which the proximal end of the femoral vessel was ligated with the popliteal vessel. A previous study showed that blood flow recovery in an ischemic region occurs initially through arteriogenesis, a process defined as the formation of functional collateral arteries from pre-existing arterio-arteriolar anastomoses [32]. This process begins by 1 week after femoral vessel occlusion [33]. After arteriogenesis, blood flow of the ischemic region is further improved by angiogenesis, which is characterized by the sprouting of new capillaries from pre-existing vessels.
One day after ligation of femoral artery, each type of MSC was transplanted intramuscularly into the ischemic region. Blood flow was measured at the ankle of the ischemic limb over 14 days (Fig. 4A). The blood perfusion of the ischemic hindlimb was evaluated by comparing values from the ischemic and nonischemic limbs. Greater improvement of hindlimb ischemia was observed in the AT-MSC-treated groups as compared with the PBS-injected control group. At day 7 and 10 after the transplantation, the relative blood flow was significantly higher in AT-MSC groups than in PBS groups (day 7, PBS: 0.5±0.1; AT-MSC: 0.8±0.1, day 10, PBS: 0.7±0.1; AT-MSC: 0.9±0.1, *P<0.05, **P<0.01). In addition, recovery of the hindlimb blood flow was higher in AT-MSC groups as compared with DT-MSC groups at day 5 and 7 (day 5, DT-MSC: 0.3±0.1; AT-MSC: 0.6±0.2, day 7, DT-MSC: 0.5±0.1; AT-MSC: 0.8±0.1, *P<0.05, **P<0.01). On day 14 following the vascular occlusion, blood flow of the ischemic region improved and resembled the blood flow at the nonischemic site, even in control mice [34].
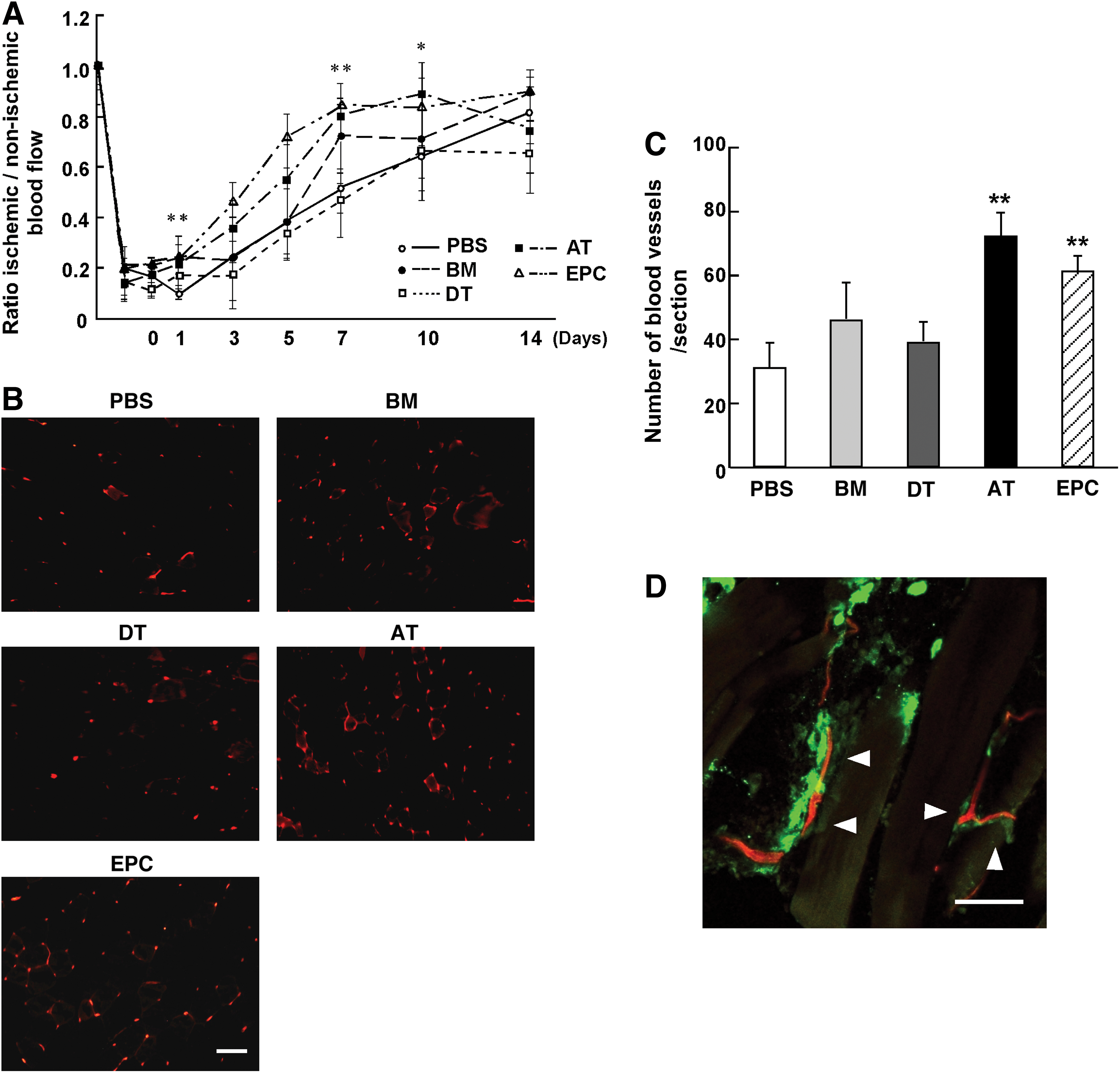
Analysis of the angiogenic effects of MSC.
A higher number of lectin-binding EC was observed at the femoral region in the AT-MSC and endothelial progenitor cell (EPC) [31] (positive control) groups on day 14 (Fig. 4B). In contrast, EC were sparsely distributed in BM-MSC, DT-MSC, and PBS groups (PBS: 29.4±7.3; BM-MSC: 46.9±10.2; DT-MSC: 36.9±5.9; AT-MSC: 68.3±7.0; EPC: 57.9±4.9, **P<0.01). The EC of the femoral region are considered to contribute to angiogenesis by supplying the blood flow to the peripheral region. Overall, these data strongly indicate that AT-MSC possess a higher ability to support re-endothelialization than do BM-MSC and DT-MSC.
To investigate how MSC affect recovery of ischemic tissue, PKH67-labeled AT-MSC were injected into the muscle. One week after the injection, a higher frequency of AT-MSC survived in the muscle than BM-MSC and DT-MSC (BM-MSC: 0.16%±0.11%; DT-MSC: 0.75%±0.24%; AT-MSC: 3.89%±0.16%, P<0.05: BM-MSC vs. DT-MSC, P<0.01: BM-MSC vs. AT-MSC and DT-MSC vs. AT-MSC). In addition, AT-MSC were closely located around the capillary; however, they were not incorporated into the vessels (Fig. 4D).
Migration of monocyte and macrophage
Monocyte extravasation is an early event in inflammatory responses [35]. Circulating monocytes are recruited in response to distress signals generated by the hypoxia of ischemic tissues. Monocytes and macrophages migrate along defined chemotactic gradients. They accumulate within avascular and necrotic hypoxic areas of ischemia and localize to the hypoxic environment [36 –38]. The local hypoxic microenvironment influences migration and recruitment of monocytes and macrophages by upregulating the expression of EC adhesion molecules and chemoattractants, including chemokines [39]. The migrated monocytes and macrophages also secrete several angiogenic factors and promote angiogenesis in the ischemic region [39].
To investigate how MSC affect the migration of monocytes and macrophages in ischemic regions, we first examined monocyte and macrophage migration at day 7 after ligation and transplantation of MSC using anti-mouse CD45 and anti-mouse F4/80 antibody (Fig. 5). A higher number of CD45-positive cells migrated to ischemic regions in the AT-MSC groups than in PBS, BM-MSC, and DT-MSC groups (PBS: 10.8±1.7; BM-MSC: 19.7±6.0; DT-MSC: 9.7±5.0; AT-MSC: 29.0±8.1; EPC: 52.2±2.3, Fig. 5A, C, *P<0.05). This was also confirmed by FACS analysis (BM-MSC: 24.8%±2.4%; DT-MSC: 14.6%±2.0%; AT-MSC 43.9%±2.2%, P<0.01: BM-MSC vs. DT-MSC, BM-MSC vs. AT-MSC, and DT-MSC vs. AT-MSC). In addition, the number of F4/80-positive cells was the higher in the AT-MSC groups than in other groups (PBS: 5.7±2.4; BM-MSC: 9.9±1.6; DT-MSC: 3.0±1.5; AT-MSC: 11.4±3.4; EPC: 14.0±3.5, Fig. 5B, D, *P<0.05, **P<0.01). These results suggest that AT-MSC might be able to promote angiogenesis by better facilitating recruitment and migration of monocytes and macrophages.
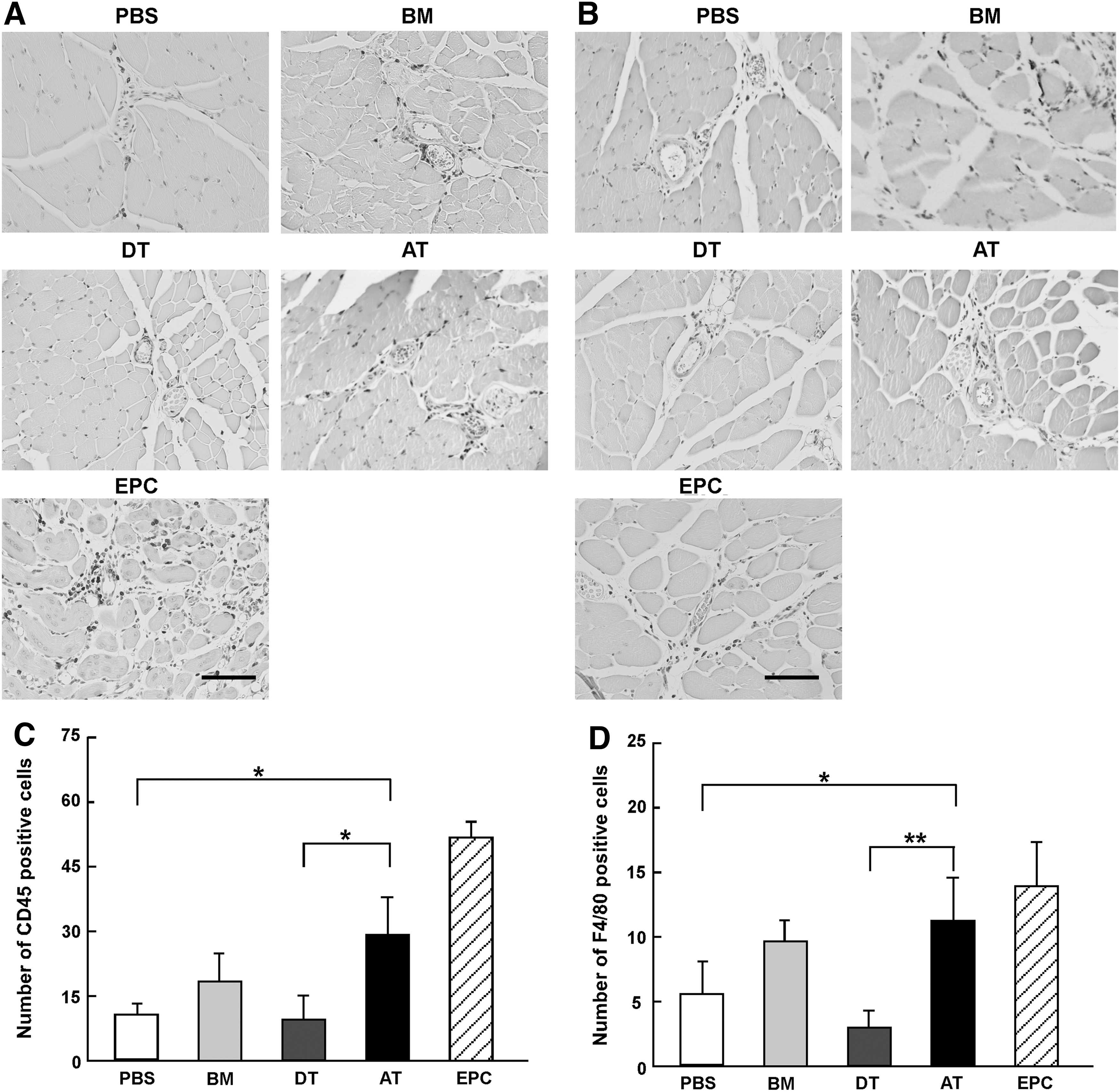
Migration of monocytes and macrophages into the ischemic region.
CCL5 secretion of AT-MSC related to migration of monocyte/macrophage and angiogenesis
The expression levels of angiogenic factors and chemokines were evaluated by semiquantitative PCR after MSC were cultured under normoxic and hypoxic conditions (Fig. 6). We found that, under hypoxic conditions, AT-MSC expressed higher levels of insulin-like growth factor (IGF) and FGF mRNA as compared with BM-MSC (IGF; BM-MSC: 0.8±0.3; AT-MSC: 4.6±1.9; FGF; BM-MSC: 1.0±0.02; AT-MSC: 2.5±0.8, Fig. 6A, *P<0.05). The expression of VEGF mRNA was upregulated equally in each MSC under hypoxic conditions. C-X-C chemokine receptor type 4 (CXCR4) and matrix metalloproteinase (MMP)9 mRNA expression levels were significantly higher in AT-MSC than in BM-MSC or DT-MSC under hypoxic conditions (CXCR4; BM-MSC: 0.8±0.3; DT-MSC: 13.3±7.9; AT-MSC: 50.0±5.5; MMP2; BM-MSC: 1.2±0.02; DT-MSC: 0.5±0.4; AT-MSC: 2.0±0.4; MMP9; BM-MSC: 1.0±0.01; DT-MSC: 3.1±1.2; AT-MSC: 17.3±2.0, Fig. 6B, **P<0.01). In addition, the mRNA expression of CCL5 was significantly higher in AT-MSC than in other types of MSC (BM-MSC: 1.9±0.8; DT-MSC: 4.8±2.5; AT-MSC: 17.9±1.6, Fig. 6B, **P<0.01). Of note, we found the injection of recombinant CCL5 intramuscularly shows significant recovery of blood flow on day 5 (PBS: 8.8±2.2; CCL5: 17.1±5.4, P<0.05) and day 7 (PBS: 12.8±3.0; CCL5: 20.7±4.2, P<0.05) compared with the control (PBS alone).
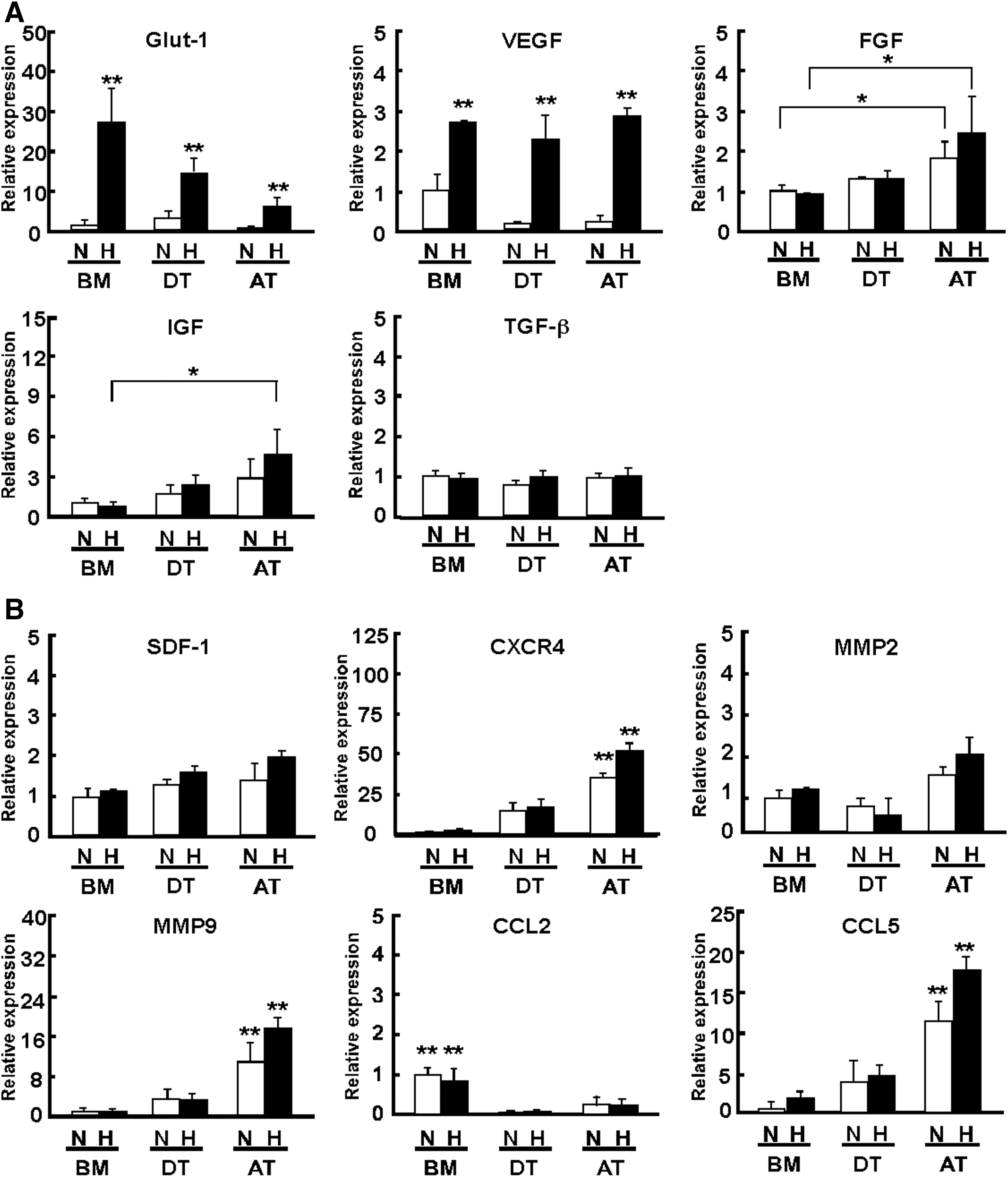
Expression of angiogenic factors and migration factors.
The chemokine CCL2, also known for its functional role in inducing the migration of monocytes and macrophages to sites of injury [38], was found to be more highly expressed in BM-MSC than in AT-MSC or DT-MSC (BM-MSC: 0.8±0.3; DT-MSC: 0.03±0.03; AT-MSC: 0.2±0.2, Fig. 6B, **P<0.01).
To determine whether CCL5 plays an important role in migration of monocytes and macrophages, we knocked down CCL5 in AT-MSC using shRNA. AT-MSC transfected with CCL5 shRNA (AT-MSC shCCL5) showed an ∼50-fold reduction of CCL5 mRNA expression (Mock: 1.0±0.3; CCL5 shRNA: 0.02±0.01, Fig. 7A, **P<0.01). We assessed the role of CCL5 using the hindlimb ischemic model; AT-MSC with CCL5 shRNA could not rescue blood flow (day 5, Mock: 0.6±0.2; CCL5 shRNA: 0.4±0.1, day 7, Mock: 0.8±0.1; CCL5 shRNA: 0.6±0.1, Fig. 7B, *P<0.05, **P<0.01). The number of migrated CD45-positive cells was significantly reduced in AT-MSC shCCL5 as compared with control (Mock: 29.0±8.0; CCL5 shRNA: 11.1±0.6, Fig. 7C–F, **P<0.01). In addition, we found that AT-MSC shCCL5 transplanted into the bone fracture model decreased the level of bone calcification compared with the control (Mock: 1.0±0.2; CCL5 shRNA: 0.6±0.1, Fig. 7G, **P<0.01).
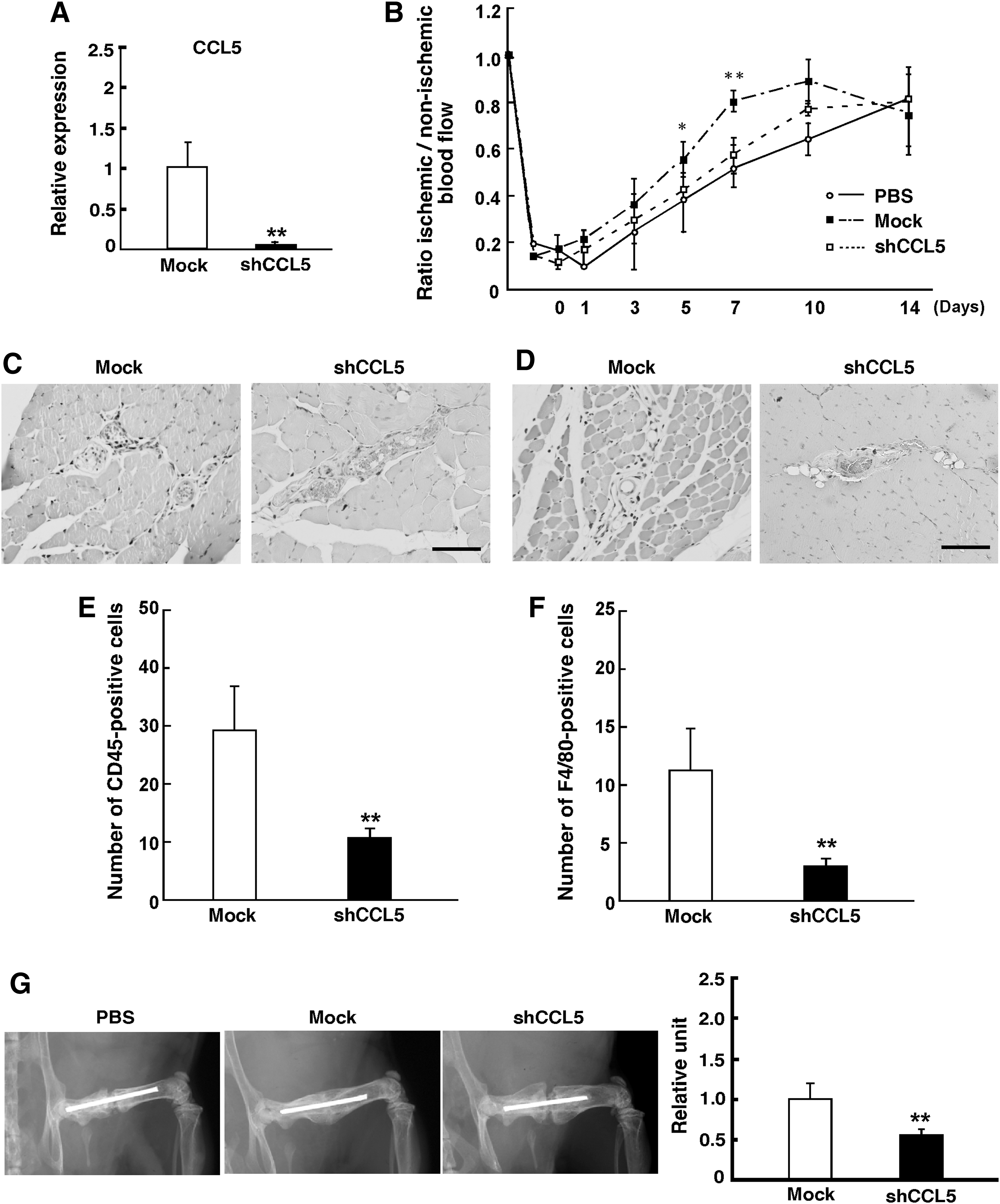
Decreased CCL5 expression in AT-MSC.
In summary, CXCR4, MMP9, and CCL5 were highly expressed in AT-MSC as compared with BM-MSC and DT-MSC. These factors in AT-MSC might be associated with arteriogenic or angiogenic effects. Of note, silencing experiments with shCCL5 clearly demonstrated that the recruitment of monocytes and macrophages by CCL5 was key to the superior ability of AT-MSC to repair limb ischemia and bone fracture.
Discussion
In this study, MSC were harvested from BM, AT, and DT to compare their function. These cells had fibroblastic morphology and similar expression profiles of cell surface markers. We examined differentiation ability in vitro; we found no significant difference in capacities for osteogenesis and chondrogenesis, but DT-MSC had less ability than BM-MSC or AT-MSC for differentiation into adipocytes. This result is in agreement with previous reports that DT-MSC are able to differentiate into osteocytes, adipocytes, and chondrocytes, but have lower adipogenic potential than BM-MSC [40].
Angiogenesis plays an important role in bone restoration [41]. When a mechanical stimulus and an inflammatory signal are received at the site of a fracture, bone restoration will begin [42]. At that time, angiogenesis begins at the site of healing and induces proliferation and differentiation of osteoblasts, promoting bone formation [43]. We analyzed the capacity for osteogenic differentiation using a mouse bone fracture model. At 4 weeks post-transplantation, each type of MSC had bone repair potential (Fig. 3). Furthermore, AT-MSC promoted osteogenic differentiation to a greater extent than did BM-MSC or DT-MSC. In addition, abundant lamellar bone formed in the central region of the joint in AT-MSC recipient. From these results, we identify that AT-MSC are a more useful cell type for fracture treatment than BM-MSC and DT-MSC.
Remarkably, AT-MSC showed significant effects on new vessel formation in ischemic tissue following vascular occlusion (Fig. 4). Although we and other groups have shown that engraftment of MSC facilitates collateral blood flow in ischemic regions, the mechanism by which MSC contribute to new vessels is still unknown [44,45]. It has been reported that MSC are able to differentiate into ECs [46,47]. However, there is no evidence that MSC-derived EC contribute to new vessel formation in vivo. On the other hand, MSC have been reported to have a paracrine function secreting angiogenic factors [48,49]. This paracrine function might explain the significant effects of AT-MSC on blood flow recovery observed in this study. VEGF mRNA expression was significantly upregulated under hypoxic conditions in each type of MSC (Fig. 6A). Under hypoxic conditions, expression of FGF and IGF mRNA were higher in AT-MSC than in BM-MSC (Fig. 6A). FGF and IGF might play specific roles in the relatively high ability of AT-MSC to promote formation of new vessels, leading to early recovery from ischemia.
Importantly, the migration of monocytes is fundamentally associated with angiogenesis and arteriogenesis in the ischemic environment [50,51]. Monocytes are recruited in response to hypoxia, migrate along defined chemotactic gradients, and accumulate within the hypoxic regions of ischemia [36 –38]. The oxygen gradient is a critical factor for monocyte migration to sites of inflammation [52]. Accumulated monocytes and macrophages secrete angiogenic factors, thereby supporting proliferation infiltration of EC and promoting new vessel formation [53].
Moreover, chemokines such as CXCL12, CCL2, and CCL5 as well as VEGF, endothelin, and Ang-2 have also been reported to be associated with migration of monocytes and macrophages [38,54,55]. Besides supporting migration of monocytes and macrophages, CCL2 also contributes to new vessel formation [56]. CCL5 is expressed by EC both in vitro and in vivo [57]. CCL5 supports migration of monocytes and macrophages that secrete MMP9 and MMP19 [58,59] and induces the influx and activation of leukocytes [29,60]. Moreover, expression of CCL5 may play a role in chronic inflammation [29,61]. Notably, CXCR4, MMP2, MMP9, and CCL5 mRNA expressions were higher in AT-MSC than in BM-MSC or DT-MSC (Fig. 6B). MMP2 and MMP9 disassemble extracellular matrix and directly support sprouting of blood vessels [62]. Since these factors were highly expressed in AT-MSC (Fig. 6B), it is likely that AT-MSC support ischemic improvement in a paracrine fashion.
Ultimately, higher numbers of monocytes and macrophages migrated in the AT-MSC recipient at the ischemic regions (Fig. 5). Transplanted AT-MSC secreted chemokine CCL5, thereby recruiting monocytes and macrophages that promoted angiogenesis, an important step for bone repair [17]. CCL5 shRNA suppressed the ability of AT-MSC to recruit monocytes and macrophages, causing impairment of the AT-MSC ability to support bone repair (Fig. 7). CCL5 has also been identified as a MSC-derived metastasis promoting factor [63]. Secretion of CCL5 from MSC is highly associated with both the physiologic and pathologic features of metastasis. Therefore, to clarify the mechanism of interaction between MSC and CCL5 would be an important step in the development of new medical treatments.
In conclusion, these results indicate that AT-MSC are useful not only for bone fracture treatment but also for healing ischemia. AT-MSC are highly promising for immediate autologous cell transplantation in many fields of cell therapy.
Footnotes
Acknowledgment
The authors thank Ms. Kaneko for excellent work on immunohistochemistry.
Author Disclosure Statement
The authors indicate no potential conflicts of interest.