
Abstract
We recently reported that mouse bone marrow stromal cells, also known as bone marrow (BM)-derived mesenchymal stem cells (MSCs), seeded onto a scaffold and implanted in vivo, led to an ectopic bone deposition by host cells. This MSCs capacity was critically dependent on their commitment level, being present only in MSCs cultured in presence of fibroblast growth factor-2. Taking advantage of a chimeric mouse model, in this study we show that seeded MSCs trigger a cascade of events resulting in the mobilization of macrophages, the induction of their functional switch from a proinflammatory to a proresolving phenotype, and the subsequent formation of a bone regenerative niche through the recruitment, within the first 2 weeks of implantation, of endothelial progenitors and of cells with an osteogenic potential (CD146+CD105+), both of them derived from the BM. Moreover, we demonstrated that, in an inflammatory environment, MSCs secrete a large amount of prostaglandin E2 playing a key role in the macrophage phenotype switch.
Introduction
B
To evaluate the cascade of cellular events activated by the implantation of scaffolds seeded with the FGF-2 expanded murine MSCs, we developed and implemented two models of ectopic bone formation. The first model involved the implantation of porous ceramic cubes seeded with mouse red fluorescent protein (RFP)-labeled MSCs into syngenic immunocopetent mice, with the aim of isolating and characterizing the inflammatory cells, such as macrophages, recruited within the pores of the scaffolds. In the second mouse model, lethally irradiated immunocompetent mice were hematopoietically reconstituted with green fluorescent protein-positive (GFPpos) syngeneic BM nucleated cells and used as hosts for transplants of combinations of bioceramic scaffolds and RFP-positive (RFPpos) syngeneic MSCs. This chimeric mouse model allowed us to distinguish between donor implanted and recipient cells originated from the BM or from the surrounding tissues. Macrophage populations were initially mobilized as a consequence of the scaffold implantation, but the cascade of events that led to bone formation was started by MSCs secreting large amounts of prostaglandin E2 (PGE2). This prostaglandin was responsible of the functional switch of phenotype from proinflammatory (M1) to anti-inflammatory (M2) macrophages, which, in turn, led to the recruitment of endothelial progenitors (CD133pos VEGFR2pos TLR2pos) and cells with an osteogenic potential (CD146posCD105pos), both of them derived from the BM.
Materials and Methods
Mice
C57Bl/6 (MHC H2b haplotype) mice were purchased from Charles River Laboratories. GFP-transgenic mice [genotype C57BL/6-Tg (CAG-EGFP)1Osb/J] and RFP-transgenic mice [B6.Cg-Tg (CAG-DsRed*MST)1Nagy/J] were purchased from The Jackson Laboratory. Mice were used between 5 and 8 weeks of age. Mice were bred and maintained at the Institution's animal facility. The care and use of the animals were in compliance with the laws of the Italian Ministry of Health and the guidelines of the European Community.
BMSCs (MSCs) isolation and culture
Mice were euthanized, and BM cells were collected by flushing nucleated cells out of the femurs and tibiae with cold phosphate buffered saline (PBS), pH 7.2. Cells were cultured (15×106 nucleated cells/10 cm Petri dish) in Coon's modified F12 medium (Biochrom AG), supplemented with 10% fetal bovine serum (FBS; Lonza), 2 mM of
BM-derived macrophages isolation and culture
BM-derived macrophages were isolated from C57Bl/6 mice by flushing the BM with 5 mL of PBS. Obtained cells were washed in alpha-minimum essential medium (α-MEM) and plated in a 10 mm diameter cell culture dish for 2 h. The supernatant was replated in a nontreated cell culture dish and α-MEM supplemented with 10% FBS, 2 mM
CD146posCD105posGFPpos cells isolation and culture
CD146posCD105posGFPpos cells were isolated 11 days after implantation by cell sorting from MSC-seeded implants. Isolated cells were cultured in fibronectin-coated plates (BD Biosciences) and Coon's modified F12 medium supplemented with 10% FBS, 2 mM of
Surgical procedures
Bioceramic scaffolds were 100% synthetic calcium phosphate multiphase biomaterials containing 67% silicon-stabilized tricalcium phosphate (TCP) (Skelite™). These scaffolds had 60–65% porosity and were produced by Octane Medical Group (Kingston). All scaffolds were cubes with a dimension of about 4×4×4 mm. After 3 or 4 weeks expansion, MSCs were detached from the dishes with 0.05% trypsin/ethylenediaminetetraaceticacid, washed in serum-free medium, and resuspended in aliquot at 2.5×106 cells per 20 μL fibrinogen (2.5 mg/mL) (Baxter). Each aliquot was seeded onto a scaffold to which 20 μL of murine thrombin (500 IU/mL) (Baxter) were then applied.
Recipient WT C57Bl/6 (nine mice/each considered time point; each mouse received four ectopic implants) or chimeric mice (12 mice/each considered time point; each mouse received four ectopic implants) were subcutaneously implanted with MSCs/bioscaffold constructs. Experiments were repeated at least three times. Some experiments included also groups of animals implanted with nonseeded, empty scaffolds. Groups of four animals were sacrificed after 3, 7, 11, and 15 days postimplantation, respectively, and the implants removed for further analysis.
Chimeric mouse model
Seventy-two C57Bl/6 recipient mice that have received a total body lethal radiation with 10 Gy were intravenously injected with 5×106 BM-nucleated cells derived from syngeneic GFP-Tg mice. Engraftment levels were assessed by the determination of the percentage of GFPpos cells in the peripheral blood and BM of chimeric mice, 5 weeks post-transplantation.
Histological and immunohistochemical analysis
Formalin-fixed scaffolds were processed as previously reported [4]. Briefly, samples were decalcified with Osteodec (Bio-Optica) and embedded in paraffin using standard histological techniques. Four-micrometer serial sections were cut. Sections were stained with hematoxylin and eosin to reveal bone tissue. To detect the presence of GFPposCD146posCD105pos cells within the pores of the implants, 4-mm sections were treated with rabbit polyclonal anti-GFP antibody (clone A11122) (Molecular Probes Europe BV), followed by biotinylated goat anti-rabbit secondary antibody (Dako Cytomation). To detect the presence of RFPpos MSCs within the pores of the implants, 4-mm sections were treated with rabbit polyclonal anti-RFP antibody (Molecular Probes Europe BV), followed by the same biotinylated goat anti-rabbit secondary antibody. Negative controls with preimmune serum and positive controls were run in parallel. Images were captured by transmitted light microscopy with an Olympus C3030 digital camera and Camedia Master Olympus software.
Cell sorting and flow-cytometric analysis
Cells, obtained by enzymatic digestions of RFPpos MSC-seeded scaffolds from both wild-type (WT) and chimeric recipient mice, were washed twice and suspended in 500 μL PBS, and separated into RFPpos and RFPneg fractions at different times (1, 3, 7, 11, 15 days). Cells recovered after 11 days from implants conducted in chimeric mice underwent an additional sorting passage to separate GFPpos CD146posCD105pos populations. All the experiments were performed using the cell sorter FACSAria (Becton Dickinson). CD146posCD105posGFPpos cells were cultured in the standard medium used for BMSC cultures. The immunophenotype of freshly-isolated RFPneg cells obtained from both MSC-seeded and empty scaffolds and the immunophenotype of BM-derived macrophages cultured for 3 days in the presence of MSC-conditioned medium were analyzed by flow-cytometry using specific monoclonal antibodies. Antibodies for the flow-cytometric analysis were: monoclonal antibodies to CD11b (M1/70), CD31 (390), CD45 (30-F11), CD51 (RMV-7), CD86 (GL1), IAb (AF6-120.1) (BD Pharmingen), monoclonal antibodies to CD36 (No.72-1), CD40 (1C10), CD133 (13A4), TLR2/CD282 (6C2) (eBioscience), monoclonal antibodies to Ly6C (1G7.G10), NK1.1 (PK136) (Miltenyi Biotec), monoclonal antibodies to CD105 (MJ7/18), CD206 (MR5D3), VEGFR2/CD309 (89B3A5) (BioLegend), monoclonal antibody to CD146 (P1H12) (Santa Cruz Biotechnology), and rabbit polyclonal to RAGE (Abcam).
All flow-cytometric analyses reported in this study were performed using the cell sorter FACSAria (Becton Dickinson). Data are expressed as percentages of positive cells and as ratio between mean fluorescence intensity (MFI) of cells stained with a specific antibody/MFI of correspondent isotype control (relative MFI).
Western blot analysis
Western blot analysis was performed as previously described [11]. Briefly, 5×105 cells were plated, the protein concentration of the lysate was measured using the Bradford method and equal protein amounts were used for western blot. Actin was blotted as an internal control. Equivalent amounts of corresponding conditioned media were analyzed. Sample were separated on NuPAGE™ 4–12% Bis-Tris gels (Invitrogen) and transferred to a Protran BA83 nitrocellulose membrane (Whatman GmbH). The blot was incubated with specific primary antibodies obtained from various sources.
Positive bands were revealed by an enhanced chemi-luminescence substrate mixture (GE Healthcare) and exposed to an X-ray film (GE Healthcare) to capture the image. Images were then scanned using the Epson perfection 1260 scanner, and band densities were quantified using the image J software (
PGE2 and 15ΔPGJ2 quantitation
Confluent MSCs were incubated in serum-free basal medium supplemented or not supplemented with 100 U/mL IL-1α for 24 h. Cells were then extensively washed with PBS to remove residual stimulant factor and incubated in serum-free medium for an additional 24 h after which media were collected, centrifuged to remove particulate matter and stored at −80°C. Aliquots were assayed for PGE2 content using a PGE2-specific competitive EIA kit-Monoclonal (Cayman Chemical) according to the manufacturer's instructions. A 15ΔPGJ2 specific competitive EIA kit (Enzo Life Sciences, Inc.) was used according to the manufacturer's instructions. Each sample was measured in triplicate in two dilutions. Statistical analysis of the data was performed.
Nuclear factor-kappa B activation
Binding of the nuclear factor-kappa B (NF-kB) p65 subunit to the NF-kB binding consensus sequence 5′-GGGACTTTCC-3′ was measured with the enzyme linked immunosorbent assay-based Trans Am NF-kB kit (Active Motif) using whole cell lysates prepared from MSC cultures, as recommended by manufacturer. All measurements were performed in triplicate. The Trans-Am kit employs 96-well microtiter plates coated with an oligonucleotide containing the NF-kB binding consensus sequence. The active form of p65 subunit in the whole cell lysates was detected using antibodies specific for an epitope that is accessible only when the subunit is activated and bound to its target DNA. Specificity was checked by measuring the ability of soluble WT or mutated oligonucleotides to inhibit binding. Results are expressed as specific binding, that is, the absorbance values observed in the presence of the mutated oligonucleotide minus those observed in the presence of the WT oligonucleotide.
Statistical analysis
In this study, results are given as the mean values±SD. All statistical analyses were performed using GraphPad software. The two-tailed Student's t-test was performed. A value of P<0.05 was considered significant.
Results
Different macrophage populations infiltrate the implanted scaffolds
We previously reported the enhancement of some biological processes, including inflammation and innate immunity in MSCs grown in FGF-2 [10]. In particular, chemokines implicated in monocyte recruitment from the blood to inflamed tissues, such as CCL8 (monocyte chemoattractant protein 2) and CCL9 (macrophage inflammatory protein-1 gamma), were oversecreted. Monocytes/macrophages (MC/Mph) led the inflammatory cascade reaction guiding revascularization and tissue repair/regeneration at injury sites [12 –14]. To investigate whether implanted MSCs could trigger the mobilization of macrophages and could alter the balance between M1 (CD86+/CD40+) and M2 (CD206+/CD51+) phenotypes, we analyzed the nature of the host macrophages recruited within the pores of both RFPpos MSC-seeded and control empty scaffolds after implantation in vivo in C57Bl/6 immunocompetent WT mice. Implants were harvested at different times, and collagenase-digested to generate single cell suspensions. Recovered cells were sorted, based on RFP expression, to separate implanted RFPpos MSCs and recruited RFPneg cells.
The flow-cytometric analysis of host derived RFPneg cells extracted from the scaffolds at 1, 3, and 7 days after implantation showed that macrophages infiltrated both types of ectopic implants, although the balance between M1 (CD86+/CD40+) and M2 (CD206+/CD51+) populations was directly correlated to the presence of the seeded MSCs.
The analysis of the immunophenotype of the host cells recovered from control empty scaffolds 1 and 3 days after implantation evidenced an increased relative MFI of the typical M1 markers compared to host cells extracted at the same times from the MSC-seeded scaffolds (Supplementary Fig. S1A, B; Supplementary Data are available online at
MFI, mean fluorescence intensity; SD, standard deviation; MSC, mesenchymal stem cell.
The inflammatory milieu decreased after 7 days of implantation, when a predominant fibrous connective tissue was observed (Supplementary Fig. S2B) in agreement with the decrease in the percentages of macrophage populations analyzed at this time point (Supplementary Fig. S1C).
After 11 days, the fibrous tissue was the main component within the pores of the scaffolds (Supplementary Fig. S2C).
To better define the balance between macrophages with M1 and M2 phenotypes at different implantation times, we performed two consecutive sorting passages. Implants, harvested at different times, were collagenase-digested to generate single cell suspensions. The first sorting passage, based on RFP expression, was performed to separate donor RFPpos MSCs from recipient RFPneg recruited cells. The second sorting passage was carried out to isolate, from the bulk of RFPneg recruited cells, macrophages with proinflammatory (M1) and anti-inflammatory (M2) characteristics. CD86 and CD40 were selected as M1 markers, CD206 and CD51 as M2 markers. M1 (CD86posCD40pos) and M2 (CD206posCD51pos) macrophages were isolated from the host cells recovered from empty scaffolds and from the RFPneg populations extracted from MSC-seeded scaffolds (Supplementary Fig. S3). We detected a higher percentage of M1 cells infiltrating the empty scaffolds compared to the MSC-seeded ones. This difference was statistically significant 1 and 7 days after implantation in vivo (Fig. 1A). Conversely, implanted MSCs induced a significantly augmented infiltration of M2 cells at day 3 and 7 (Fig. 1B). Moreover, the ratio between M2 and M1 cells (M2/M1) progressively increased from day 1 to 7 in the populations recovered from the scaffolds seeded with MSCs, whereas it remained constant in the populations isolated from the nonseeded implants (Fig. 1C).
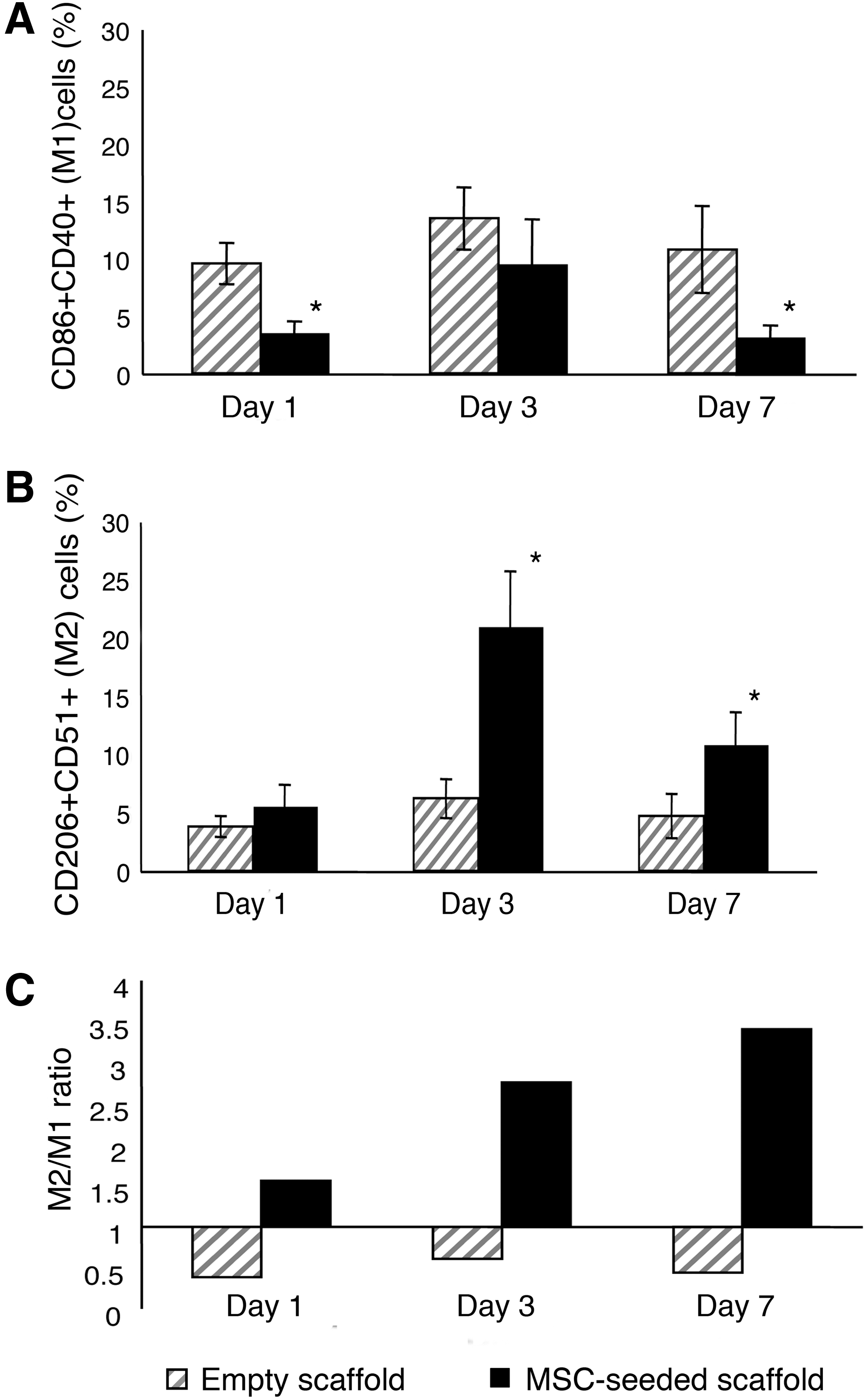
Quantification of M1 and M2 macrophages recovered from empty and bone marrow stromal cell (BMSC)-seeded scaffolds. Percentage of M1 (CD86 + CD40+)
Quantification of cytokine expression by real-time polymerase chain reaction in M1 and M2 cells isolated from both types of implants confirmed the nature of the two recovered cell populations. Being the differences relative to the expression of M1 and M2 markers more pronounced 3 days after in vivo implantation, we performed this analysis on the cells sorted at day 3. Compared to the M2 counterparts, M1 cells isolated from both types of implants had higher expression of both the proinflammatory cytokine tumor necrosis factor-α (TNF-α) (M1 Empty scaffold vs. M2 Empty scaffold: P<0.005; M1 MSC-seeded scaffold vs. M2 MSC-seeded scaffold: P<0.025) and HMGB1 (M1 Empty scaffold vs. M2 Empty scaffold: P<0.0001; M1 MSC-seeded scaffold vs. M2 MSC-seeded scaffold: P<0.0001) (Supplementary Fig. S4A, B). HMGB1 is a nuclear protein that can act as a cytokine to regulate different biological processes, such as inflammation, cell migration and metastasis. HMGB1 directs the migration of vascular and transformed cells. It has been described that RAGE, as well as the members of the toll-like receptors TLR-2 and TLR-4, binds HMGB1, causing the activation of different cell pathways, such as NF-kB, ERK1/2, and p38.
A higher expression of IL-10, an anti-inflammatory cytokine, was instead detected in the M2 populations isolated from both empty and MSC-seeded scaffolds (M1 Empty scaffold vs. M2 Empty scaffold: P<0.05; M1 MSC-seeded scaffold vs. M2 MSC-seeded scaffold: P<0.001) (Supplementary Fig. S4C).
MSC-secreted PGE2 promotes the switch of proinflammatory macrophages to an alternatively activated phenotype
MSCs are very sensitive to inflammatory stimuli [16,17]. Since the injury generated by the ectopic implant induces per se a strong inflammatory response, we mimicked in vitro the inflammatory environment surrounding the implanted MSCs to investigate whether the activated microenvironment affected the release of factors possibly involved in the macrophage functional switch. MSC confluent monolayers, were treated either with SF medium or SF medium supplemented with 100 U/ml IL-1α (IL-1). After 24 h, culture media were replaced with fresh medium with no supplements and cells were incubated for additional 24 h before media collection. To evaluate the direct effect of MSC-released factors on the macrophage phenotype, collected conditioned media were used to treat cultures of BM-derived MC/Mph. The flow-cytometric analysis of the MC/Mph isolated before and after the treatments showed that, when the cells were cultured in standard α-MEM (no conditioned medium treatment), they expressed high levels of the proinflammatory immature markers CD11b and Ly6C (48.2%+9.7 and 61.6%+18.5, respectively; Supplementary Fig. S5, upper panels). In these culture conditions, the expression of protein markers of classically activated M1 macrophages, such as CD40 and CD86, was less than 2% (Supplementary Fig. S5, middle panels). Moreover, the BM-MC/Mph did not express any of the typical M2 markers, such as the scavenger receptor CD36, the mannose receptor CD206, or the αvβ3 integrin CD51 (Supplementary Fig. S5, lower panels). The frequency of Ly6C+ macrophages decreased after 3 days of treatment with MSC-conditioned medium (SF) compared to treatment with the control standard medium (α-MEM), although this difference was not statistically significant. The percentage of Ly6C+ cells decreased significantly when MC/Mph were cultured with the conditioned medium obtained from IL-1-stimulated MSCs (P<0.05; Fig. 2). Interestingly, the percentage of CD206-expressing macrophages significantly increased after the cell treatment with both SF and IL-1 conditioned media (P<0.025 and P<0.025; Fig. 2).
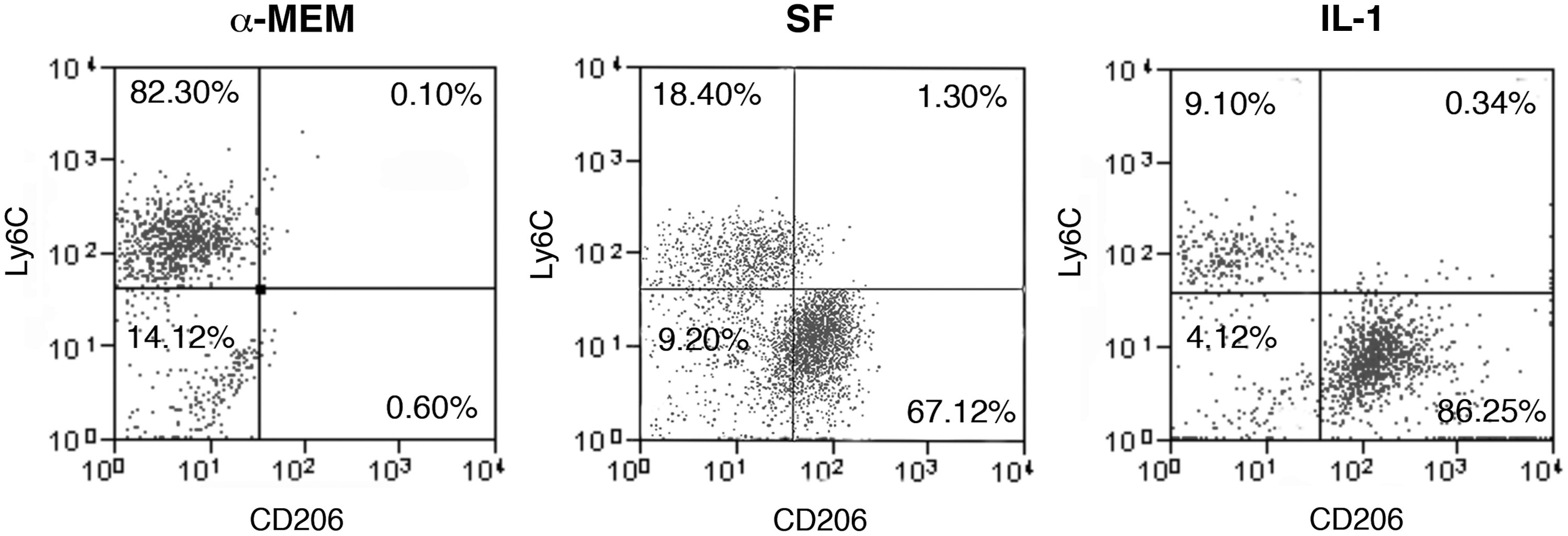
Phenotypic characterization of cultured BM-derived macrophages treated with MSC-conditioned medium. Flow-cytometric analysis of BM-macrophages cultured for 3 days in control standard medium alpha-minimum essential medium (α-MEM), in MSC-conditioned medium [serum free (SF)], and in conditioned medium derived from interleukin (IL)-1α stimulated MSCs (IL-1). CD206 was selected as marker of anti-inflammatory macrophages, Ly6C as marker of proinflammatory macrophages. Results show one representative experiment, and five independent experiments were performed.
A characterization of the pathways activated in MSCs cultured in inflammatory conditions was performed and we searched for bioactive molecules released in the culture media capable of turning the macrophage phenotype into a proresolving profile.
We observed that the IL-1 treatment strongly increased NF-kB activity in the MSCs compared to the no supplement control (P<0.0001; Fig. 3A). In parallel, we checked the synthesis and secretion of keratinocyte chemoattractant (KC) and Lcn-2. The amount of both KC and Lcn-2 present in the conditioned media of IL-1-stimulated MSCs was significantly increased (P<0.002 and P<0.0001; Fig. 3B, C). Moreover, IL-1 treatment induced in the MSCs the expression of COX-2 and the downstream enzyme mPGE synthase, whose products play a key role in the inflammatory cascade leading to the acute phase response [11]. It is noteworthy that PGD synthase expression level was unaffected (Fig. 3D).
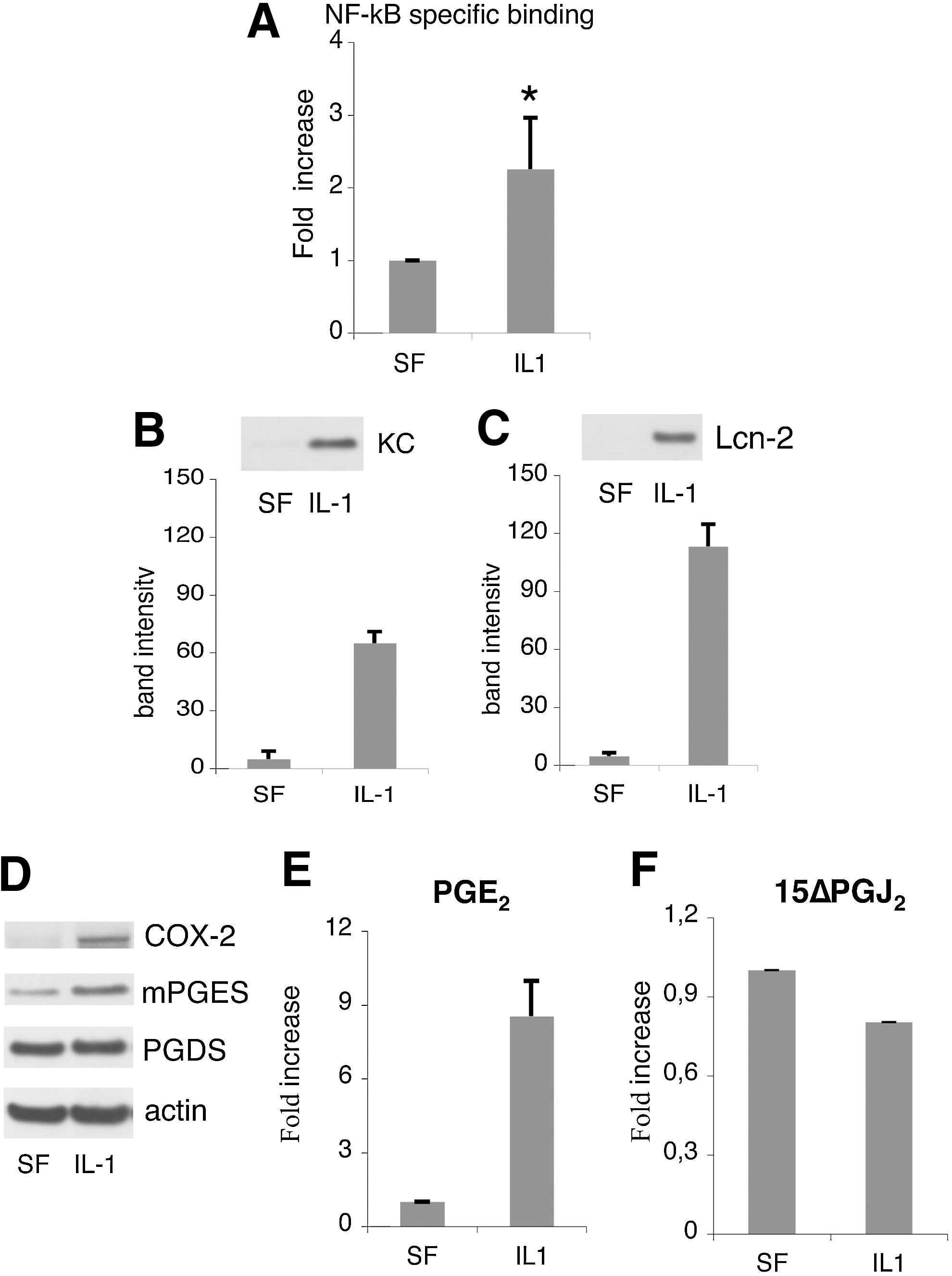
Activation of nuclear factor-kappa B (NF-kB)-dependent inflammatory pathways in MSCs in response to IL-1α.
In agreement with these findings, PGE2 production and secretion in MSC-culture medium was strongly induced by the IL-1 treatment, increasing from 3.7+1.1 ng/mL in SF medium up to 29.5+11 ng/mL in the medium of cells maintained in inflammatory conditions (P<0.0001; Fig. 3E). The production and secretion of the PGD2 metabolite 15 ΔPGJ2 was only slightly reduced (Fig. 3E). PGE2 has been described to modulate the cytokine profile of macrophages [18,19]. To establish a possible involvement of MSC-secreted PGE2 in the macrophage functional switch from a proinflammatory to an anti-inflammatory phenotype, BM-derived MC/Mph were stimulated with conditioned media from: (1) MSCs cultured in SF medium without any further supplement (SF); (2) supplemented with IL-1α (IL-1); (3) supplemented with the COX-2 specific inhibitor NS-398 to block PGE2 production (NS-398); (4) supplemented with IL-1α and NS-398 (IL-1+NS-398). MSC-conditioned media from cells incubated in the presence of the inhibitor NS-398 yielded less than 1% of CD206pos macrophages; thus, indicating that a COX-2 product was involved in the macrophage switch (Fig. 4). The involvement of PGE2 in the BMSC-induced macrophage polarization was confirmed by the observation that the percentage of recovered CD206pos cells progressively augmented when macrophages were cultured in (IL-1 + NS-398)-MSC conditioned medium supplemented with increasing concentrations of PGE2. The PGE2 concentrations tested ranged from 1 to 100 nM, being the intermediate concentration (15 nM) roughly equivalent to the concentration of PGE2 found in the SF condition, and the highest dose (100 nM) equivalent to the concentration found in the medium of IL-1-treated MSCs (Fig. 3E, left panel). Less than 3% of CD206pos cells were detected when PGE2 was added at the lower concentration (1 nM) (Fig. 4), whereas a statistically significant increase in the percentage of CD206pos cells was observed when PGE2 was used at 15 and 100 nM (P<0.02 and P<0.005) (Fig. 4).
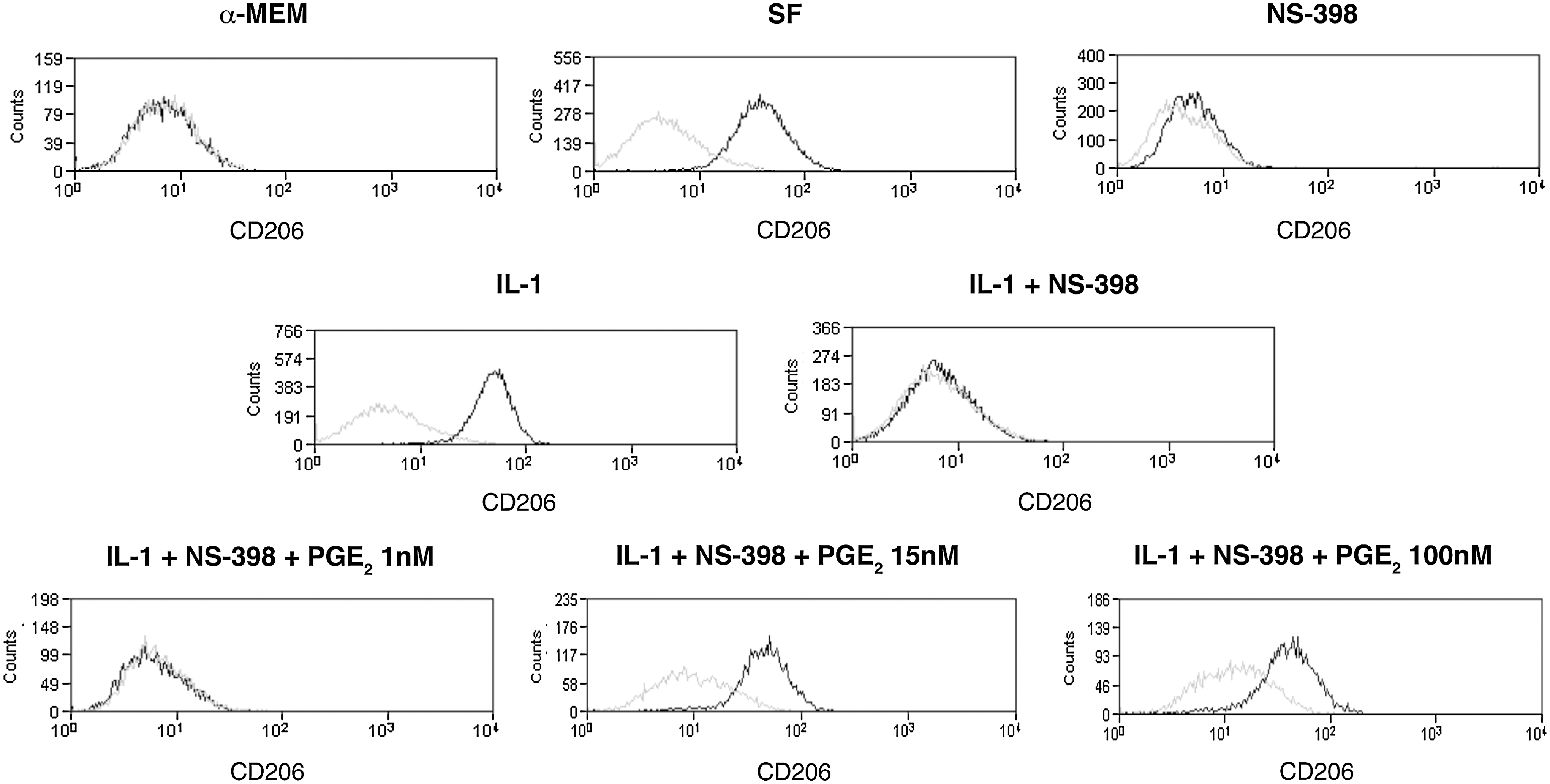
Effect of the PGE2 content in MSC-conditioned culture media on macrophage switch from M1 to M2 profile. Flow-cytometric analysis of BM-macrophages cultured for 3 days in: control standard medium (α-MEM); MSC-conditioned medium (SF); MSC-conditioned medium supplemented with the COX-2 specific inhibitor NS-398 (NS-398); conditioned medium derived from IL-1α stimulated MSCs (IL-1); conditioned medium derived from IL-1α stimulated MSCs and supplemented with NS-398 (IL-1 + NS-398); and (IL-1 + NS-398) conditioned medium supplemented with different concentrations of PGE2 (1, 15, and 100 nM) (IL-1 + NS-398 + PGE2 1 nM, IL-1 + NS-398 + PGE2 15 nM, IL-1 + NS-398 + PGE2 100 nM, respectively). CD206 was selected as marker of M2 macrophages. The black line represents the expression of the specific marker on tested cell populations. The gray line indicates nonreactive immunoglobulin of the same isotype, which was included as a negative control. Presented data refer to one representative experiment.
Taken together, these data demonstrate that MSCs responded to the inflammatory environment activating NF-kB and secreting PGE2, in turn, triggering the functional switch of macrophages from the inflammatory M1 to the anti-inflammatory M2 phenotype.
Mobilization of BM-derived endothelial progenitor cells into MSC-seeded scaffolds
We previously reported that the neovascularization of the implants was one of the first host responses to the graft and that cells with endothelial features could be recovered from the scaffolds 7 days after implantation in vivo [5]. By flow-cytometric analysis, we checked for the presence of endothelial progenitor cells (EPCs) in the host cell population migrated into scaffolds either seeded or not seeded with BMSCs after 7 days of implantation. Implants were performed in chimeric mice lethally irradiated and reconstituted with GFPpos donor BM cells to identify the possible BM origin of the mobilized cells. We harvested the implants, recovered the cells through an enzymatic digestion, and sorted out the RFPpos cells originally seeded on the scaffold (Fig. 5A). We observed the presence of GFPpos BM-derived cells within the RFPneg recruited host cells from both empty scaffold and MSC-seeded scaffold (gate R6), although to a different extent [27.5%+6.8 and 49.8+12.6, respectively; Fig. 5B (upper panel), C (upper panel)]. Since EPCs express the HMGB1 receptor TLR2 [20], we searched for TLR2-expressing cells within the recruited populations. All the TLR2pos cells were BM derived, being also GFPpos [Fig. 5B (bottom-left panel), C (bottom-left panel)]. Among the TLR2posGFPpos populations (gate R11), observed in both types of implants, we searched for cells coexpressing CD133 and VEGFR2, whih are specific markers of EPCs. A 4.3% of CD133posVEGFR2pos EPCs was detected within the cells from MSC-seeded scaffold (gate R16) (Fig. 5C, bottom-right panel). No EPCs were observed in the cells from the control empty scaffold (Fig. 5B, bottom-right panel); thus, indicating that the paracrine activity mediated by implanted MSCs was crucial in triggering the specific recruitment of BM-derived EPCs, the main responsible for vasculogenesis.
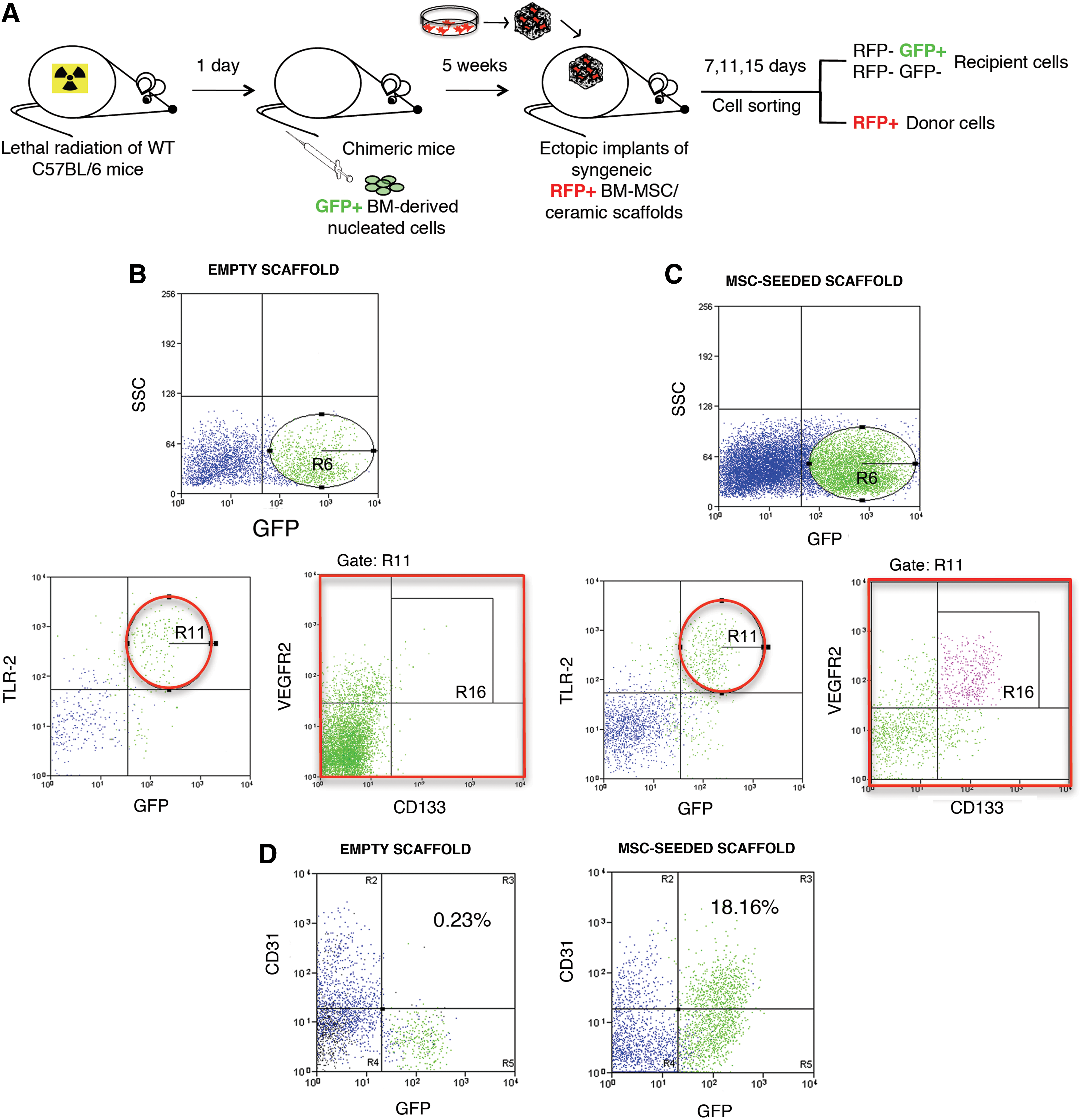
Phenotypic characterization of host-derived endothelial cells recovered from scaffolds implanted for 7 days in chimeric mice.
To evaluate whether the recruited EPCs could differentiate into mature endothelial cells (ECs), we checked for the presence of CD31-expressing cells in the bulk of the host-derived population after 15 days from implantation in vitro. CD31posGFPneg ECs were recovered from both implanted empty scaffolds and MSC-seeded scaffolds, indicating that a sprouting phenomenon from surrounding pre-existing vessels occurred in both types of implants (Fig. 5D). On the contrary, CD31posGFPpos ECs were present only in the MSC-seeded scaffolds, confirming the presence of BM-derived mature ECs possibly differentiated from the previously described recruited EPCs (Fig. 5D).
BM-derived CD146posCD105pos cells are recruited into the bone regenerative niche
We previously reported that CD146pos host cells with an osteogenic potential were mobilized into the MSC-seeded scaffolds and that the bone observed within the pore of the scaffold after 60 day implantation was derived from host cells [5] (see also Supplementary Fig. S6). Microvascular and BM pericytes are characterized by the expression of the cell surface marker CD146 [7,21], and in addition to play an important role in the stabilization of nascent capillaries and prevention of vascular regression [22], they possess mesenchymal plasticity and robust mesodermal developmental potential [21]. However, CD146 antigen is expressed by a variety of mouse cells, including myeloid, ECs, and natural killer T cells (NKT). To investigate whether tissue resident/local pericytes or BM-derived pericyte-like cells migrated toward the bone regenerative niche, scaffolds, either seeded or not seeded with MSCs, were implanted in the chimeric mouse model, harvested after 11 days, and enzymatically digested to recover migrated cells (Fig. 5A). To evaluate the authentic contribution of CD146pos cells in the regenerative process, we checked the sorted RFPneg host cells for the presence of cells coexpressing CD146 with: (1) the mesenchymal marker CD105; (2) the myeloid marker CD45; (3) the typical NKT cells marker NK1.1. The flow-cytometric analysis of the host-derived cell population isolated from control empty scaffolds revealed that 17.6% of the cells migrated into the ectopic implants were BM-derived GFPpos cells (Fig. 6A, upper-left panel). Nevertheless no CD146posCD105pos cells were present, while 13.9% of CD146pos cells expressed the nonspecific marker CD45 and the 3.1% coexpressed the marker NK1.1 (Fig. 6A, upper-right, bottom-left, and bottom-right panels, respectively).

Phenotypic characterization of host derived CD146+ recovered from scaffolds implanted for 11 days in chimeric mice.
Isolated host-derived RFPneg cells obtained from MSC-seeded scaffolds underwent an additional sorting passage to separate BM-derived GFPpos from GFPneg cells. Both RFPneg GFPpos and RFPneg GFPneg populations were cytofluorimetrically analyzed to confirm the successful outcome of the sorting steps. Since some GFPneg contaminating cells were present in the initially sorted RFPneg GFPpos population, only confirmed GFPpos cells (Gate R6 in Fig. 6B, upper-left panel) were considered for further analysis. Instead, no contaminant GFPpos cells were present in the sorted RFPneg GFPneg population (Fig. 6C, upper-left panel).
Only the GFPpos population was characterized for the presence of a large number of cells coexpressing the myeloid marker CD45 and the CD146, whereas the GFPneg population was characterized by [Fig. 6B (bottom-left panel), C (bottom-left panel)]. In both populations, less than 1% of CD146pos cells coexpressed the NKT marker NK1.1 [Fig. 6B (bottom-right panel), C (bottom-right panel)]. Interestingly, the 5.6% of BM-derived GFPpos cells coexpressed the mesenchymal markers CD146 and CD105 (Fig. 6B, upper-right panel), whereas no CD146posCD105pos cells were observed in the GFPneg counterpart (Fig. 6C, upper-right panel).
CD146posCD105posGFPpos cells were isolated by cell sorting from the MSC-seeded scaffold and culture-expanded utilizing the same medium used for MSC cultures (Fig. 7A). After 2 months of culture, cells still expressing high levels of GFP, CD105, and CD146 (Fig. 7B) were tested for their osteogenic differentiation potential in vivo. Cell seeded scaffolds were subcutaneously implanted into immunocompetent syngeneic recipient mice. The histological analysis of transplants harvested after 60 days revealed the presence of a forming bone within the pores of the scaffolds (Fig. 7C). Immunohistochemical analysis revealed that the cells entrapped in the bone matrix were GFPpos; thus, indicating that the bone tissue was of donor origin (Fig. 7C).

In vitro and in vivo analysis of cultured CD146posCD105posGFPpos cells isolated from MSC-seeded scaffolds 11 days after implantation in vivo.
Discussion
Although the improvement observed in injured tissues after administration of exogenous stem/progenitor cells could be due to their direct engraftment and differentiation to replace injured cells, it became evident that in many situations the exogenous progenitors acted by producing bioactive factors without a significant engraftment and differentiation [2,23,24]. We recently reported that in an ectopic bone formation model, BM-derived stromal cells implanted in vivo triggered regenerative mechanisms by recruiting specific host cells in the bone regenerative niche [4,5,25]. Since we used BM cell cultures of passage 1 or 2 for the implantation experiments, to rule out the possibility that a massive presence of contaminating macrophages could have played an important role in eliciting the effects attributed exclusively to MSCs, we performed experiments in which we implanted scaffolds loaded with CD45neg MSCs further depleted of the CD11b+ and CD14+ components. Also in that case, we observed a new bone tissue of host origin formed within the implants 60 days after implantation [4].
In this study, we show that implanted MSCs generated a cascade of events resulting in the mobilization of cells of the innate immune system, such as macrophages, the induction of their functional switch from a proinflammatory to a proresolving phenotype, and the recruitment of BM-derived specific progenitors with vasculogenic and osteogenic properties. Moreover, we demonstrated that MSCs, in an inflammatory environment, secreted a large amount of PGE2 playing a key role in the macrophage polarization.
Among the several biological functions hypothesized as related to the trophic effects of MSCs [26], the most significantly upregulated by FGF-2 were immune response, inflammatory response, response to wounding, and chemotaxis; thus, suggesting that a “wound” signature was induced in MSCs in presence of this factor [10]. Indeed, the bulk of cytokines and chemokines upregulated in treated cells contributed to orchestrate a microenvironment typically activated during wound healing and repair. We tested the hypothesis that the appearance of a localized host-derived bone tissue in grafts conducted with MSCs cultured in presence of FGF-2 could be mediated by cellular events also occurring in wound healing, and involving the recruitment of competent cells otherwise not found in the local tissue environment. The immuno-ablated strains of mice, such as nude mice, normally employed, could not represent the optimal model for this type of studies. In our reported experiments, the employment of immunocompetent recipient mice was possible because we used scaffold seeded with syngeneic MSCs.
Cells belonging to the innate immunity and, in particular, MC/Mph lead the inflammatory cascade reaction guiding revascularization and repair/regeneration of tissue at injury sites [12,13,27]. They exert this function by secreting inductive cytokines responsible for progenitor cell migration, a critical step in tissues that heal by regenerative processes rather than by a mere repair [28]. Macrophages are a heterogeneous subset of the mononuclear cell population that comprises multiple phenotypes that are induced in response to local stimuli during the wound healing process [29]. Classically activated (M1) macrophages exhibit potent antimicrobial properties, high capacity to present antigen, and consequent activation of Th1 responses. Alternatively activated (M2) macrophages possess the capacity to facilitate tissue repair and regeneration [30].
Consistent with the MSC-mediated anti-inflammatory capacities already described by different authors [19] and with previous studies conducted using different experimental approaches [27,31], we showed that implanted MSCs caused an increased percentage of alternatively activated (CD206posCD51pos) M2 macrophages infiltrating the ectopic implants, and a decrease in the percentage of infiltrating proinflammatory (CD86posCD40pos) M1 macrophages. The secretory pattern of MSCs is highly influenced by their microenvironment, and, in particular, they are very sensitive to inflammatory stimuli [2,16,17]. To gain more insight in the contribution of MSCs to the tissue regeneration, we mimicked in vitro the inflammatory environment surrounding the implanted cells and we studied whether it could affect the activation of intracellular pathways and the release of factors possibly involved in the macrophage functional switch. It has been described that MSCs express high levels of IL-1Rα, enabling them to modulate the expression profile of M1 activated macrophages [32]. We here reported that, in response to the IL-1α, MSCs activated NF-kB-dependent inflammatory pathways leading to the enhanced expression of COX-2 and acute phase proteins, such as KC/CXCL1, related to angiogenesis [33], and Lcn-2, controlling the expression of the SDF-1, a chemokine playing a major role in both tissue repair and mobilization of progenitor cells [34]. We focused on COX-2 downstream enzymes since prostaglandins play an important role in modulating the macrophage cytokine profile [19]. Our in vitro data demonstrated that the MSC-mediated secretion of PGE2 was responsible for the macrophage switch to the proresolving phenotype, and suggested that the enhanced percentage of alternatively activated macrophages in the MSC-seeded implants was related to the secretion of PGE2. This bioactive molecule plays also a role in the control of angiogenesis and vasculogenesis, inducing the mobilization of EPCs [35].
Moreover, the polarization of macrophages skews the secretion of DAMPS, including HMGB1, as well as TNF-α, vascular endothelial growth factor (VEGF), and MMP-9 (metalloproteinase-9), all molecules involved in the regulation of cell diapedesis and migration.
In a regenerative microenvironment, both angiogenesis and vasculogenesis can occur [36]. We previously showed that the vascularization of the implant was one of the first host reactions to the graft [5,25]. To define the compartmental origin of the ECs recruited within the pores of the scaffolds, and to evaluate whether cells present in the scaffold (still present seeded MSC and host-derived macrophages) were effective at inducing BM-derived EPCs migration within the scaffold, we took advantage of the chimeric mouse model. The empty scaffold implantation induced the migration into the scaffold, of only locally resident mature CD31posGFPneg cells. On the contrary, seeded MSCs triggered the mobilization toward the implanted scaffold of both mature locally resident CD31posGFPneg cells and BM-derived TLR2posCD133posVEGFR2posGFPpos EPCs, indicating that the vasculature of the cell-seeded scaffolds originated from both the sprouting of pre-existing vessels and the recruitment of circulating BM progenitor cells.
In our in vivo model, donor cells were dismissed from the graft within 2–3 weeks, whereas host cells competent to form bone were recruited to a direct osteogenic function at a later time [4,5]. In principle, this phenomenon can reflect two conceivable events. Local mesenchymal cells residing in proximity of the graft site can be reprogrammed to an osteogenic fate/commitment by factors known to act as key osteogenic inducers. Alternatively, cells competent to form bone can be recruited from the circulation and/or mobilized from the BM.
Our previous published data indicated that a population of CD146pos cells could be recovered from the MSC-seeded scaffold, but not from the empty scaffolds, 11 days after implantation [5]. Taking advantage of the chimeric mouse model, we could determine that a significant percentage of BM derived CD146posCD105pos cells were recovered from the MSC-seeded scaffolds, but not from the empty scaffolds. These cells were isolated, expanded in vitro and shown to maintain an osteogenic potential after 2 months in culture. When these cells were investigated for the expression of markers characteristic of mesenchymal lineages, they were shown to express markers expressed also by pericytes. Pericytes are important in the stabilization of nascent capillaries and in the prevention of vascular regression. In fact, during angiogenesis pericytes are recruited to growing microvessels, where they directly inhibit EC proliferation and migration through contact and secretion of growth inhibitory factors [37,38]. In an osteogenic environment, pericytes can become osteoblasts and participate in the bone deposition. Taken together, our data indicate that the role of MSCs as factories of bioactive molecules could explain many of the beneficial effects observed with administration of these cells for tissue repair. The recruitment of endogenous cells toward an induced regenerative niche may represent a new strategy for tissue repair.
Footnotes
Acknowledgments
This work was supported partially by funds from the European Union FP7 (Project “Angioscaff” no. 214402) and from UPMC-Pittsburgh. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Disclosure Statement
No competing financial interests exist
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.