
Abstract
Vascular calcification is a severe consequence of several pathological processes with a lack of effective therapy. Recent studies suggest that circulating and resident mesenchymal stem cells (MSC) contribute to the osteogenic program of vascular calcification. Molecular mechanisms underlying MSC osteogenic potential and differentiation remain, however, sparsely explored. We investigated a role for the complement receptor C5aR in these processes. We found that expression of C5aR was upregulated upon differentiation of human MSC to osteoblasts. C5aR inhibition by silencing and specific antagonist impaired osteogenic differentiation. We demonstrate that C5aR expression upon MSC differentiation was regulated by the multifunctional urokinase receptor (uPAR). uPAR targeting by siRNA resulted in complete abrogation of C5aR expression and consequently in the inhibition of MSC-osteoblast differentiation. We elucidated the NFκB pathway as the mechanism utilized by the uPAR-C5aR axis. MSC treatment with the NFκB inhibitor completely blocked the differentiation process. Nuclear translocation of the p65 RelA component of the NFκB complex was induced under osteogenic conditions and impaired by the inhibition of uPAR or C5aR. Dual-luciferase reporter assays demonstrated enhanced NFκB signaling upon MSC differentiation, whereas uPAR and C5aR downregulation lead to inhibition of the NFκB activity. We show involvement of the Erk1/2 kinase in this cascade. In vivo studies in a uPAR/LDLR double knockout mouse model of diet-induced atherosclerosis revealed impaired C5aR expression and calcification in aortic sinus plaques in uPAR−/−/LDLR−/− versus uPAR+/+/LDLR−/− control animals. These results suggest that uPAR-C5aR axis via the underlying NFκB transcriptional program controls osteogenic differentiation with functional impact on vascular calcification in vivo.
Introduction
I
Materials and Methods
Animal experiments
Animal experiments were approved by the local animal research committee and carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Academy of Science. Low-density lipoprotein-receptor-deficient (LDLR−/−) (Jackson Laboratories) and urokinase receptor-deficient (uPAR−/−) mice [14] were crossbred into the C57Bl/6J-background (F10). Double heterozygous offspring were bred to obtain uPAR−/−/LDLR−/− mice and uPAR+/+/LDLR−/− control mice. Genotypes were monitored by polymerase chain reaction. Mice were housed at the Centre for Animal Studies at the University-Hospital Münster, Münster, Germany. Gender matched 6-week-old mice were fed a high-fat diet (1.4% cholesterol, 12% cocoabutter, 3% oil, TD95046; Harlan) for 10 weeks before animals were re-anesthetized, euthanized by exsanguination from the inferior vena cava, and perfused from the abdominal aorta using 4% paraformaldehyde at physiological pressure for fixation. Heart base including aortic sinus with the aortic valve and ascending aorta was dissected and embedded in Tissue-Tek® OCT Compound and stored at −80°C for immunohistochemistry.
Cell culture
Human bone marrow MSC were obtained from Lonza (Lonza Walkersville, Inc.). These cells have been well characterized by the manufacturer for their surface markers and differentiation potential. MSC were cultured and expanded in the MSC growth medium (GM) as recommended by the supplier and were used between passages 6 and 7. To induce MSC osteogenic differentiation, GM was replaced with the osteogenic condition medium (OM) when MSC reached 90% of confluence. OM consisted of GM with 100 nM dexamethasone (Merck pharma GmbH), 5 mM β-glycerophosphate (Sigma-Aldrich Co.), and 5 mM L-ascorbic acid (Sigma-Aldrich Co.). The medium was changed every 3 days and induction of cell differentiation was carried out for 3 to 12 days.
To elucidate involvement of C5aR, Erk, and NFκB in osteogenic differentiation of MSC, cells were treated with 10 nM of a C5aR antagonist (C5aRA) (EMD Millipore), 500 ng/mL of pertussis toxin (Calbiochem), 10 μM of ERK inhibitor (Cell Signaling), and 1–10 μM of NFκB Inhibitor (Enzo Life Sciences), respectively. The substances were added to OM each third day with each change of the medium.
Monitoring of osteogenic differentiation
To demonstrate the osteogenic differentiation of MSC induced to differentiate into osteoblasts, standard markers of osteogenic differentiation, such as calcium deposition and alkaline phosphatase (ALP) activity, were monitored. The inclusion of calcium phosphate by osteoblast-like cells was determined by using an alizarin red-S assay (Sigma-Aldrich Co.) according to the manufacturer's instructions. To measure the ALP activity, MSC were seeded and cultured as described above. After 6 days the cultures were washed with phosphate-buffered saline (PBS, pH 7.3) and lysed with RIPA buffer containing 50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM phenylmethylsulfonyl fluorid, 1 mg/mL aprotinin,1 mg/mL leupeptin, 1 mM Na3VO4, and 1 mM NaF. Fifty micrograms of cell lysate was mixed with 150 μL ALP substrate buffer containing 5 mM p-nitrophenylphosphate (Sigma-Aldrich Co.), 0.1 M glycin, 1 mM MgCl2, adjusted to pH 9.8. After 1 h, the reaction was stopped by adding 50 μL of 1 M NaOH, pH 8.0. Absorption was measured at 405 nm using the GENios Multiplate reader (Tecan Groupd Ltd.).
Lentiviral transfection and nucleofection
Lentiviral small interfering RNA (siRNA) vectors were designed and cloned, and lentiviral vectors were produced by transient transfection of human embryonic kidney-293T cells and used for MSC transfection as described previously [8]. For uPAR overexpression, lentiviral vector pWPTS-GFP (kindly provided by Didier Trono, Department of Genetics and Microbiology, Faculty of Medicine, University of Geneva, Switzerland) was modified by ligating synthetic oligonucleotide duplex in SalI and BamHI restriction sites. The resultant vector was designated as pWPTS-Ad. Final pWPTS-uPAR lentiviral vector was generated by ligating SalI and NotI-digested pWPTS-Ad together with uPAR cDNA.
SiRNA for downregulation of C5aR and control siRNA were obtained from Santa Cruz Biotechnology and used for MSC transfection using Amaxa Nucleofector™ (Lonza). A basic primary smooth muscle cell nucleofector kit (Lonza) was used according to the manufacturer's instructions.
Cell fractionation
Cell fractionation was performed as described [15]. Cultured cells were washed in ice-cold PBS, pH 7.4, scraped on ice, and collected in 1.5-mL microcentrifuge tubes in 1 mL of ice-cold PBS. After centrifugation for 10 s, supernatants were removed from each sample and cell pellets were resuspended in 900 μL of ice-cold 0.1% NP40 (Calbiochem) in PBS and mixed well. Three hundred microliters of the lysate was taken as “whole cell lysate” and kept on ice until the sonication step. The remaining material was centrifuged for 10 s in 1.5-mL microcentrifuge tubes and 300 μL of the supernatant was taken as the “cytosolic fraction.” After the remaining supernatant was removed, the pellet was resuspended in 1 mL of ice-cold 0.1% NP40 in PBS and centrifuged as above for 10 s and the supernatant was discarded. The pellet (∼20 μL) designated as “nuclear fraction.”
Western blotting
Cultured cells were lysed in RIPA buffer directly on culture dishes. Lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The protein was transferred onto a polyvinylidene fluoride membrane (Roche Diagnostics). The membranes were developed with antibodies against C5aR, RUNX-2, p-Erk1/2 (Santa Cruz Biothechnology, Inc.), and uPAR (R&D Systems). Chemiluminescent images were captured using VersaDoc-3000 (Bio-Rad Laboratories) and quantified using Quantity One software (Bio-Rad Laboratories).
Quantitative real-time polymerase chain reaction analysis
Total RNA was isolated from MSC using QIAGEN QiaSpin miniprep kit (QIAGEN) and DNAse kit (QIAGEN), respectively, according to the manufacturer's suggested protocol. Real-time quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed on a LightCycler 480 Real-Time PCR System using LightCycler 480 RNA Master Hydrolysis probes (Roche Diagnostics GmbH). Primer sequences were for C5aR: 5′-TGTGGGTGACAGCCTTCGA-3′ (sense), 5′-CCGCCAGATTCAGAA ACCAG-3′ (antisense), 6-FAM-CCAGACGGGCCGTCAAACGC-TAMRA (probe); uPAR: 5′ACCACCAAATGCAACGAGG-3′ (sense); 5′-GTAACACTGGCGGCCATTCT-3′ (antisense); 6-FAM-CAATCCTGGAGCTTGAAAATCTGCCG-TAMRA (probe); GUSB: 5′-GTGGTGCTGA GGATTGGCA-3′ (sense); 5′-TAGCGTGTCGACCCCATTC-3′ (antisense); 6-FAM-TGCCCAT TCCTATGCCATCGTGTG-TAMRA (probe).
Luciferase assay
The control vector GAPDH-PG04 was purchased from GeneCopoeia. pGL3-promoter plasmid (Promega) was digested by NheI and XhoI restriction enzymes (NEB) and ligated with oligonucleotide duplex:
5′-ctagcgggactttccgaattcgggactttccggaaagtcccc
5′-tcgaggggactttccggaaagtcccgaattcggaaagtcccg
The duplex coded three copies of the NFκB-responsive sequences. Resulting plasmid was designated as pGL-promoter-NFκB. GAPDH promoter from GAPDH-PG04 plasmid was replaced by promoter and NFκB-responsive sequences from pGL-promoter-NFκB in NheI and HindIII sites. Final plasmid pPG04-NFκB coded Gaussia Luciferase (GLuc) under control minimal SV40 promoter and NFκB-responsive sequences and Secreted Alkaline Phosphatase (SEAP) under control constitutive promoter.
Transient transfection of MSC with plasmid constructs has been performed using Nanofectin kit (PAA) due to the manufacturer's instructions. Efficiency of transfection was proved with GFP. Twenty-four hours after transfection, cells were stimulated with OM for 5 h, and secreted Gaussia Luciferase was measured by Luciferase Assay Kit (New England BioLabs, Inc.) due to the manufacturer's instructions.
Chemotaxis assay
MSC migration was studied in a modified Boyden chamber with polyvinylpyrrolidone-free polycarbonate filter membranes, 8-μm pore size, as described previously [16]. 25,000–30,000 cells in a MSC basal medium (MSCBM) with 2% FCS serum were added to the upper well of the Boyden chamber. Chemoattractants diluted in MSCBM with 2% FCS serum were added to the lower well of the Boyden chamber and migration was allowed for 5–6 h. All experiments were performed at least in triplicates. Quantification of cell migration was performed by densitometry using VersaDoc 3000 (BioRad).
Immunofluorescent confocal microscopy
MSC were seeded on 13-mm-diameter microscope cover glasses with 4,000 cells/cm2. After 2–3 days, the cells were stimulated with OM for 3 h. Then, they were fixed with 2% formaldehyde, permeabilized in 0.1% Triton X-100 for 3 min, and blocked with 3% (w/v) BSA/PBS at 4°C overnight. After 24 h, the cells were labeled with polyclonal NFκB p65 antibody (Santa Cruz Biotechnology, Inc.) and subsequently with corresponding Alexa Fluor® 488 (Invitrogen) secondary antibody for 1 h at room temperature. Hoechst 33258 (Invitrogen) was applied as nuclear staining. For negative controls, the primary antibody was substituted with rabbit immunoglobulin (Santa Cruz Biothechnology, Inc.). Unspecific binding was blocked with 2% BSA in PBS. Cells were then mounted with the mounting medium (Aqua-Poly-Mount; Polysciences) and analyzed on a Leica TCS-SP2 AOBS confocal microscope.
Histology and immunohistochemical analysis
Serial transverse frozen sections (7 μm) of the aortic valve were stained for immunohistochemistry. Double stainings were prepared for C5aR/CD88 (rabbit anti-mouse CD88, H-100; Santa Cruz) and α-smooth muscle actin (αSMA, clone 1A4; Sigma). Adjacent sections were labeled for C5aR/CD88 and Von-Willebrand-Faktor (vWF; rat anti-human vWF). For detection, Cy3-coupled secondary antibodies (goat anti-rat and goat anti-mouse; Jackson Immuno Research) and a Cy5-coupled secondary antibody were used (Goat anti-rabbit IgG; Jackson Immuno Research). Nuclei were stained by 4′,6-diamidino-2-phenylindole labeling. Photomicrographs to assess C5aR-positive endothelial cell (EC) and vascular smooth muscle cell (VSMC) content were acquired at room temperature using an Olympus IX81 microscope (Olympus) equipped with a Retiga EXi camera (QImaging, Surrey) and QCapture Pro Software, (version 6.0.0.412; QImaging). A color threshold mask for immunostaining was defined to detect the green or yellow color. The same threshold was applied to all samples. Adjacent sections were stained using the von Kossa method using 5% silver nitrate and 5% sodium thiosulfate to identify calcification. Deposition of hydroxyapatite was visualized by bright field microscopy as dark brown staining. Animals were considered positive when deposits were observed in one or more slices. Morphometry was done using CellF life science fluorescence imaging software (version 3.1; Olympus). At least six atherosclerotic lesions were analyzed per animal. To quantify C5aR-positive cells, a region-of-interest was drawn around the plaque and the double positive area was measured and expressed as percentage of the overall EC and VSMC area, respectively.
Statistical analysis
All the experiments were performed in triplicates unless otherwise stated. Data from all experiments are presented as mean±standard deviation. Statistical analysis was performed using Student's t-test. Values of P<0.05 were regarded as statistically significant. Immunohistochemical data were analyzed using GraphPad Prism 5.0b (GraphPad Software). Data did follow Gausian distribution (Shapiro–Wilk test). Unpaired Students t-test was used for comparisons between the two groups. Data are presented as mean±SEM. P<0.05 was considered significant. Calcification data were analyzed by Fishers's exact test.
Results
MSC undergo differentiation to osteoblasts
MSC are multipotent progenitor cells with the capacity to differentiate into different tissue cell types, such as chondrocytes, osteocytes, adipocytes, and VSMC-like cells [17 –22]. To address the question of a probable involvement of C5aR and uPAR in MSC osteogenic differentiation, experimental conditions for this process have been first established. Several conditions may drive MSC differentiation to osteoblasts depending on MSC origin, and other parameters varying in experimental protocols. We tested MSC differentiation according to different experimental conditions and monitored osteogenic differentiation by cell morphological changes, ALP activity, RUNX-2 expression, and calcium deposition. MSC stimulation with dexamethasone, β-glycerophosphate, and ascorbic acid in the osteogenic medium was the most effective approach in our hands and was finally selected and used in all experiments. Under these conditions, MSC revealed reliable osteogenic differentiation. Thus, MSC underwent morphological changes with related calcium deposition typical for osteoblasts (Fig. 1A) and revealed strongly upregulated ALP activity (Fig. 1B) and RUNX-2 expression (Fig. 1C).
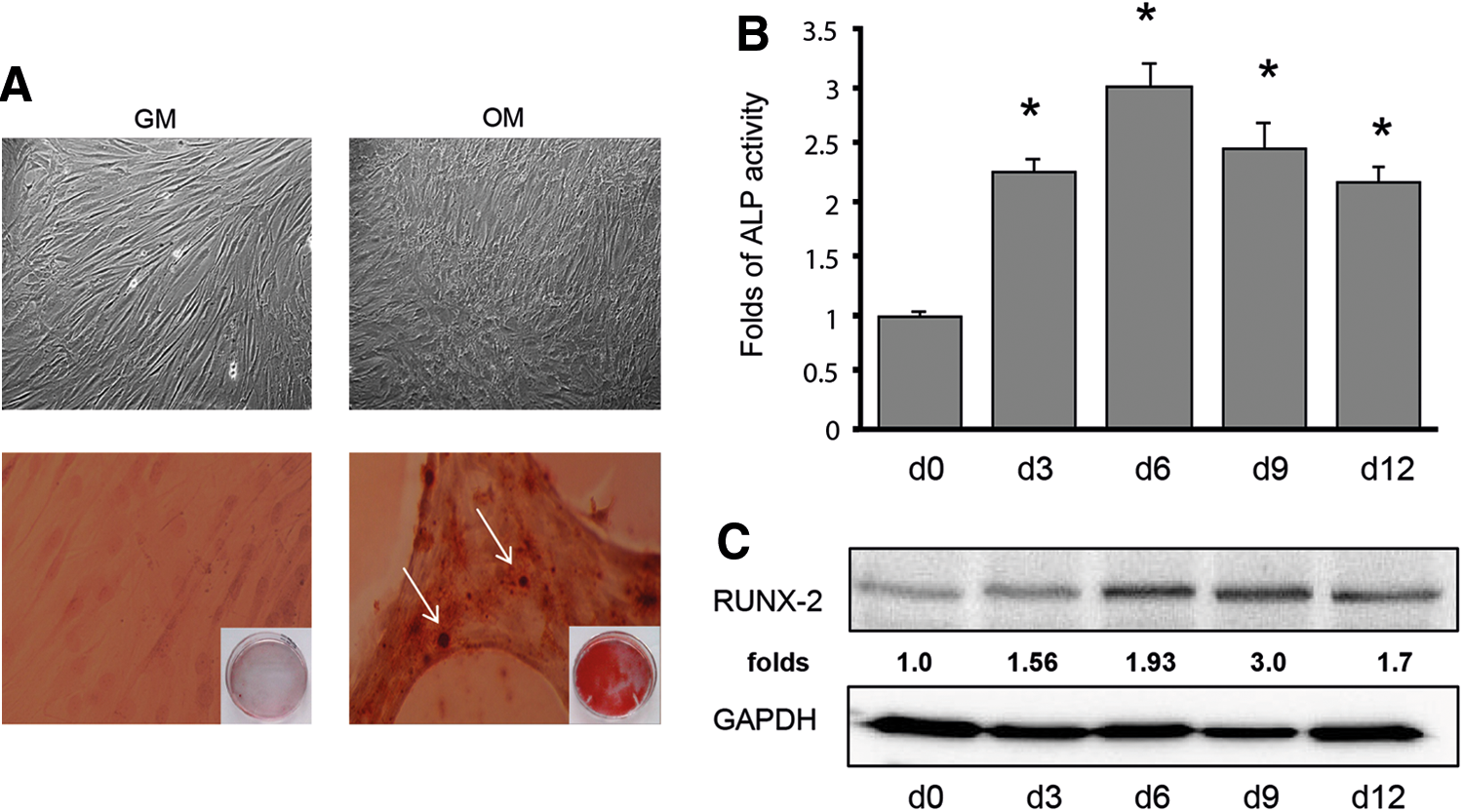
Mesenchymal stem cells (MSC) undergo differentiation into osteoblast-like cells.
C5aR mediates MSC osteogenic differentiation
The key central complement receptors C5aR and C3aR were identified in bone cells, including osteoblasts, MSC, and osteoclasts [4]. To examine whether C5aR is expressed in MSC under our experimental conditions and might undergo changes upon osteogenic differentiation, C5aR expression was analyzed. Indeed, we observed that C5aR expression, although being less pronounced in undifferentiated MSC, underwent strong upregulation upon MSC–osteoblast differentiation at both protein and mRNA levels (Fig. 2A). In a separate set of experiments, we proved C5aR functional competence in MSC examining and confirming the C5a-induced chemotactic activity of these cells in Boyden chamber assay (Supplementary Fig. S1A; Supplementary Data are available online at

C5aR mediates osteogenic differentiation of MSC.
uPAR mediates C5aR expression and osteogenic differentiation
As mentioned already above, the multifunctional GPI-anchored receptor uPAR has been recognized as an important functional participant in the differentiation of MSC to VSMC and in VSMC phenotypic modulations [8,23]. Our previous studies point to uPARs ability to interfere with the complement system and, in particular, to control C5aR expression and to affect by this way C5aR-directed functional effects [6,7]. To examine whether uPAR might control C5aR upregulation in MSC during osteogenic differentiation, uPAR was downregulated in MSC by means of a lentivirus-based interfering RNA (Supplementary Fig. S1C). As shown in Fig. 3A, uPAR silencing completely abrogated the increase in C5aR expression induced by osteogenic conditions.
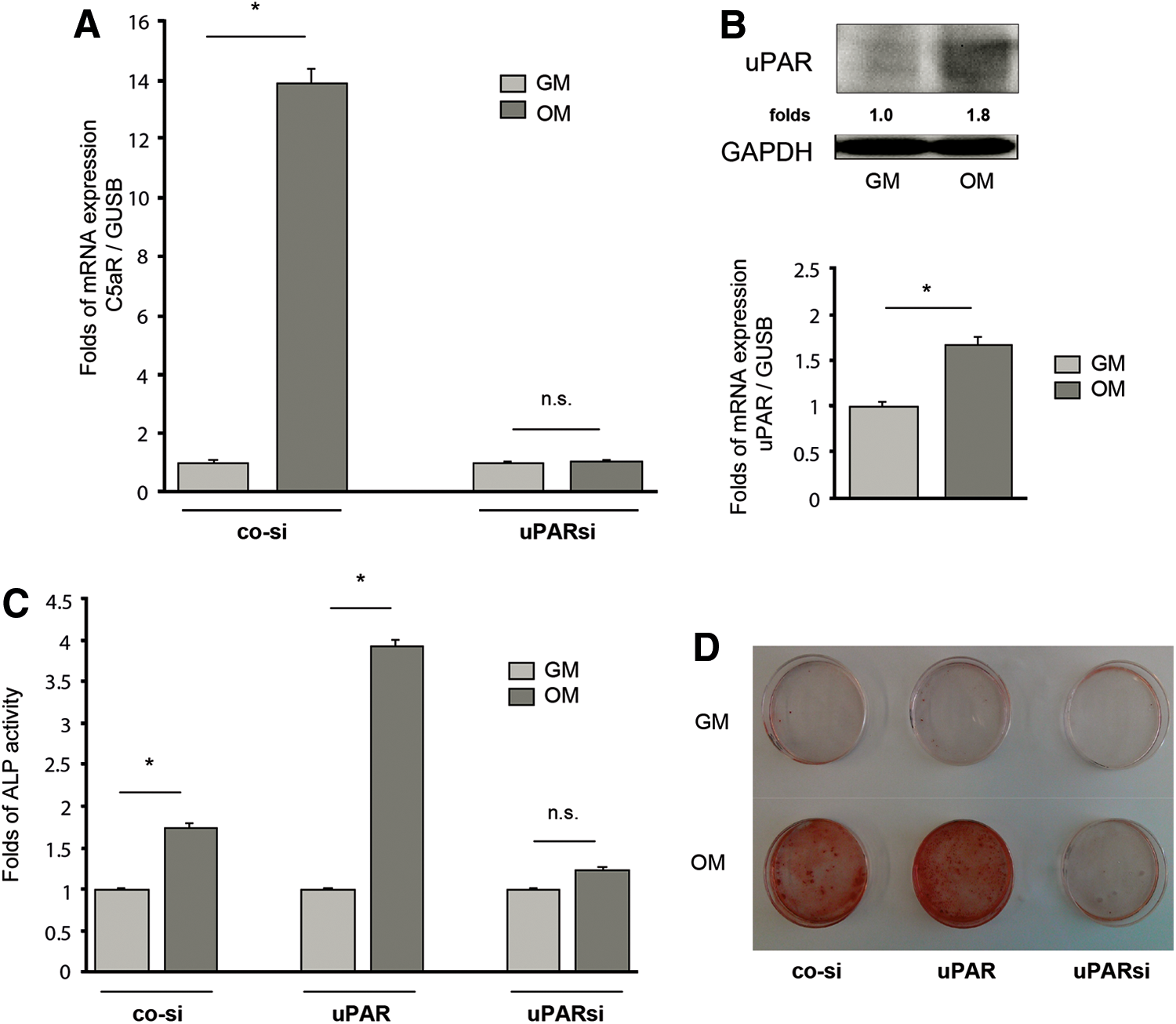
uPAR regulates osteogenic differentiation of MSC via C5aR.
uPAR expression analysis demonstrated uPAR upregulation in MSC stimulated for differentiation to osteoblasts (Fig. 3B). Targeting uPAR in MSC by means of silencing or overexpression resulted in inhibition or stimulation of osteogenic program correspondingly (Fig. 3C, D).
uPAR-C5aR axis activates Erk and NFκB upon MSC osteogenic differentiation
There is evidence that NFκB signaling pathway is required to trigger and propagate osteogenic program and vascular calcification [24 –26]. Further studies point to involvement of the NFκB transcriptional activity and the Erk (Extracellular-signal Regulated Kinase) pathway for C5aR-related functional responses [2,27]. We addressed these issues in our signaling studies and found that MSC respond to osteogenic conditions with a strong and transient Erk1/2 phosphorylation (Fig. 4A). Cell treatment with the Erk-inhibitor leads to impaired ALP activity under osteogenic conditions but not in control cells, thus providing further evidence for Erk requirement for the osteogenic process (Fig. 4B). C5aR is a G-protein coupled receptor (GPCR) transducing downstream cellular signals via G-proteins [28]. To examine whether the observed Erk1/2 activation might be mediated via this way, we treated MSC with pertussis toxin (PT) blocking Gi-directed signaling. As expected, Erk1/2 phosphorylation was completely blocked by this treatment (Fig. 4C).

Erk1/2 and NFκB signal MSC osteogenic differentiation downstream of uPAR-C5aR axis.
To examine whether NFκB signaling might underlie MSC–osteoblast differentiation and the observed interference with the C5aR-uPAR axis, we first performed inhibitory studies. MSC were pretreated with the NFκB-specific inhibitor and then stimulated for differentiation. We observed that the osteogenic process was strongly impaired after NFκB blockade, as monitored by the ALP activity and calcium deposition (Fig. 4D). We next investigated intracellular distribution of the p65 RelA component of the NFκB complex in MSC upon osteogenic conditions. In biochemical experiments on cellular fractionation, we found a pronounced RelA enrichment in nuclear fractions of cells stimulated for osteogenic differentiation that was abolished in cells treated with the NFκB inhibitor (Fig. 4E).
To provide direct evidence for changes in NFκB transcriptional activity in our experimental settings, we performed a dual-luciferase reporter assay optimized by us for primary MSC. We observed that the NFκB promoter activity was upregulated in MSC stimulated for differentiation, whereas the inhibiton of Erk in these cells completely abrogated this effect (Fig. 4F). We further observed that the inhibition of uPAR or C5aR affected the NFκB pathway. In agreement with the cell fractionation experiments, our confocal microscopy studies showed that MSC osteogenic differentiation was associated with upregulated nuclear translocation of the RelA. However, uPAR silencing or C5aR inhibition impaired this effect (Fig. 5A). Similar results came from the luciferase assay where downregulation of uPAR and C5aR resulted in abrogation of NFκB transcriptional activity under osteogenic conditions (Fig. 5B).

uPAR, C5aR, and NFκB cross talk during osteogenic differentiation.
uPAR mediates C5aR expression and vascular calcification in vivo
To elucidate whether our findings have a functional role in vivo, we performed studies using an animal model reflecting vascular calcification. We crossbred LDLR−/− and uPAR−/− mice in the C57Bl/6J-background and bred double heterozygous offspring to obtain uPAR−/−/LDLR−/− mice and uPAR+/+/LDLR−/− control mice. Animals were fed a western high-fat diet to develop complex atherosclerotic plaques in the sinus of Valsalva of the aorta. Previously, it has been shown that apolipoprotein E deficient and LDLR−/− mice have significantly more calcified aortic areas than nontransgenic mice [29] and that uPAR is atherogenic in nontransgenic LDLR−/− mice [30]. Furthermore, it was demonstrated that uPAR deficiency results in impaired vascular inflammation and remodeling [23,31]. Therefore, in our experimental settings, we focused on the requirement for uPAR in vascular calcification and C5aR expression. We used von Kossa staining to verify vascular mineralization. Generally, uPAR−/−/LDLR−/− mice revealed much less vascular calcification than uPAR+/+/LDLR−/− control mice. As shown in Fig. 6, even in advanced atherosclerotic plaques, von Kossa-positive areas were significantly less pronounced in uPAR−/−/LDLR−/− versus uPAR+/+/LDLR−/− mice. Next, we analyzed atherosclerotic lesions for C5aR expression by immunohistochemistry. Specific vascular markers, such as αSMA and vWF, were used in parallel to C5aR staining, and the double positive areas in plaques were quantified. Analysis of these data identified C5aR within the plaques and revealed remarkably strong inhibition of C5aR expression in uPAR−/−/LDLR−/− mice compared with uPAR+/+/LDLR−/− animals (Fig. 7).

uPAR mediates vascular calcification in vivo.

uPAR mediates C5aR expression in atherosclerotic plaques in mice.
Discussion
Vascular calcification, a manifestation of calcium–phosphate salt deposition in vascular wall, is associated with atherosclerosis and related cardiovascular risk and mortality. The pathophysiology of vascular calcification is of complex nature and the underlying cellular and molecular mechanisms remain poorly understood. Consequently, no effective therapies are available. Increasing body of recent evidence has challenged the traditional consideration of vascular calcification as a passive aging-related process and points to specific mechanisms of its regulation depending on disease-related environmental cues [32,33]. A role of circulating calcifying cells in this process is emerging [34]. Although sources of cells contributing to vascular calcification have not been clearly defined yet, convincing in vivo genetic fate mapping studies imply VSMC and bone marrow-derived MSC as major calcifying participants [12]. Our study identifies uPAR as a key factor regulating MSC–osteoblast differentiation and atherosclerosis-related vascular mineralization. Our findings further suggest uPAR-directed C5aR expression and activation of the downstream Erk-NFκB signaling as a potential underlying mechanism.
The uPA/uPAR system is a critical participant in cardiovascular diseases and related processes of pathological vascular remodeling [35,36]. Its contribution to vascular calcification, however, has never been addressed. In this study, we used uPAR−/−/LDLR−/− and uPAR+/+/LDLR−/− mice as a model for vascular calcification. We show convincingly that uPAR deficiency results in a decrease of calcified areas in the aortic root. It has been reported earlier that uPAR-deficient mice revealed increased bone mass compared with wild-type animals [37]. These data together with our findings are in agreement with clinical studies providing evidence that bone mass is conversely associated with vascular calcification [38,39]. Our observations are also in line with previous findings from our groups and those of others pointing to uPAR proinflammatory [40] and proatherogenic potential [41]. They also strengthen the previously demonstrated interference of uPAR with the complement cascade controlling C5aR expression and related functional effects [6,7]. We found that C5aR expression in atherosclerotic lesions was significantly impaired in uPAR−/−/LDLR−/− mice. Our in vitro data are in agreement with these observations and demonstrate uPAR-directed C5aR regulation in MSC upon their osteogenic differentiation.
Recent evidence indicates that the complement system, which is an essential component of innate immunity, serves as important regulator of tissue remodeling in damaged organs [27]. Some recent reports imply involvement of the complement cascades in mobilization and trafficking of stem cells including MSC [2] and suggest complement involvement in bone development, disorders, and regeneration [1]. We examined the differentiation of human bone marrow-derived MSC to osteoblasts and provide evidence that C5aR was indispensible for this process. C5aR was upregulated upon MSC osteogenic differentiation and its inhibition by means of silencing or specific antagonist resulted in abrogation of differentiation program. Elucidation of the C5aR-directed signaling pathway triggered in MSC by osteogenic conditions revealed that phosphorylation of Erk1/2 and activation of NFκB transcriptional program served as downstream components of this cascade. It has been reported earlier that uPAR-deficient osteoblasts showed higher levels of Erk phosphorylation compared with wild-type cells [37], whereas we observed uPAR-related Erk activation. There is no discrepancy between these studies because different cell types have been used, namely human MSC in our experiments and osteoblasts isolated from mouse calvaria in the study performed by others. Involvement of Erk through its phosphorylation in transmission of C5a/C5aR-induced responses, such as cytokine expression in epithelial cells and chemotactic behavior of MSC has been demonstrated [2,42]. Our data add MSC osteogenic differentiation as one further C5aR-directed physiological process where this pathway may be functionally utilized by the complement system. We further show the important role of NFκB in C5aR-directed MSC differentiation. Thus, we demonstrate that (i) MSC osteogenic markers were strongly impared after NFκB blockade; (ii) MSC osteogenic differentiation was associated with nuclear translocation of the p65 RelA in a C5aR-dependent manner; (iii) NFκB promoter activity was upregulated in MSC stimulated for differentiation while C5aR inhibition completely abrogated this effect.
Coronary artery calcification is believed to convey biphasic effects on atherosclerotic lesion stability [43]. A limitation of our study is that we can only speculate how the protective effects observed in our murine model relate to human disease. Compared to rodents, human plaques show more complex lesions and specifically more calcification. Experimental characterization of plaque instability is challenging as mouse models for atherosclerotic plaque rupture are scarcely available [44 –46] and not well characterized [47]. Kossa-stainable calcium deposition is nevertheless considered an acceptable surrogate for human complex lesion calcification [48]. We clearly demonstrate that uPAR deficiency reduces vascular calcification in murine atherosclerosis, a phenomenon that is much more prominent in human plaques and plays a major role in lesion destabilization. Recent studies suggest that bone marrow-derived MSC account for about 20% of osteochondrogenic (Runx-2/Cbfa1)-positive cells in calcified atherosclerotic vessels of ApoE−/− mice, whereas VSMC were found to be a major contributor to vascular calcification reprogramming its lineage toward osteogenic differentiation [12]. Further reports provide evidence that ECs may also participate in vascular calcification [49] and that MSC serve as the main source for both VSMC and EC in atherosclerotic plaques [50]. Our findings could be explained in several ways. uPAR function in cell differentiation seems to be of a general nature. As shown previously, uPAR governs VSMC phenotypic modulations from contractile to proinflammatory phenotype and regulates MSC differentiation to VSMC [8,23]. Therefore, a scenario could be suggested that in atherosclerotic vessels uPAR may initiate osteogenic differentiation not only in MSC but in VSMC and most likely in EC as well. Our preliminary data on VSMC–osteoblast differentiation speak in favor of that possibility. It could not be excluded that MSC may acquire VSMC properties upon osteogenic differentiation in vessel wall and could not be recognized by lineage-specific antibodies. αSMA-positive areas of atherosclerotic plaques examined for C5aR expression in our immunohistological studies may therefore include several lineages, primarily VSMC and MSC. We have reported recently that uPAR deficiency results in a strong inhibition of MSC mobilization from the bone marrow and their engraftment at the site of vascular injury [8]. These abilities of uPAR may additionally contribute to the observed impaired ossification of aortic sinus wall in uPAR−/−/LDLR−/− versus uPAR+/+/LDLR−/− mice.
MSC have been recognized as more beneficial for cellular therapy and tissue repair than other stem cells because of their multipotent differentiation capacities and immunomodulatory properties. A growing body of animal and clinical studies reports on beneficial therapeutic potential of MSC in cardiovascular diseases [51]. In parallel, another growing body of evidence points to critical issues of MSC therapeutic application that should be kept in mind cautioning clinical use of these cells [10]. It has been documented that MSC therapy is related to the development of atherosclerosis. In particular, it was shown recently that transplantation of bone marrow-derived MSC induced vascular remodeling and calcification after balloon angioplasty in hyperlipidemic rats [52]. Another previous study showed increased size of atherosclerotic lesion as a result of MSC homing [53]. Also severe intramyocardial calcification was observed after transplantation of unselected bone marrow cells after myocardial infarction [54]. Our findings confirm strong calcifying properties of MSC that may contribute to adverse effects of these cells on the cardiovascular system. The mechanisms underlying MSC-triggered vascular calcification are less explored. Further intensive research is required to elucidate and understand these mechanisms in depth. This novel knowledge may help to develop disease-orientated therapeutic strategies to affect MSC either to enhance or to inhibit their osteogenic potential. We have identified uPAR and C5aR as potential targets to control pathological processes during vascular calcification.
Footnotes
Acknowledgments
We express our grateful acknowledgments to Frank Hausadel and Kerstin Reher for technical assistance and to Yulia Kiyan for helpful advices and discussions throughout the work. This work was supported by Grant DU 344/7-1 from the Deutsche Forschungsgemeinschaft to I.D., by the PhD program “Molecular Medicine” of the Hannover Medical School and by IZKF Münster-project C21 to G.T.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.