
Abstract
Various types of somatic cells can be reprogrammed to induced pluripotent stem (iPS) cells. Somatic stem cells exhibit enhanced reprogramming efficiency by fewer factors, in contrast to fully differentiated cells. Nuclear LaminA is highly expressed in differentiated cells, and stem cells are characterized by the absence of LaminA. Granulosa cells (GCs) and cumulus cells in the ovarian follicles effectively and firstly generated cloned mice by somatic cell nuclear transfer, and these cells lack LaminA expression. We tested the hypothesis that GCs could be effectively used to generate iPS cells with fewer factors. We show that iPS cells are generated from GCs at high efficiency even with only two factors, Oct4 and Sox2, like the iPS cells generated using four Yamanaka factors. These iPS cells show pluripotency in vitro and in vivo, as evidenced by high expression of pluripotency-associated genes, Oct4, Nanog, and SSEA-1, differentiation into three embryonic germ layers by embryoid body formation and teratoma tests, as well as high efficient generation of chimeras. Moreover, the exogenous genes are effectively silenced in these iPS cells. These data provide additional evidence in supporting the notion that reduced expression of LaminA and stem cells can improve the reprogramming efficiency to pluripotency.
Introduction
T
Thus far, iPS cells have been generated from many cell types, including fetal and adult fibroblasts [1,9], hepatocytes [10], stomach cells [10], peripheral blood [11], keratinocytes [12], cord blood [13,14], dental pulp cells [15 –17], and even fully differentiated lymphocytes (T and B cells) [18 –23]. It is also recognized that stemness facilitates reprogramming, as shown by more efficient reprogramming of progenitor stem cells to iPS cells than of differentiated cells [24]. Notably, some precursor cells already express high levels of pluripotent factors. The advantage of expression of endogenous Yamanaka factor genes in precursors could be taken to reduce the number of reprogramming factors during iPS induction [8,25 –27]. For example, neural progenitor cells with endogenous Sox2 expression [28] could be reprogrammed into authentic iPS cells with only Oct4 and Klf4 transduction, but with lower efficiency [8,26]. Also, umbilical cord cells with endogenous expression of Klf4 and c-Myc were found to form iPS cells with high efficiency using only two factors [27].
Embryonic stem cells are truly pluripotent stem cells that lack the expression of LaminA in their nuclear envelope, in addition to the expression of pluripotent genes Oct4, Nanog, and Sox2. Consistently, somatic stem cells with reduced expression of LaminA or artificial reduction of LaminA by shRNA facilitate iPS induction [29]. During folliculogenesis in mammals, oocytes grow while surrounded by an increasing number of granulosa cell (GC) layers [30]. There are two types of GCs: those which surround the oocyte are cumulus cells and those which surround the antrum are mural GCs [31], and they possess characteristics of multipotent stem cells [30,32,33]. We recently also found that mouse and monkey GCs and cumulus cells from adult ovarian sections are negative for LaminA by immunocytochemistry. This further confirms that GCs and cumulus cells exhibit stemness to some extent.
Cumulus cells were the first to successfully produce clone mice, Cumilina [34]. GCs also were effectively used to clone animals [35 –39], including the first cloned piglets by somatic cell nuclear transfer [37], indicating that both cell types from ovarian follicles are valuable cell sources for successful cloning and amenable to reprogramming. In addition, GCs and cumulus cells are often byproducts from in vitro fertilization (IVF) clinic or animal industry where they are removed off from oocytes and discarded. We tested the hypothesis that iPS cells can be efficiently induced from GCs including cumulus cells with fewer factors.
Materials and Methods
Isolation of mouse embryonic fibroblasts and mouse granulosa cells
Mouse embryonic fibroblasts (MEFs) were derived from E13.5 embryos from C57BL/6 mice isolated by caesarean section and washed in Hank's balanced salt solution (HBSS). Heads and visceral tissues were removed, and remaining tissue was washed in fresh phosphate-buffered saline (PBS), then submerged in 0.05 mM trypsin/1 mM EDTA HBSS solution, and incubated at 37°C for 10 min. Tissue was pipetted repeatedly to aid in tissue dissociation, then added to MEF medium containing 10% fetal bovine serum (FBS) and plated (Passage 0).
For the isolation of mouse GCs (mGCs), pregnant mare serum gonadotropin was injected into the abdominal cavity of C57BL/6 mice 48 h before isolating GCs. The mice were sacrificed and the ovarian were dissected. GCs were aspirated from ovarian using a Pasteur pipette under a stereomicroscope. Then, the cells were washed in fresh PBS and cultured in the mouse embryonic stem cell (mES) medium, and the medium was changed 48 h after incubation. Use of mice for this project was approved by the Nankai Institutional Animal Care and Use Committee.
iPS cell induction
Derivation of iPS cells from GCs or MEFs has been described previously [40,41]. iPS cells were induced by transduction with Yamanaka factors or fewer factors using a standard protocol [4], with slight modification of induction medium for some experiments.
The day before virus packaging, Plat-E cells were seeded at 5×106 cells per 100 mm dish. On the next day, pMXs-based retroviral vectors (pMXs-Oct4, Sox2, Klf4, c-Myc) were introduced into Plat-E cells using lipo-2000 transfection reagent according to the manufacturer's recommendations. Viruses were collected with 0.45 μm membrane at 48 h and 72 h, respectively, after transfection.
For iPS induction, 5×104 MEF cells (P2) were seeded in a six-well dish 24 h or ∼5×105 freshly isolated primary GCs seeded 48 h before transduction when they adhered to the dish. GCs grew much slower and their adherence to the dish required extra time. To compare the transduction efficiency, MEF or GCs were transfected with Oct4, Sox2, Klf4 and c-Myc (OSKM) and green fluorescent protein (GFP) and collected 48 h after transduction. GFP-positive cells were estimated by flow cytometry.
After infection twice, the cells were cultured in 2 mL ES cell medium containing knock-out Dulbecco's modified Eagle medium (DMEM) medium (Invitrogen), added with 20% knock-out serum replacement (Invitrogen), 1000 U/mL leukemia inhibitory factor (LIF; Millipore), 0.1 mM β-mercaptoethanol (Sigma), 1 mM
iPS and ES cell culture
N33 ES cell lines as control were derived from C57BL/6 mice [40]. The ES and iPS cell culture medium consisted of the knock-out DMEM added with 20% FBS (Hyclone), 1000 U/mL LIF, 0.1 mM nonessential amino acids, 0.1 mM β-mercaptoethanol, 1 mM
RT-qPCR and conventional PCR
Total RNA was purified using a RNA mini kit (Qiagen), treated with DNase I (Qiagen), and the cDNA was generated from 2 μg RNA using Oligo(dT)18 primer (Takara) and M-MLV Reverse Transcriptase (Invitrogen). For the cDNA used for detecting Xist, random primer (Takara) was used. Primers were confirmed their specificity with dissociation curves. All data are normalized using β-actin as internal control. Reverse transcription quantitative PCR (RT-qPCR) was carried out on a MyiQ Detection system (BIO-RAD) using Fast Start Universal SYBR Green Master (Roche). All reactions (in duplicate) were carried out by amplifying target genes and internal control in the same plate. The amplification was performed for primary denaturation at 95°C for 10 min, then 40 cycles of denaturation at 95°C for 15 s, annealing and elongation at 58°C for 1 min, and the last cycle under 55°C–95°C for dissociation curve. Relative quantitative evaluation of target gene was determined by comparing the threshold cycles. Most primers were designed using IDT DNA website. For the conventional PCR, the samples were performed using Ex-Taq polymerase (Takara) and the products of PCR reactions were subject to electrophoresis in 1% agarose gel. Primers used in this study are listed in Table 1.
Primers for exogenous transcription factor were based on the plasmid and can be used for both RT-qPCR analysis and transgenes integration detection.
RT-qPCR, reverse transcription quantitative PCR.
Western blot
Cells were washed twice in PBS, collected, and lysed in sodium dodecyl sulfate (SDS) sample buffer on ice for 30 min and then sonicated for 1 min at 60 of amplitude with 2 s intervals. After centrifugation at 10,000g, 4°C for 30 min, 100 μL supernatant was transferred into new tubes. The concentration of the protein sample was measured by bicinchoninic acid. Twenty micrograms of total proteins of each cell extracts was resolved by 10% Bis-Tris Sodium dodecyl sulfate-polyacrylamide gelelectrophoresis and transferred to polyvinylidinedifluoride membranes (Millipore). Nonspecific binding was blocked by the incubation in 5% skin milk in Tris-buffered saline and Tween 20 at room temperature for 2 h. Blots were then probed overnight at 4°C with anti-LaminA (Rabbit polyclonal; Abcam, ab26300) and anti-β-tubulin (Mouse monoclonal, AbM59005-37-PU). Immunoreactive bands were then probed for 2 h at room temperature with the appropriate horseradish peroxidase (HRP)-conjugated secondary anti-Rabbit IgG-HRP (GE Healthcare, NA934V) and goat anti-Mouse IgG (H+L) / HRP (ZSGB-BIO, ZB-2305). Protein bands were detected by Chemiluminescent HRP substrate (Millipore, WBKLS0500) and imaged using a Tanon western blot imager.
Immunofluorescence microscopy
Immunofluorescence staining was performed as described [40]. Cells were washed in PBS, then fixed in freshly prepared 3.7% paraformaldehyde in PBS (pH 7.4), permeabilized in 0.1% Triton X-100 in PBS for 30 min, washed in PBS for one time, and left in blocking solution (3% goat serum in PBS) for 2 h. Cells were incubated overnight at 4°C with primary antibodies against Oct4 (sc5279; Santa Cruz), Nanog (AB80892; Abcam), SSEA-1 (MAB4301; Millipore), β-III-tubulin (CBL412; Chemicon), smooth muscle actin (SMA, ab5694-100; Abcam), alpha 1-fetoprotein (AFP, DAK-N150130; Dako), washed three times with blocking solution, and incubated for 2 h with secondary antibodies at room temperature. Goat anti-mouse IgG (H+L) FITC (115-095-003; Jackson) and goat anti-rabbit IgG (H+L) AlexaFluor® 594(111-585-003; Jackson), diluted 1:200 with blocking solution, were used. Samples were washed and counterstained with 0.5 μg/mL Hoechst33342 in the Vectashield mounting medium. Fluorescence was detected and imaged using a Zeiss fluorescence microscope.
Embryoid body formation test
iPS cells were removed off feeder cells twice based on their differences in the adherence to the bottom of dish. The cells were diluted to 4×104 per milliliter. Every 30 μL was pipetted to form a hanging drop on the cover of the 100-mm dish, and embryoid bodies (EBs) were formed by day 4. EBs were fixed for immunofluorescence staining using markers of three embryonic germ layers 14 days after EB formation.
Teratoma test
Approximately 1×106 cells were injected subcutaneously into 4-week-old immunodeficient nude mice. At 4 weeks after injection, the mice were humanely sacrificed; the teratomas were excised, fixed in 3.7% paraformaldehyde, washed in 70% ethanol, embedded in paraffin, and sectioned for histological examination by haematoxylin and eosin staining.
Chromosome spread and karyotyping
iPS cells were arrested in 0.3 μg/mL nocodazole for 2.5 h to enrich cells at metaphase. Cells were collected and treated with 75 mM KCl for 30 min. After centrifugation at 300g for 8 min, cell pellets were fixed three times in 1:3 cold acetic acid: methanol for 30 min. Fixed cells were dropped onto precooled slides and dried overnight. More than 30 chromosome spreads were counted, and the normal karyotype percentage was recorded. Cell line with more than 70% diploid karyotyping was considered normal.
Chimera generation
Blastocysts were collected from the uterus of E3.5 superovulated ICR females mated with Institute of Cancer Research (ICR) males and maintained in the KSOM medium with amino acids until iPS cell injection. For the blastocyst injection, cells were trypsinized, resuspended in mouse ES cell medium, and kept at 4°C. A flat tip microinjection pipette with an internal diameter of 12–15 μm was used for the injection. Approximately 10–15 iPS cells were injected into the blastocyst cavity. The blastocysts were placed in KSOM until embryo transfer. Ten to fifteen injected blastocysts were transferred to each uterine horn of 2.5 days postcoitum pseudopregnant ICR females. Chimeric mice were born 17 days after the transfer. Pups were identified by coat color.
Statistics
Data were analyzed by the analysis of variance and means compared by Fisher's protected least-significant difference using the Stat View software from SAS Institute, Inc. Significant differences were defined as P<0.05, 0.01, or lower.
Results
LaminA expression in GCs within ovarian follicles and in cultured GCs
Mouse ovarian sections were processed by immunocytochemistry using LaminA antibody. Many ovarian cells were stained strongly positive for LaminA, including germinal vesicle of oocytes (Oo). In contrast, GCs and cumulus cells in the growing follicles were almost negative for LaminA (Fig. 1A). We compared the relative expression levels of LaminA by RT-qPCR analysis of GCs (mGC), which may have been mixed with cumulus cells, ES, and MEF that were originally and routinely used for iPS induction. Notably, LaminA level of GCs was similar to that of ES (N33) from the same genetic background, but much lower than that of MEF (Fig. 1B). Western blot confirmed that the protein level of LaminA was relatively low in GCs, like ES cells, and became higher when GCs were cultured in vitro, but still lower than those of MEF and tail tip fibroblasts (TTF) (Fig. 1C). Under mES culture condition, the cultured GCs showed clonal colony formation, in morphology similar to clones of stem cells, with high nuclei/cytoplasmic ratio (Fig. 1D), but these cells could hardly be passaged further. Consistent with the western blot, the cultured GCs showed nuclear LaminA staining to low degree compared with MEF by immunofluorescence microscopy (Fig. 1E).
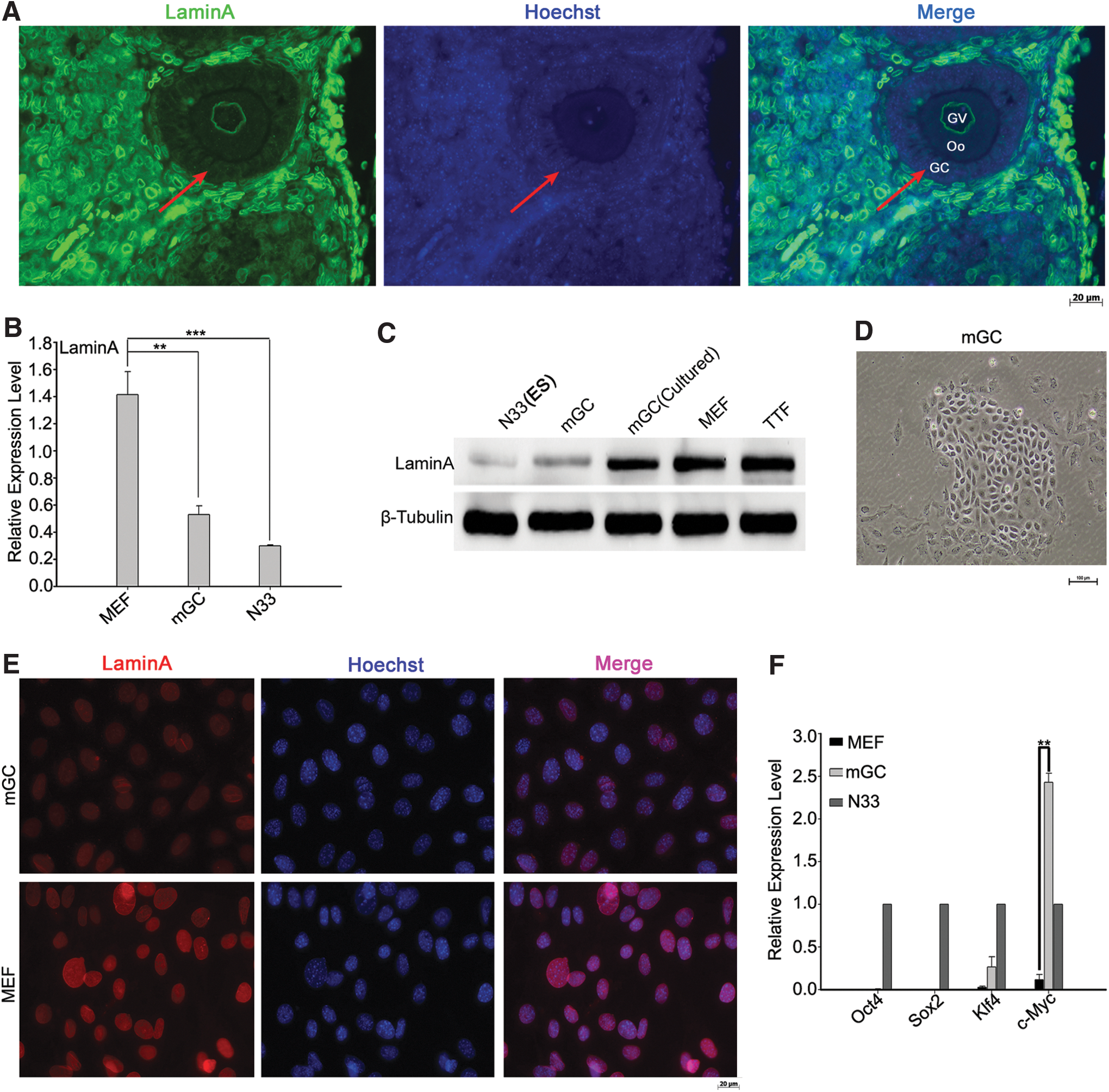
Low expression levels of LaminA in granulosa cells (mGC) of growing follicles from mouse ovaries.
In addition, GCs also expressed relatively high level of Klf4 and c-Myc endogenously, although they did not express Oct4 and Sox2 (Fig. 1F). We speculated that GCs might have higher potential to the induction of iPS.
Reprogramming of mouse GCs with Oct4 and Sox2
Since mouse GCs express endogenous Klf4 and c-Myc, we attempted to generate iPS from GCs using only two factors, Oct4 and Sox2 (OS), and compared with the induction of iPS from GCs using standard OSKM or Oct4, Sox2 and Klf4 (OSK). MEF were induced to iPS as controls under the same conditions. After transfected with OSKM and GFP, transfected cells with green fluorescence were observed in both GCs and MEF (Supplementary Fig. S1A; Supplementary Data are available online at
The morphologies of the cells began to change and the cells began to aggregate by day 3 after transduction for iPS induction. Both GCs and MEF formed colony aggregates as early as day 6 with OSKM, and the colony appeared more compact in GCs than in MEF (Fig. 2A, B). At day 6, the cells were replated on MEF feeders, cells induced by OSKM formed colonies rapidly, and the colonies became apparent by day 9. OSK and OS also induced changes in cell morphology. GCs and MEF induced by OSK and GCs by OS showed cell clusters but did not form colonies by day 9 of induction. Unlike GCs, MEF failed to form any distinct cell aggregation or clusters at day 9 by the induction with OS. At day 12 (after culture on feeders for 6 days), compact and domed clones with clear boundaries formed on the feeder cells in MEF and GCs using OKSM or OSK. Notably, OS induced iPS primary colonies from GCs but not from MEF (Fig. 2A, B). All of the iPS clones showed AP-positive staining (Fig. 2C). iPS primary colonies were efficiently achieved by the induction with OSKM, and the proportion of AP-positive clones of GCs (8%) was higher than that of MEF (Fig. 2D). The induction efficiency was much lower with OSK (<0.5%), and the efficiency did not differ between MEF and GCs (Fig. 2D). GCs formed clones by the induction with OS despite at reduced efficiency shown by AP-positive staining (about 0.1%), whereas MEF with OS failed to form any clones. Together, the efficiency of iPS generation from GCs is comparatively higher than that of MEF using four factors OSKM. Furthermore, GCs can be reprogrammed to iPS formation by only two factors, Oct4 and Sox2.
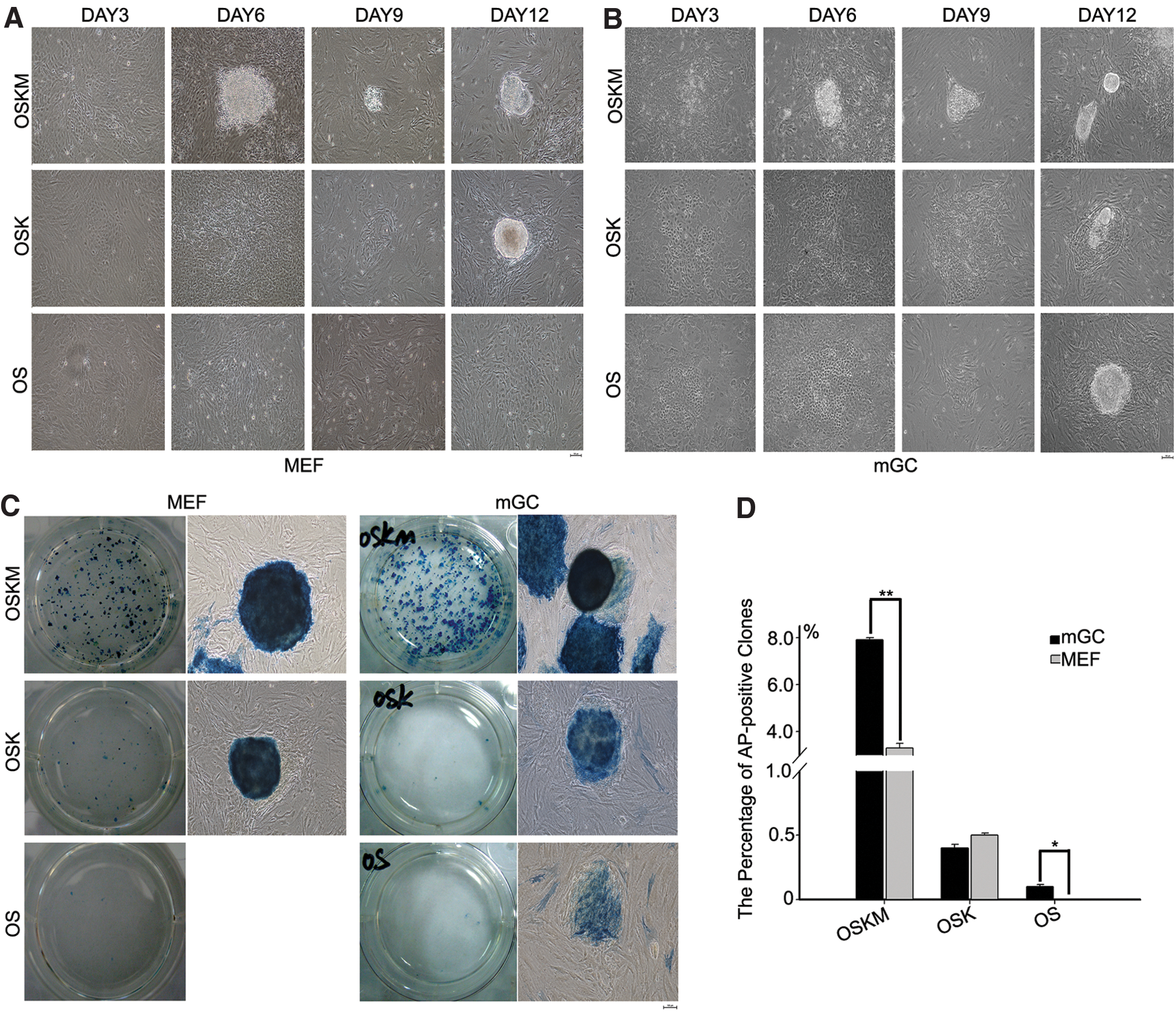
Derivation of iPS cells from MEF and mGC.
Characterization of iPS cells induced from GCs
We obtained 12 iPS cell lines (mGCiPS) generated from GCs by OSKM, OSK, or OS and these iPS cells were stably passaged. These stable iPS cells resembled typical ES cell colonies in morphology, with large nuclei and clear nucleoli and compact clonal boundaries, distinct from feeder fibroblasts (Fig. 3). The iPS clones have been propagated for more than 20 passages. The iPS cells induced from GCs, regardless of using OSKM, OSK, or OS, expressed multiple pluripotent stem cell markers Oct4 and Nanog in the nuclei and SSEA-1 on cell surface by immunofluorescence, common ES cell markers (Fig. 3).
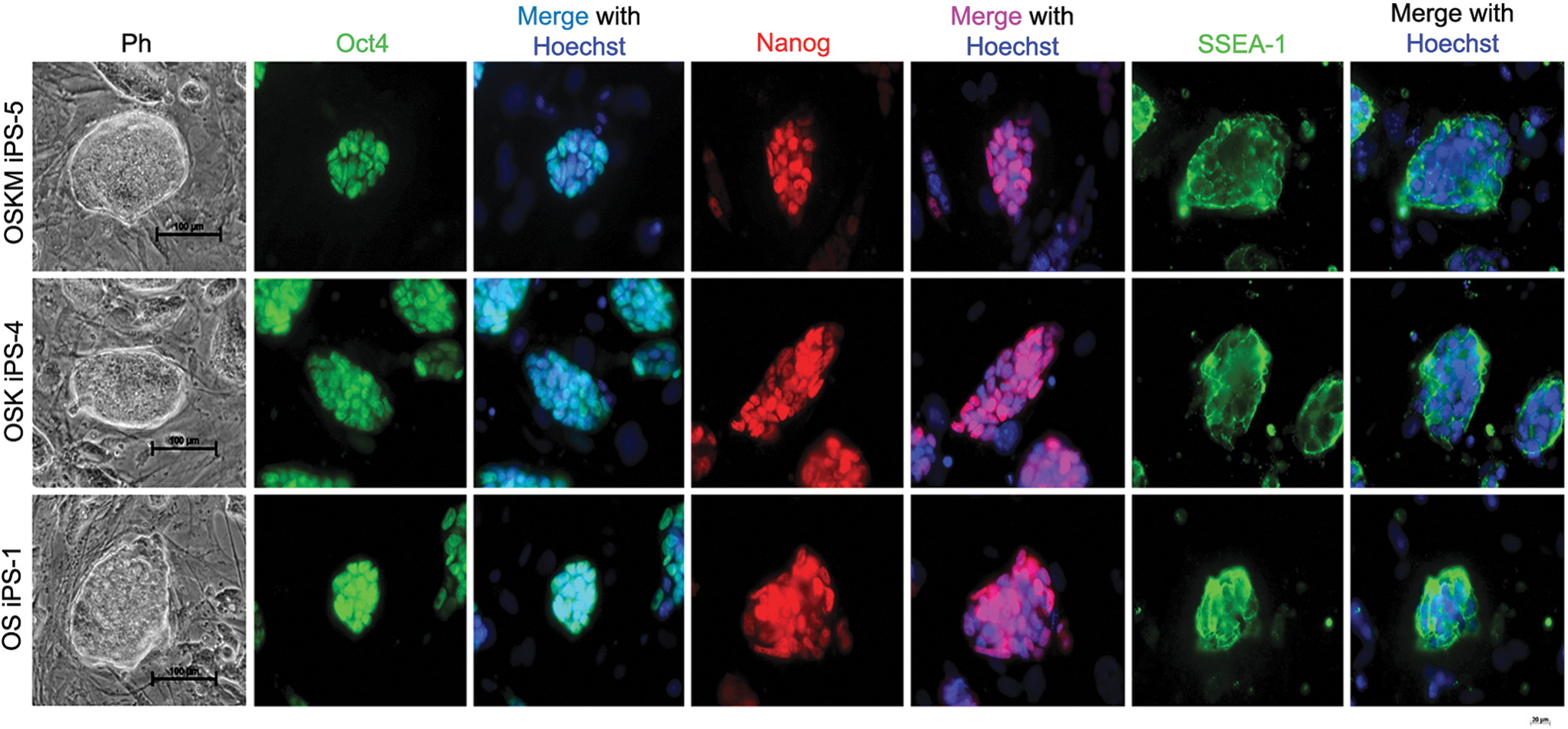
Characterization of iPS generated from mGC by immunofluorescence staining with ES markers, Oct4, Nanog and SSEA1. Clones by phase-contrast optics (Ph) of iPS from mGC (scale bar=100 μm) express strong Oct4, Nanog, and SSEA1 by immunofluorescence (scale bar=20 μm). Nuclei stained with Hoechst 33342 (blue). Color images available online at
RT-qPCR analysis of GCs, mGCiPS, and mES revealed that mGC iPS cells were transcriptionally highly similar but not exactly identical to mES cells, and expression levels of Nanog, Oct4, Sox2, and Klf4 in both iPS and ES cells were dramatically higher than those of GCs (Fig. 4A). mGC after cultured in vitro did not express endogenous Klf4 (Fig. 4A). Reverse transcription-PCR analysis also confirmed the reactivation and expression of endogenous Nanog, Oct4, Sox2, Klf4, and c-Myc (Fig. 4B). Transgenes from the integrated retroviral vectors were silenced in the established iPS cells from GCs, using transgene-specific PCR primers (Fig. 4B). The expression of c-Myc did not vary markedly among mGC, mGCiPS, and mES, except for OSKM iPS (Fig. 4A, B). PCR of genomic DNA extracted from iPS using transgene-specific primers validated stable and corresponding integration of retroviral Oct4, Sox2, Klf4, and c-Myc by two, three, or four factors in combination (Fig. 4C).
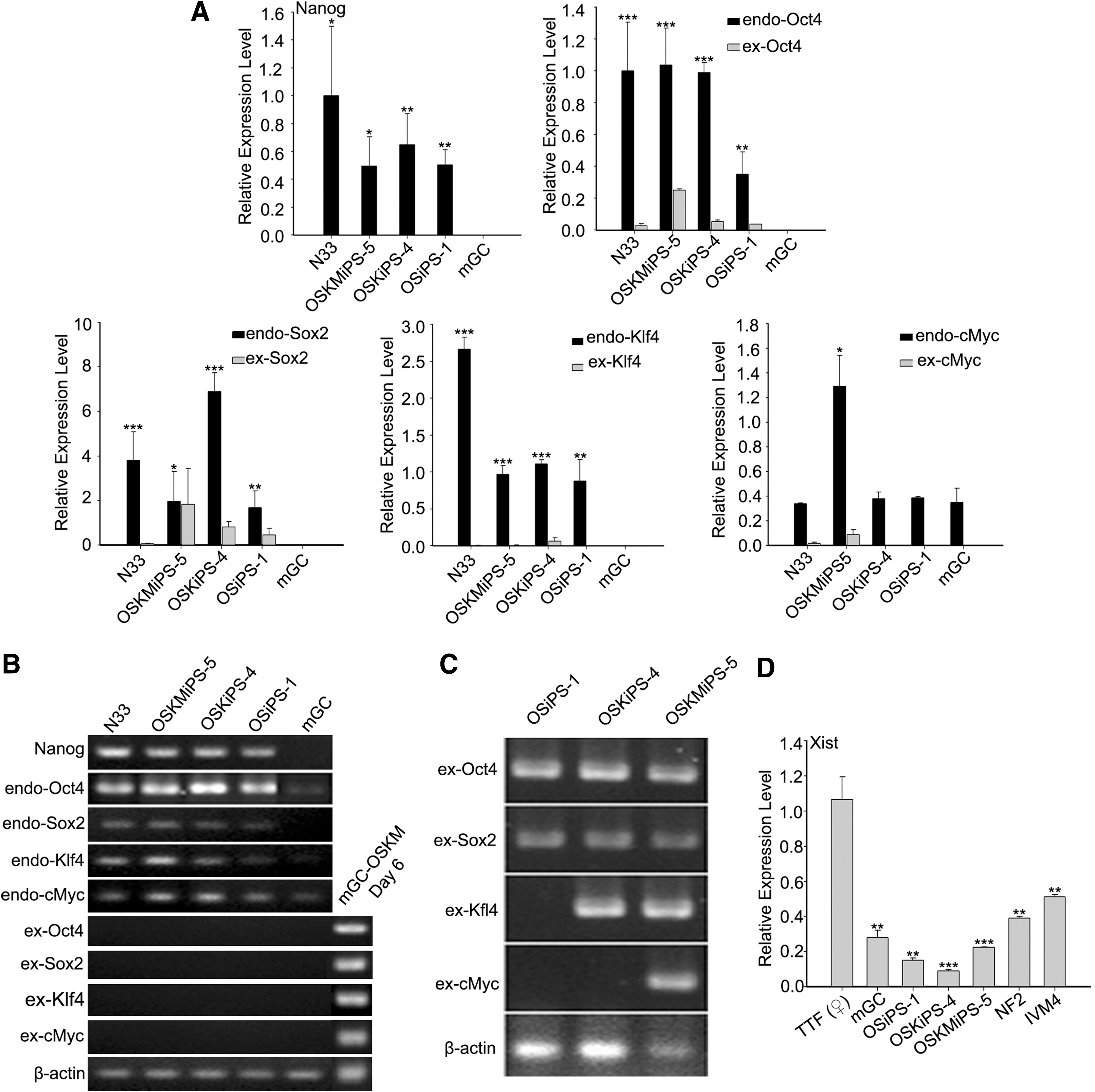
Expression of genes associated with pluripotency and integration of exogenous factors in iPS cells generated from mGCs.
In female mammals, one of the X chromosomes is inactivated [42]; however, the inactive X chromosome is reactivated in reprogrammed iPS cells [4,43,44]. Given that GCs were isolated from female mouse, we checked whether the inactive X chromosome of mGC iPS is reactivated. By measuring the expression of Xist, which is associated with X chromosome silencing [45], we found the expression levels of Xist in all of the mGC iPS lines were much lower than those of female TTF (differentiated somatic cells), and similar to or slightly lower than those of female mESC NF2 and parthenogenetic ES cell IVM4 (Fig. 4D). Interestingly, mGC also expressed Xist at a considerable low level (Fig. 4D), suggesting that GCs may have two active X chromosomes, additional characteristic of female stem cells.
Pluripotency of iPS cells generated from GCs
To examine the developmental potential of iPS cells generated from GCs, we performed in vitro differentiation by standard EB formation assay (Fig. 5A). Differentiation of iPS cells via EB formation yielded cells representing three embryonic germ layers indicated by tissue-specific immunofluorescence staining of SMA (cardiac muscle, mesoderm), β-III-tubulin (neurons, ectoderm), and AFP (liver, endoderm) (Fig. 5A).
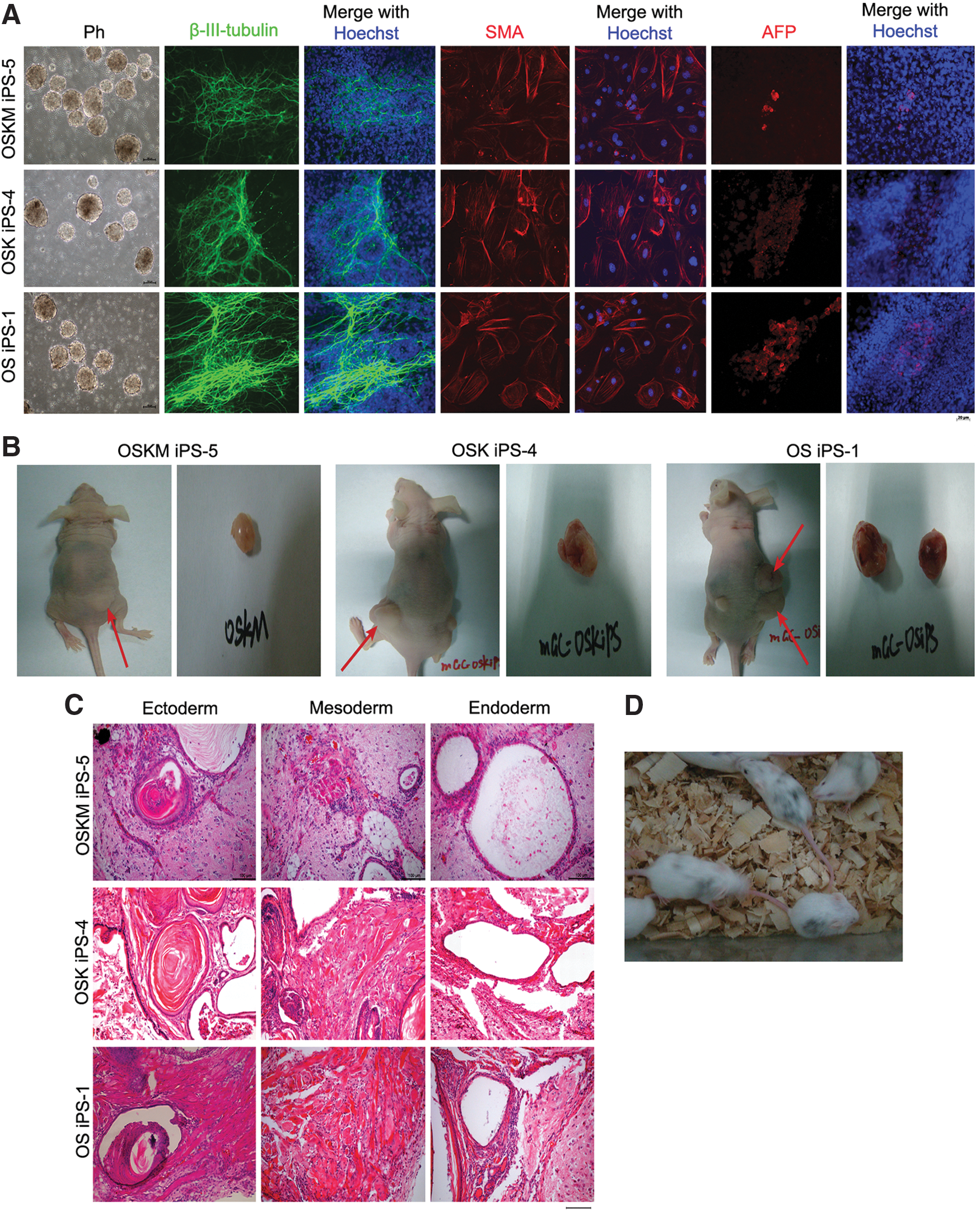
Pluripotency of iPS induced from mGC.
The in vivo differentiation test by transplantation into nude mice showed that all iPS cell clones tested formed teratomas (Fig. 5B), consisting of representative derivatives of three germ layers, including epidermis (ectoderm), muscle (mesoderm), and gland epithelium (endoderm) (Fig. 5C). These in vitro and in vivo characterizations demonstrated that iPS cells generated from GCs closely resemble ES cells in terms of pluripotency, marker expression, and differentiation potential.
To further test the pluripotency of iPS cells generated from GCs using two factors OS, we injected the iPS cells into blastocysts to test their capacity to form chimeras. Before injection, the karyotypes of mGC-OS iPS-1 were performed and the results showed that they had normal karyotype (>70%). Following injection of iPS with C57BL/6 background into blastocysts from ICR albino mice and embryo transfer, 19 pups were delivered and five were chimeras based on coat color (Fig. 5D). The chimeric mice formed by mGC-OS iPS-1 were generated successfully, and further experiments are required to test their germline capacity. Together, the iPS cells induced from GCs by two factors, Oct4 and Sox2, exhibit developmental and differentiation competence.
Discussion
We show that GCs can efficiently generate iPS cells with fewer transcription factors, Oct4 and Sox2. Notably, GCs express endogenous Klf4 and c-Myc, but not nuclear LaminA, allowing efficient reprogramming during iPS induction.
Our results also support the mathematical model that faithfully predicts the activation of OKSM targets in the course of reprogramming [46]. Previously, c-Myc is required for early-stage reprogramming [47] and Sox2 for late requirement during reprogramming [6,48]. Klf4 plays an unanticipated dual function by predominantly repressing somatic targets early and activating pluripotency targets late in iPS cell formation [46].
Further motivation is to direct reprogramming with fewer factors. There have been concerns about the integration of introduced factors into the genome that may negatively affect potential cell replacement therapy in humans. Certain cell types, however, can be reprogrammed with fewer factors. Neuronal stem cells, a type of multipotent adult stem cells, for example, can be sufficiently reprogrammed to iPS cells by overexpression of one single gene, Oct4, presumably because the other three reprogramming genes are already active in the cells [49].
During last several years since Yamanaka's discovery of iPS from MEF [1], iPSC lines have been generated from a variety of somatic cell types [10 –23]. More cell types that are amenable and efficiently to be reprogrammed remain to be explored. Here, we have been able to generate iPSCs from GCs with only two transcription factors, Oct4 and Sox2, by using two single retroviral vectors without any additional chemical compounds. Likewise, cord blood cells and renal proximal tubular cells can be reprogrammed by Oct4 and Sox2 transcription factors with no additional chemical compounds [14,50]. As Klf4 and c-Myc are both carcinogenic, the removal of them would improve the safety of iPS in clinical application. Previously, the chimeric and progeny mice derived from iPS cells that are devoid of Myc transgene appear to be normal [51 –53]. Our data show that GCs express higher endogenous levels of Klf4 and c-Myc when compared with other somatic cell types such as fibroblasts, suggesting that higher endogenous levels could compensate for the omission of Klf4 and c-Myc in the reprogramming cocktail. Thus, iPS cells can be generated from GCs in the absence of the oncogenes, Klf4 and c-Myc.
iPS cells can be induced from GCs using only two factors, Oct4 and Sox2, although at lower efficiency. The efficiency will likely be improved greatly by the addition of small chemicals that have been shown to facilitate reprogramming. For instance, BIX-01294, a histone methyltransferase inhibitor, was shown to enable the reprogramming of MEFs under Oct4 and Klf4 two-factor conditions [8]. 5-azacytidine (5-AZA), a DNA methyltransferase inhibitor, improves the reprogramming efficiency in MEFs up to fourfold by transiting partially reprogrammed cells to become fully pluripotent [54,55]. Histone deacetylase inhibitors such as valproic acid can reprogramme human fibroblasts with only Oct4 and Sox2 [56].
Together, we show here for the first time the generation of iPS cells from a new cell source GCs using two factors, Oct4 and Sox2. GCs are byproducts from human IVF clinic and also from IVF and embryo biotechnology routinely performed for large domestic animals, thus the generation of iPS cells using GCs may show great potential in future clinical applications, and in animal production and transgenesis.
Footnotes
Acknowledgments
We thank Jihong Yuan, Jiaojiao Li, and Mengyuan Liu for helping with the experiments. This work was supported by the China MOST National Major Basic Research Program (2009CB941004, 2011CBA01002, 2010CB94500), the National Natural Science Foundation of China (31271587), and the Natural Science Foundation of Tianjin (12JCZDJC24800).
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.