
Abstract
Recently, the involvement of PIN1, a peptidyl-prolyl cis/trans isomerase, has been reported in age-related bone homeostasis and adipogenesis. However, the role of PIN1 during odontogenic and adipogenic differentiation remains to be fully understood, particularly regarding human dental pulp stem cells (HDPSCs). Thus, in the present study, we have investigated the role of PIN1 in odontogenic and adipogenic differentiation of HDPSCs and signaling pathways possibly involved. PIN1 mRNA and protein level were upregulated in a time-dependent manner during adipogenic differentiation, increasing until 1 day of odontogenic induction and then steadily declined during odontogenic differentiation. Treatment of a known PIN1 inhibitor, juglone, significantly increased odontogenic differentiation as confirmed by alkaline phosphatase (ALP) activity, calcium deposition, and mRNAs induction of odontogenic markers [ALP, osteopontin (OPN), osteocalcin (OCN), dentin sialophosphoprotein (DSPP), and dentin matrix protein 1 (DMP-1)]. On the contrary, adipogenic differentiation was dramatically reduced upon juglone treatment, with concomitant downregulation of lipid droplet accumulation and adipogenic marker genes [peroxisome proliferation-activated receptor gamma (PPARγ), lipoprotein lipase (LPL), and adipocyte fatty acid-binding protein (AP2)]. In contrast to PIN1 inhibition, the overexpression of PIN1 via adenoviral infection (Ad-PIN1) in HDPSCs inhibited odontogenic differentiation but increased adipogenic differentiation, in which stem cell property markers such as stage-specific embryonic antigen-4 (SSEA-4) and STRO-1 were upregulated during odontogenic differentiation but downregulated in adiopogenic differentiation. Consistently, juglone-mediated inhibition of PIN1 augmented the osteogenic medium (OM)-induced activation of bone morphogenetic protein (BMP), Wnt/β-catenin, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and nuclear factor-kappa B (NF-κB) pathway, which response was reversed by Ad-PIN1. Moreover, juglone blocked the adipogenic induction medium-induced activation of PPARγ, C/EBPα, C/EBPβ, ERK, and NF-κB pathways, which was rescued by Ad-PIN1 infection. In summary, the present study shows for the first time that PIN1 acts as an important modulator of odontogenic and adipogenic differentiation of HDPSCs and may have clinical implications for regenerative dentistry.
Introduction
R
Phosphorylation of proteins on Ser/Thr residues is an important cellular signaling mechanism to induce their functional activity [7]. The peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 (PIN1) has been identified as a regulator of the phosphorylation-mediated signaling that catalyzes the conversion of specific phosphorylated motifs between the two completely distinct conformations in a subset of proteins [8]. PIN1 regulates diverse cellular processes, including growth-signal responses, cell cycle progression, cellular stress responses, neuronal function, and immune responses [9,10]. For example, neuronal differentiation of primary neuronal precursor cells was impaired in the absence of PIN1 but enhanced upon forced expression of PIN1 [11]. In addition, the adipogenesis of human preadipocytes [12] and mouse embryonic fibroblasts [13] was markedly suppressed by PIN1 gene silencing, which resulted in the diet-induced obesity-resistant PIN1 knock-out (KO) mice [12]. In contrast, silencing or inhibition of PIN1 promoted skeletal muscle differentiation of C2C7 murine muscle cells and myeloid differentiation of primary acute myeloid leukemia blasts, respectively [14]. Notably, the important role of PIN1 in age-related bone homeostasis has been reported [15]. However, nothing is known about the role of PIN1 in odontogenic and adipogenic differentiation of human dental pulp cells (HDPCs).
Therefore, the purpose of the present study was to clarify the role of PIN1 in odontogenic and adipogenic differentiation of HDPCs, with its underlying signal transduction pathways.
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and penicillin/streptomycin were purchased from Gibco BRL Co. (Grand Island, NY). Juglone, ascorbic acid, β-glycerophosphate, dexamethasone, insulin, indomethacin, methyl-isobutylzanthine, and β-actin antibody were purchased from Sigma-Aldrich Chemical Co (St. Louis, MO). Smad2/3 antibody was purchased from BD Bioscience. Antibodies against phospho-extracellular signal-regulated kinase (p-ERK), ERK, phospho-c-Jun N-terminal kinase (p-JNK), JNK, phospho-Smad2 (p-Smad2), phospho-GSK3β, C/EBPα, Smad1/5/8, and phospho-Smad1/5/8 (p-Smad1/5/8) were purchased from Cell Signaling, Inc. (Beverly, MA). Smad-ubiquitin regulatory factor 1 (Smurf1), bone morphogenetic protein 2 (BMP-2), peroxisome proliferation-activated receptor gamma (PPARγ), C/EBPβ, Wnt1, Wnt3a, Wnt5a, PIN1, phospho-nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha (IκB) (p-IκB), IκB, p65, phospho-Akt (p-Akt), Akt, and donkey antigoat IgG were purchased from Santa Cruz Biotechnology (Delaware Avenue, CA).
Cell culture
Surgical pulp samples (n=10) were obtained from nine patients (four females and five males), with a mean age of 16 years, at the Department of Oral and Maxillofacial Surgery, Kung Hee Dental Hospital (University of Kung Hee, Seoul, Korea), during orthodontic treatment. Dental pulps were isolated after tooth extraction as previously reported [16]. Cells were cultured in alpha-minimum essential medium (α-MEM; Gibco, Life Technologies, Grand Island, NY) supplemented with 10% FBS (Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in 5% CO2. The fresh medium was changed every 2 days. Cells were subcultured at the ratio of 1:3 when they reached 75%–85% confluence. Experiments were performed only in the first five cell passages. All experimental procedures were approved by the Institutional Review Board of Kyung Hee University, Seoul, South Korea.
To induce odontogenic differentiation, primary HDPCs were cultured with OM (DMEM, 1% FBS, 50 μg/mL ascorbic acid, 10 mM β-glycerophosphate, and 10−7 M dexamethasone) for 7 and 14 days [17]. For adipogenic differentiation, the adipogenic induction medium (AM, high glucose DMEM, 10% FBS, 1 μM dexamethasone, 10 μg/mL insulin, 100 μM indomethacin, and 0.5 mM methyl-isobutylzanthine) was used to induce differentiation [18,19]. The medium was removed and replaced with the adipogenic maintenance medium (high glucose DMEM, 10% FBS, 10 μg/mL insulin) for 4 and 11 days [18,19].
Preparation of adenovirus
Preparation and amplification of adenovirus was performed as described previously [12]. Adenoviruses encoding PIN1 were created using the Virapower adenovirus expression system according to the manufacturer's instruction (Invitrogen, Carlsbad, CA). Site-specific recombination between entry vectors (PIN1-pENTR) and the adenoviral destination vector (pAd/PL-DEST) were established with LR clonase II (Invitrogen). Viruses from the culture supernatants of 293 cells that showed cytopathogenic effects were purified by cesium chloride banding. Virus titers were determined by a plaque assay using serial dilution.
Isolation of RNA and reverse transcription polymerase chain reaction
Total RNA was isolated using the TRIzol reagent (Invitrogen) according to the manufacturer's instructions. It was then reverse transcribed using AccuPower RT pre-mix (Bioneer, Daejeon, Korea). Polymerase chain reaction (PCR) amplication of the resulting cDNA samples was performed using a GeneAmp PCR System 2400 thermal cycler (PerkinElmer, Wellesley, MA). Primer information is summarized in Table 1. The following PCR conditions were used: 34 cycles of denaturation at 95°C for 30 s, primer annealing at 60°C for 30 s, and extension at 72°C for 30 s. The PCR products were resolved on 1.5% agarose gels and stained with ethidium bromide.
Preparation of whole cell lysates, cytosolic, and nuclear extracts
Cells (10×106) were harvested, washed once with ice-cold phosphate-buffered saline (PBS), and gently lysed for 30 min in ice-cold lysis buffer [1 mM MgCl2, 350 mM NaCl, 20 mM N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES), 0.5 mM ethylenediaminetetraacetate (EDTA), 0.1 mM ethyleneglycol-bis(β-aminoethyl ether)-N,N′-tetraacetic acid (EGTA), 1 mM dithiothreitol (DTT), 1 mM Na4P2O7, 1 mM PMSF, 1 mM aprotinin, 1.5 mM leupeptin, 1 mM Na3VO4, 20% glycerol, 1% NP40]. Cell lysates were centrifuged for 10 min at 4°C (10,000 g) to obtain the supernatants (whole cell lysate). For the cytosolic extracts, the confluent cells were washed in ice-cold PBS, suspended in ice-cold hypotonic buffer (10 mM HEPES, pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.3% NP-40, 0.1 mM EDTA, 0.1 mM EGTA, 0.5 mM DTT, 0.1 mM PMSF, 0.1% aprotinin, and lysed for 15 min on ice. The lysates were centrifuged for 10 min at 10,000 g. The supernatants were designated as cytosolic extracts. The nuclear pellet was resuspended in the high salt lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 0.1 mM EGTA, 25% glycerol, 0.5 mM DTT, 0.1 mM PMSF, 0.1% aprotinin) and incubated at 4°C for 30 min. The resulting supernatants were reserved as nuclear extracts. The protein concentrations of samples were determined by Bio-Rad (Hercules, CA) protein assay.
Western blot analysis
Proteins (30 μg per lane) were then separated by 6%–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and blotted onto Hybond-N nitrocellulose. The membrane was then rinsed once with Tris-buffered saline (TBS) and blocked for 1 h in blocking buffer (TBS with 5% nonfat dry milk and 0.05% Tween 20) and then incubated with primary antibody diluted in 2.5% blocking buffer. Unbound primary antibody was subsequently removed by washing three times in TBS containing 0.05% Tween 20, before bound primary antibody was labeled through incubation at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibody. Following three washes in TBS containing 0.05% Tween 20, the membranes were developed using the ECL chemiluminescence detection kit.
ALP assay
Incubated HDPCs were washed with PBS and then sonicated with a cell disruptor. ALP activity was measured using p-nitrophenyl phosphate (3 mM final concentration) as the substrate in 0.7 M 2-amino-methyl-1-propanol (pH 10.3) and 6.7 mM MgCl2. Absorbance was measured at 410 nm using an enzyme-linked immunosorbent assay reader (Beckman Coulter, Fullerton, CA).
Alizarin red staining
After incubation in OM for 7 and 14 days, cell mineralization was determined using Alizarin red staining. Cells were washed twice in distilled water and fixed by incubation in ice-cold 70% (v/v) ethanol for 1 h. They were then rinsed twice in deionized water and stained with Alizarin red (dissolved in deionized water, pH 4.2) for 10 min at room temperature.
Oil red O staining
The adipogenic cultures were fixed in 4% paraformaldehyde (in PBS) for 1 h. The sample was then washed with 60% isopropanol (in PBS). It was stained with fresh 60% Oil Red-O solution (in PBS) for 2 h and washed with distilled water.
Flow cytometric analysis
For stage-specific embryonic antigen-4 (SSEA-4) and STRO-1, antigens from BD Pharmingen (Buccinasco, Milan, Italy) were used. Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes, Eugene, OR) was used as the secondary antibody. Cells were fixed in 4% paraformaldehyde for 30 min. Primary antibodies were incubated in 1% bovine serum albumin (BSA)/PBS for 6 h at 4°C. Secondary antibodies were incubated in 1% BSA/PBS for 2 h at 4°C. After the final wash, a total of 10,000 events was acquired and analyzed using a FACsCalibur apparatus (Becton Dickinson, Franklin Lakes, NJ) using Cell Quest Research Software (Becton Dickinson).
Immunofluorescence assay
For the localization of p65, HDPCs were seeded onto glass coverslips in six-well plates at a density of 1×105 cells/well. Treated cells were fixed with 4% paraformaldehyde for 30 min following permeabilization with 0.1% Triton X-100. After being washed in PBS buffer, slides were blocked with 10% normal goat serum for 1 h and then incubated with mouse monoclonal anti-human p65 antibody for 2 h at a 1:100 dilution in 1% normal goat serum. The slides were washed with PBS, incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG at a 1:500 dilution in 0.5% normal goat serum for 1 h, and counterstained for nuclei with 10 μg/mL propidium iodide. Slides were imaged at 400×magnification on a confocal microscope (Olympus, Tokyo, Japan).
Mouse tooth germ and dental pulp histology and immunohistochemistry
Mandibles from 1-, 7-, and 28-day-old mice were dissected and fixed in 4% paraformaldehyde overnight at 4°C. After rinsing with 0.01 M PBS, the specimens were decalcified in 10% EDTA/PBS for 2–4 weeks, then dehydrated, embedded in paraffin, and sectioned at a thickness of 5 μm. For immunostaining, tissue sections were treated with 3% hydrogen peroxide and incubated with rabbit polyclonal anti-Pin1 antibody (1:200; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Histostain Plus Rabbit Primary (DAB) kit (Zymed Laboratories, San Francisco, CA) was used according to the manufacturer's instructions.
Statistical analysis
Differences among groups were analyzed using one-way analysis of variance combined with the Bonferroni test. All values were expressed as means±standard deviations, and differences were considered significant at P<0.05.
Results
Time course of PIN1 mRNA and protein expression during odontogenic and adipogenic differentiation of HDPCs
To investigate the expression of PIN1 mRNA and protein during odontogenic or adipogenic differentiation of HDPCs, cells were cultured in OM or AM for 14 days, and the samples were analyzed by conventional reverse transcription polymerase chain reaction (RT-PCR) and western blotting (Fig. 1A, B). Moderate PIN1 levels were detected in nonstimulated cells, and PIN1 mRNA and protein was increased early, with highest expression 1 day after OM treatment and a decrease was observed thereafter (Fig. 1A). In contrast, expression of PIN1 mRNA and protein was increased in a time-dependent manner following exposure to AM after the initiation of treatment (Fig. 1B).

Time course expression of PIN1 during odontogenic
Effects of PIN1 inhibition and activation on cell growth and stem cell characteristics in HDPCs
To examine the effects of PIN1 inhibition and overexpression on cell growth, the cell viability was analyzed by MTT assay. As shown in Figure 2A and B, juglone and adenovirus PIN1 (Ad-PIN1) did not significantly affect cell viability during odontogenic and adipogenic differentiation in HDPCs. To investigate how stem cell properties were affected by PIN1 inhibition and activation, SSEA-4- and STRO-1-positive cells were counted by flow cytometry. SSEA-4+ and STRO-1+ cells were decreased in OM- and AM-treated cells, compared with control HDPCs. Treatment with the PIN1 inhibitor, juglone, enhanced OM-induced decrease of SSEA-4+ and STRO-4+ cells but reversed AM induction in these cells (Fig. 2C, E). In contrast, pretreatment with Ad-PIN1 to activate PIN1 reversed OM-induced decrease of SSEA-4+ and STRO-1+ cells (Fig. 2D) but enhanced AM induction of these factors (Fig. 2F).

Effects of PIN1 inhibitor (juglone) and Ad-PIN1 on cell growth
Effects of PIN1 inhibition on odontogenic differentiation of HDPCs
To investigate the potential of HDPCs undergoing odontoblast-like differentiation after PIN1 inhibitor (juglone) treatment, we assessed ALP activity, Alizarin red staining, and the level of mRNA encoding ALP, OPN, OCN, DMP-1, and DSPP. ALP activity and calcified nodule formation of HDPCs were markedly stimulated by juglone in a dose-dependent manner (Fig. 3A, B). In addition, juglone dose-dependently upregulated osteogenic (ALP, OPN, and OCN) and odontogenic (DMP-1 and DSPP) mRNAs (Fig. 3C).
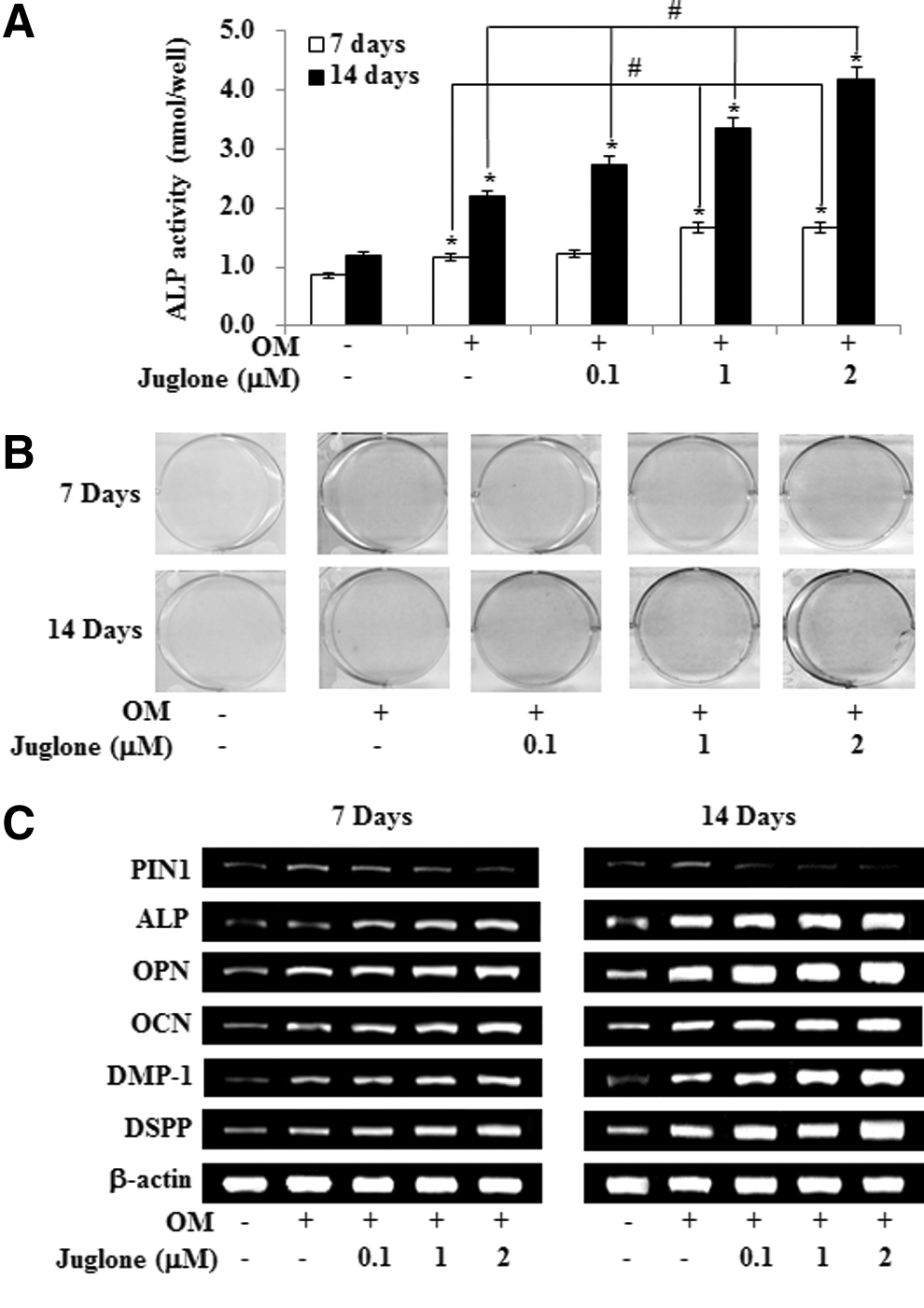
Effects of PIN1 inhibition by juglone on odontogenic differentiation in HDPSCs. Cells were treated with juglone and OM for 7 and 14 days. Differentiation was assessed by
Effects of PIN1 overexpression on odontogenic differentiation of HPDCs
To evaluate the effects of overexpression of PIN1 by Ad-PIN1 in HDPCs, HDPCs transduced with Ad-PIN1 were probed by western blot. Virus-derived activation of PIN1 could be detected in HDPCS throughout the 7-day culture period. However, decline of PIN1 expression by adenovirus was noted after 7 days in culture (Fig. 4A). We next examined the effect of exogenous Ad-PIN1 on odontogenic differentiation in HDPCs. As shown in Figure 3B, mock-infected cells showed the same level of expression with OM in ALP activity, Alizarin red stain and mRNA expression for marker genes. However, PIN1 overexpression blocked OM-induced ALP activity, mineralized module formation, and upregulation of ALP, OPN, OCN, DMP-1, and DSPP mRNA expression in HDPCs (Fig. 4B–D).
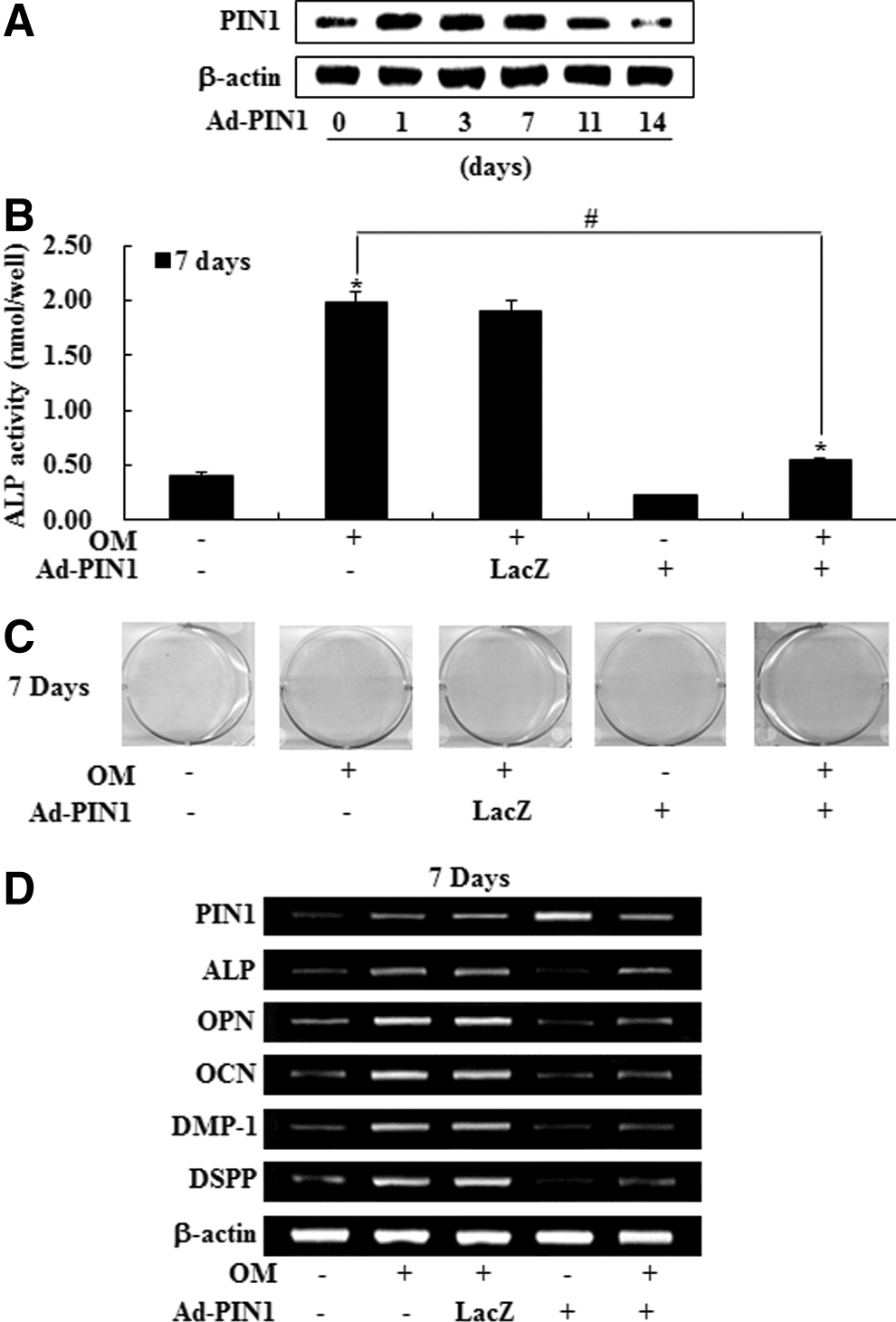
Effects of PIN1 overexpression by Ad-PIN1 on odontogenic differentiation in HDPSCs. Cells were pretreated with Ad-PIN1 for 5 h and post-treated with OM for 14 days. Time course expression of Ad-PIN1-induced PIN1 was examined by western blotting
Effects of PIN1 inhibition and overexpression on adipogenic differentiation in HDPCs
To investigate the effects of PIN1 inhibition and activation on adipogenic differentiation, numbers of adiopogenic cells were estimated by Oil Red-O staining and by assessing the expression of marker genes using RT-PCR. As shown in Figure 5A and B, Oil Red-O-positive colonies and mRNA expression of adipogenesis-related genes, such as PPARγ, lipoprotein lipase (LPL), and adipocyte fatty acid-binding protein (AP2) were decreased by juglone in a dose-dependent manner. In contrast, Ad-PIN1 increased red-colored lipid droplets in the Oil Red experiment (Fig. 5B), and mRNA expression of differentiation markers, PPARγ, LPL, and AP2, was not affected by Ad-LacZ (Fig. 5D).
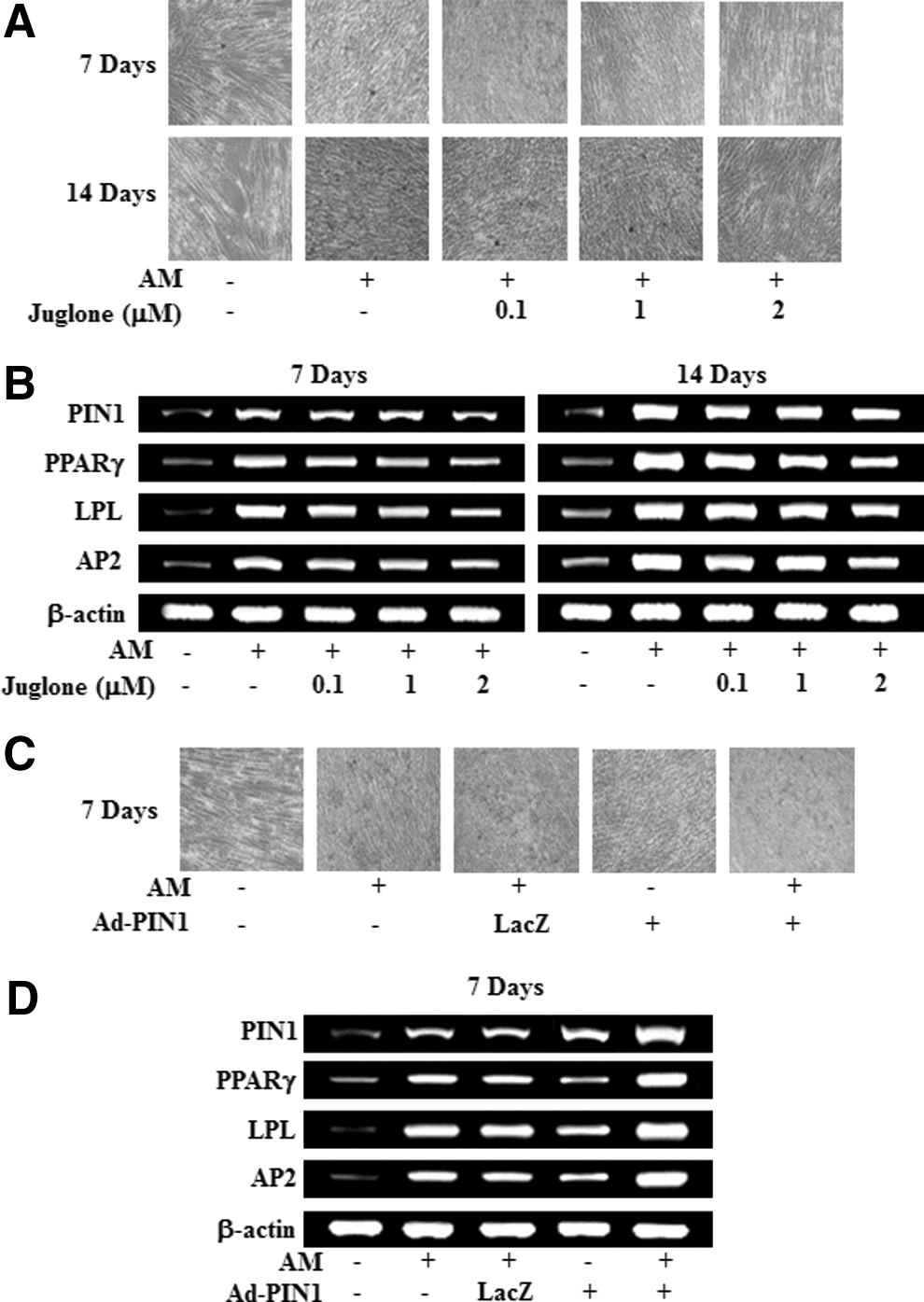
Effects of PIN1 inhibition and overexpression on adipogenic differentiation in HDPSCs. Cells were treated with juglone and AM
Effects of PIN1 inhibition and overexpression on transforming growth factor-beta, BMP, and Wnt/β-catenin pathways during odontogenic differentiation in HDPCs
To ask whether the BMP pathway may be involved in odontogenic differentiation of HDPCs, we examined the effects of OM on the expression of BMP-2 and its downstream phosphorylation (p) of Smad1/5/8 and Smurf1. OM treatment time dependently increased the expression of BMP-2 and p-Smad1/5/8 but decreased Smurf1 expression (Fig. 6A). Because the BMP pathway was activated by OM in HDPCs, we next tested whether PIN1 activation or inhibition might influence these same signaling molecules. Treatment of HDPCs with juglone enhanced OM-induced BMP-2 and p-Smad1/5/8 expression and downregulated Smurf1. In contrast, pretreatment with Ad-PIN1 and the BMP inhibitor noggin blocked OM-induced BMP-2 and p-Smad1/5/8 expression and downregulated Smurf1 (Fig. 6B).

Effects of PIN1 inhibitor (juglone) and Ad-PIN1 on BMP-2/Smad pathway expression during odontogenic differentiation in HDPSCs. Time course expression of OM-induced BMP-2/Smad pathway was examined at indicated time points by western blotting
To examine the involvement of Wnt/β-catenin signaling in the regulation of odontogenic differentiation, the effects of OM on expression levels of Wnt ligands and β-catenin protein expression were measured (Fig. 7C). We found that OM induced Wnt1 and Wnt3a expression in a time-dependent manner, whereas Wnt5a was not changed (data not shown). As a consequence, OM also enhanced total β-catenin protein levels in HDPCs (Fig. 7C). Treatment of HDPCs with juglone enhanced OM-activation of Wnt/β-catenin signaling, including Wnt1, Wnt3a, and β-catenin, whereas PIN1 activation by Ad-PIN1 showed opposing effects (Fig. 7D). Furthermore, Dickkopf-1 (DKK-1), an antagonist of canonical Wnt signaling, also blocked OM-induced Wnt and β-catenin activation.

Effects of PIN1 inhibitor (juglone) and Ad-PIN1 on the Wnt/β-catenin pathway during odontogenic differentiation in HDPSCs. Time course expression of OM-induced Wnt/β-catenin pathway protein levels as examined by western blotting
To examine whether endogenous transforming growth factor-beta (TGF-β) ligands and receptors were upregulated during odontoblastic differentiation, mRNA expression of TGF-β1, TGF-β2, TGF-β3, TGF-β type I receptor (TGF-βRI), and TGF-β type II receptor (TGF-βRII) were accessed. OM treatment time dependently increased the mRNA expression of TGF-β ligands and receptors (Fig. 8A). However, treatment with juglone and Ad-PIN1 had no effects on OM-induced TGF-β ligand and receptors activation (Fig. 8B). To investigate whether the TGF-β, BMP, and Wnt regulate PIN1 expression, HDPCs were pretreated with inhibitors of these signaling molecules. OM-induced PIN1 upregulation was not changed by the BMP inhibitor noggin, the Wnt inhibitor DKK-1, and TGF-β inhibitor SB431542 (Fig. 8C).
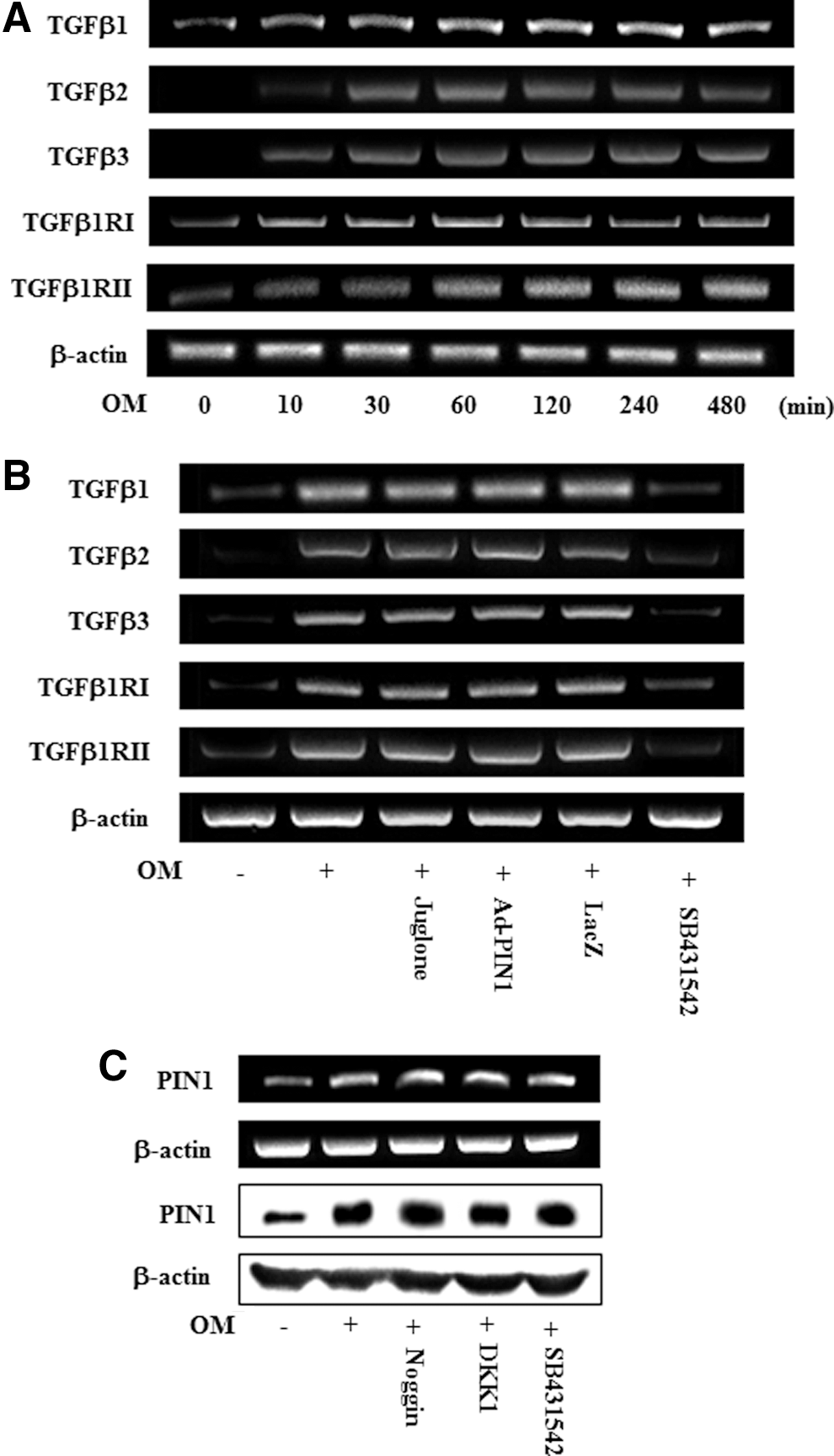
Time course expression of TGF-β ligands and receptors during odontoblastic differentiation
Effects of PIN1 inhibition and activation on overexpression of the mitogen-activated protein kinase and nuclear factor-kappa B pathways during odontogenic differentiation in HDPCs
Because the mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) pathways are involved in odontogenic differentiation in HDPCs, as we described previously [19 –21], we asked whether PIN1 activation or inhibition might influence these signaling molecules (Fig. 9). Pretreatment with juglone enhanced OM-induced phosphorylation of ERK and JNK but had no effect on the activation of p38. In contrast, pretreatment with Ad-PIN1 decreased OM-induced phosphorylation of ERK and JNK (Fig. 9A). Furthermore, PIN1 inhibition enhanced OM-induced NF-κB activation, which we determined by measuring IκBα degradation and phosphorylation, as well as nuclear translocation of p65. In contrast, Ad-PIN1 inhibited these processes (Fig. 9B). To ascertain whether p65 nuclear translocation following exposure to PIN1 activation or inhibition occurred, we used an immunofluorescence assay to confirm nuclear localization. Treatment with juglone caused the translocation of NF-κB p65 from the cytoplasm to cell nuclei. However, pretreatment with Ad-PIN1 abolished this NF-κB activation (Fig. 9C).
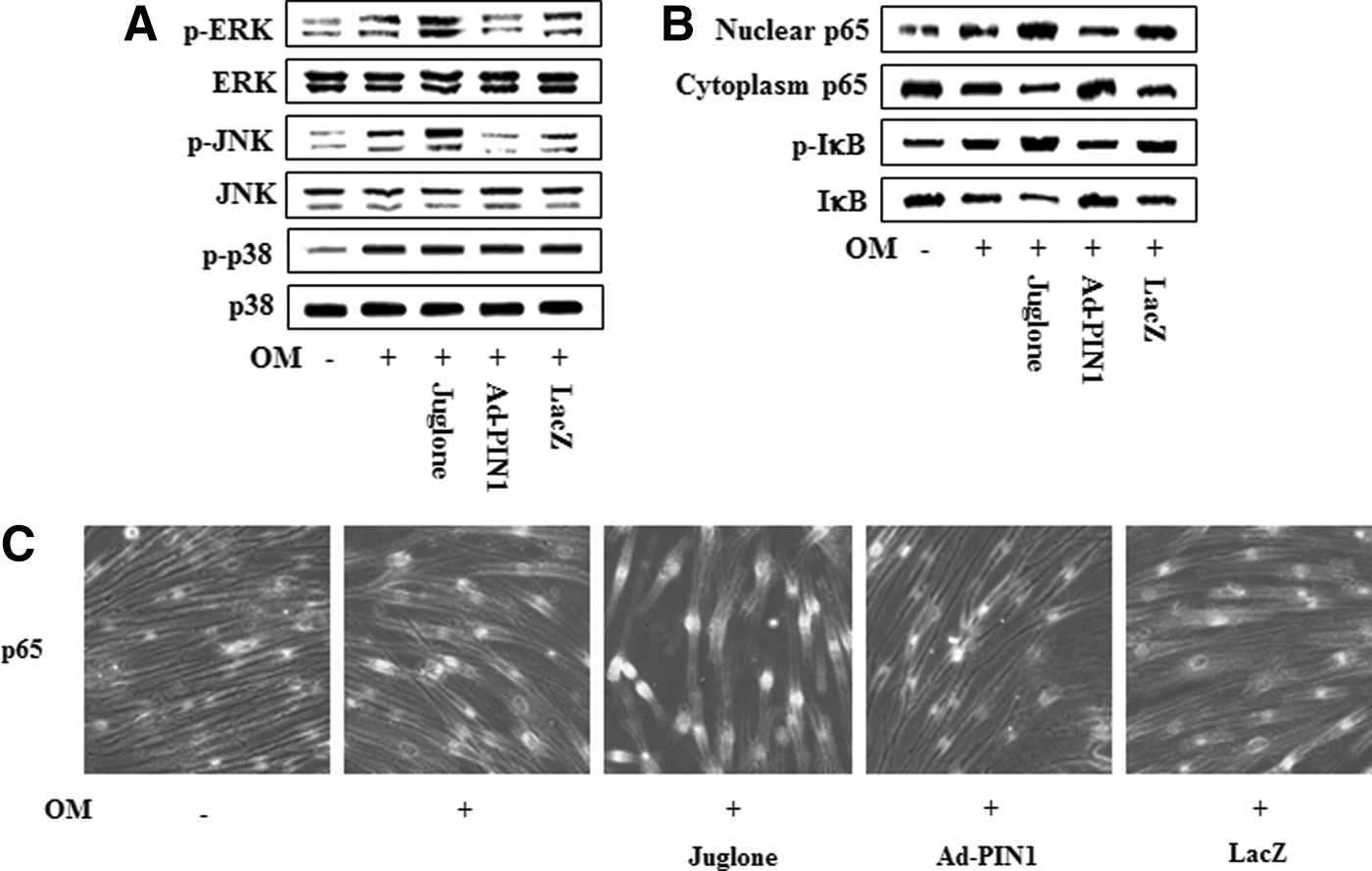
Effects of the PIN1 inhibitor (juglone) and Ad-PIN1 on MAPK and NF-κB protein levels during odontogenic differentiation in HDPSCs. Cells were pretreated with Ad-PIN1 for 5 h and treated with OM or 2 μM juglone and OM. Activation of MAPK (30 min) and NF-κB (60 min) were examined by western blotting
Effects of PIN1 inhibition and overexpression on transcriptional events during adipogenic differentiation in HPDCs
To investigate the expression of transcriptional events during adipogenic differentiation of HDPCs, cells were cultured in AM for 7 days and C/EBPβ, C/EBPα, and PPARγ protein were analyzed by western blotting (Fig. 10A). Expression of C/EBPβ protein was increased in a time-dependent manner following exposure to OM until 12 h after the initiation of treatment, after which it decreased. However, expression of C/EBPα and PPARγ protein increased in time-dependent manner for 7 days (Fig. 10A). To investigate the effects of PIN1 inhibition and overexpression on adipogenic marker expression during adipogenic differentiation, the expression of C/EBPβ, C/EBPα, and PPARγ was examined. As shown in Figure 10B, AM-induced activation of C/EBPβ, C/EBPα, and PPARγ was attenuated by juglone treatment but increased following pretreatment with Ad-PIN1.
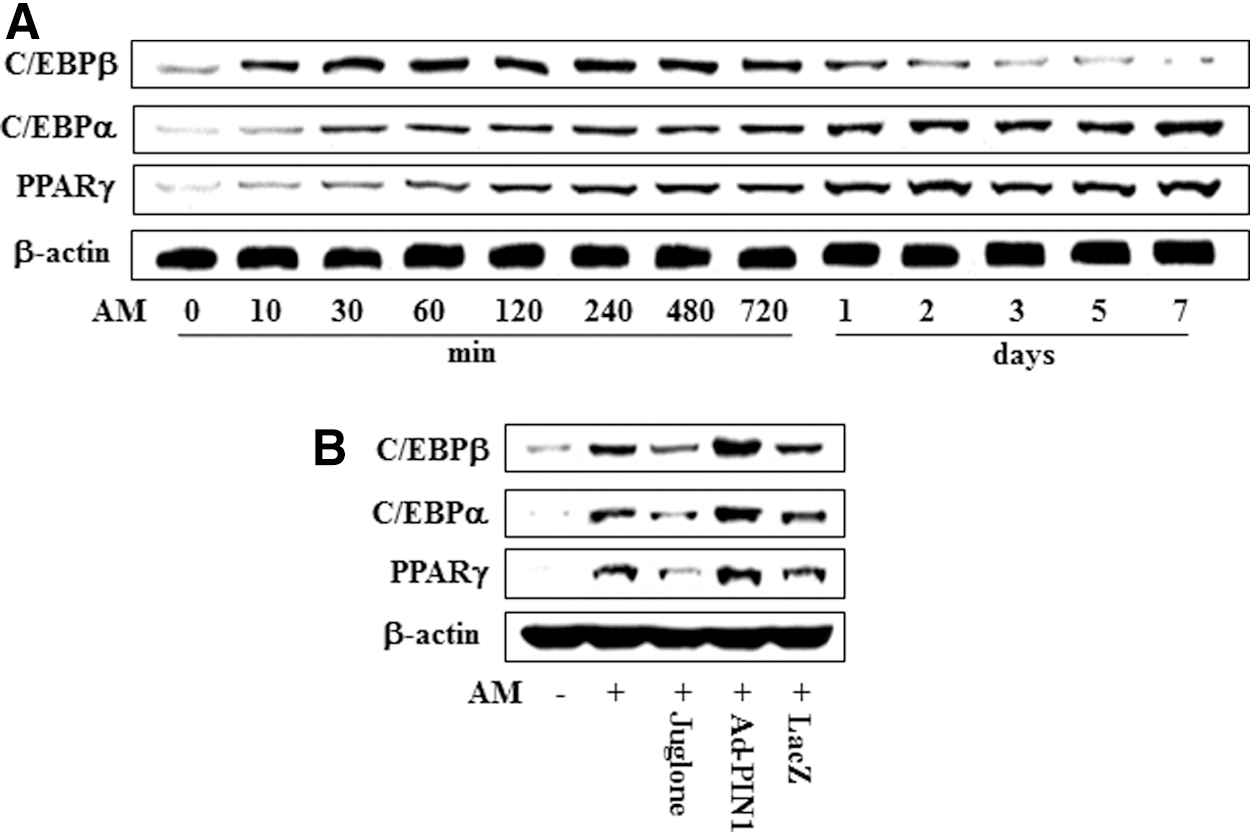
Effects of PIN1 inhibitor (juglone) and Ad-PIN1 on expression of master regulatory transcription factors for adipogenic differentiation in HDPSCs. Time course expression of AM-induced transcriptional factors were examined by western blotting
Effects of PIN1 inhibition and overexpression on the MAPK and NF-κB pathways during adipogenic differentiation in HPDCs
To investigate whether the MAPK and NF-κB signal pathways are also affected by AM in HDPCs, we examined MAPK and NF-κB activation. AM treatment increased the phosphorylation of ERK and p38 and caused the degradation or phosphorylation of IκBα, as well as the nuclear translocation of p65, but did not affect JNK (Fig. 11A). Maximal expression of p-ERK and p-p38 was detected following the incubation of HDPCs in AM for 30 min, whereas NF-κB activation levels peaked at 60 min (Fig. 11B). However, PIN1 inhibition and overexpression was not affected by phosphorylation of p38 and JNK (data not shown). Furthermore, AM-mediated activation of NF-κB and phosphorylation of ERK was abolished by juglone but was enhanced by Ad-PIN1 (Fig. 11C, D).
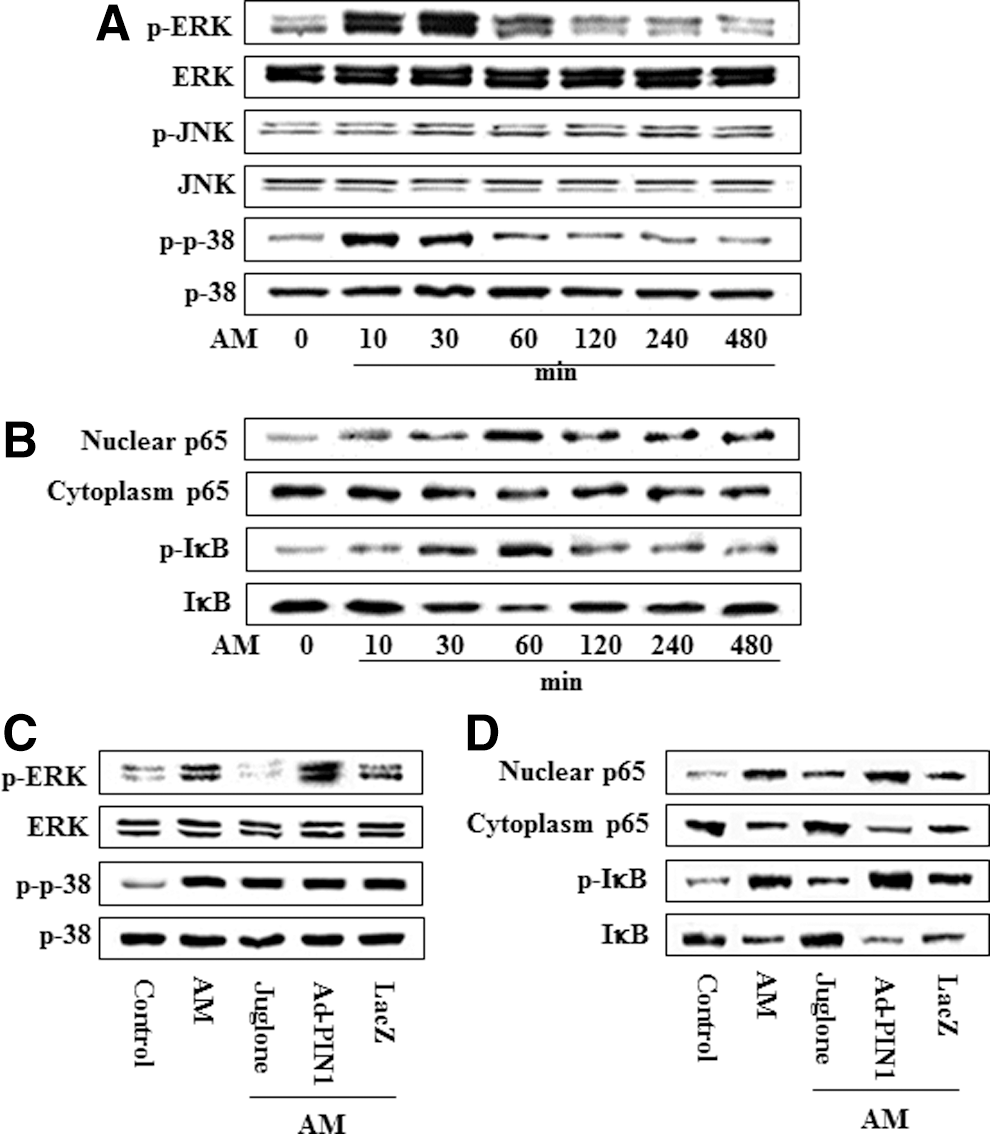
Effects of PIN1 inhibitor (juglone) and Ad-PIN1 on MAPK and NF-κB pathway protein levels during adipogenic differentiation in HDPSCs. Time course expression of AM-induced MAPK
Expression of PIN1 in mouse tooth germ and dental pulp
To gain more insights into the specific in vivo roles of Pin1 during tooth development, the expression of PIN1 was investigated in tooth germ and pulp of bell stage (neonatal mice), root-forming stage (7-day-old mice), and adult tooth stage (28-day-old mice) by immunohistochemistry. In neonatal mice, PIN1 expression was not found in the bell stage of tooth germ except the dental lamina (Fig. 12A). In developing tooth of 7-day-old mice, PIN1 was weakly expressed in odontoblasts, but not in undifferentiated cells of pulp (Fig. 12B). In the mandibular molar of 28-day-old mice, PIN1 was strongly expressed in the odontoblasts as well as cells of subodontoblast layer (Fig. 12C, D). In addition, PIN1 was expressed in the nerve fibers located in the pulp core (Fig. 12D).
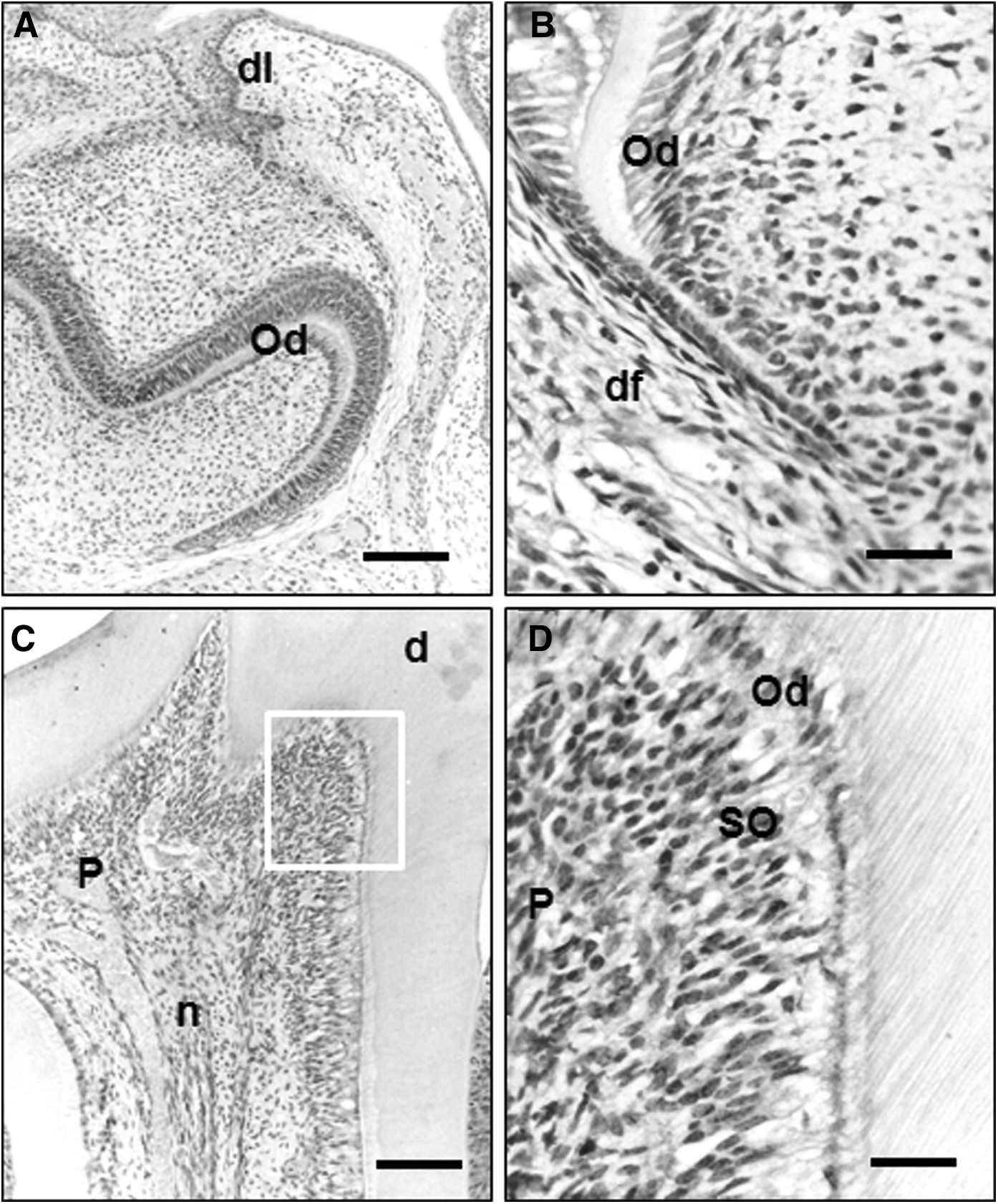
Immunohistochemical localization of PIN1 in neonatal
Discussion
Human dental pulp stem cells (HDPSCs) have been demonstrated to be a suitable cell source for dental tissue regeneration because of the clonogenic ability, rapid proliferation rate, and multiple differentiation potentials [4,5]. Thus, human dental pulp could be a desirable option as a cell source for potential therapeutic applications. Recently, we reported that heme oxygenase-1, lysyl oxidase, and SIRT1 are novel molecular targets of HDPC differentiation and may have clinical implications for regenerative endodontics [21,22 –24]. To the best of our knowledge, this is the first reported study examining the expression of PIN1 during odontogenic or adipogenic differentiation, the effects of activating PIN1 with Ad-PIN1, and inhibiting it using juglone on the differentiation of HDPCs.
In the present study, PIN1 mRNA and protein level were significantly increased after 1 day of odontogenic induction and then steadily declined, but were time dependently increased throughout the entire adipogenic differentiation in HDPCs. This adipogenic result is consistent with the study in 3T3-L1 preadipocytes, which PIN1 expression was shown to be upregulated from day 0 to 10 [12]. In contrast, PIN1 expression level does not changed during terminal differentiation of skeletal muscle cells [25]. This different expression pattern of PIN1 might be related to differences in cell or differentiation types.
HDPCs have the capacity to differentiate into adipocytes and odontoblast [4 –6]. In general, mechanisms that promote one cell fate actively suppress mechanisms that induce the alternative lineage [25]. Our studies showed that the inhibition of PIN1 by juglone promoted odontogenic differentiation of HDPCs, as evidenced by the formation of mineralized nodules, induction of ALP activity in the early stage, and upregulation of marker genes, such as ALP, OPN, OCN, DMP-1, and DSPP. These results are consistent with previous data that knockdown of PIN1 shRNA promotes skeletal muscle differentiation [25]. Furthermore, overexpression of PIN1 through transfection with Ad-PIN1 blocked OM-induced odontogenic differentiations of HDPCs, which results were similar to that of previous report that overexpression of PIN1 in myeloid cells leads to block of granulocyte differentiation [14]. Furthermore, in this study, we assessed the effect of PIN1 inhibition or activation on adipogenic differentiation in HDPCs, and our results showed that PIN1 inhibitor juglone could inhibit adipogenic differentiation in HDPCs manifested by Oil Red O staining and RT-PCR for PPARγ, AP2, and LPL. These results are consistent with the previous reports that adipose differentiation blocked by PIN1 siRNA in human preadipocytes [12]. In contrast, three transcription factors, PPARγ, AP2, and LPL, involved in adipogenic differentiation [26] and lipid droplets were increased markedly in HDPCs treated with Ad-PIN1. We found that PIN1 inhibition decreased in adipocyte differentiation but increased in odontogenic differentiation of HDPCs. Overexpression of PIN1 in HDPCs enhanced adipogenic differentiation, but concommitently suppressed odontogenic differentiation, indicating the role of PIN1 in regulating the balance between adipogenesis and odontogenesis of HDPCs.
To identify stem cell populations, it is necessary to understand the cell surface phenotype of each population. Recent studies have demonstrated that SSEA-4, an embryonic stem cell-associated antigen, can be used to identify mesenchymal stem cell (MSC) [27]. Furthermore, Kawanabe et al. [28] suggested that SSEA-4 is a specific cell surface antigen that can be used to identify DPSCs. STRO-1 is also thought to be a putative cell surface marker for the isolation of stem cells from both human and rat dental pulps [29]. Our data showed that the PIN1 inhibition increased the expression of SSEA-4 and STRO-1 with adipogenic differentiaiton but decreased these markers with odontogenic differentiaiton. These results were similar to the previous study that the progression of MSCs precusor cell population toward a mature and functional osteoblastic phenotype correlates to the loss of STRO-1 expression [29]. In contrast, PIN1 overexpression has opposite effect on the stem cell markers during odontogenic or adipogenic differentiation in HDPCs. Further studies are needed to understand the physiological significance of these subpopulations and multilineage differentiation potential of HDPCS.
BMP signaling is initiated by receptor binding, propagated by the phosphorylation of the Smad1/5/8 complex, and finally translocated into the nucleus, resulting in the regulation of target gene transcription [30]. Smurf1 and 2 are negative regulators of both BMP and TGF-β signaling pathways [30]. Furthermore, the BMP pathway can be regulated by a negative feedback loop. Noggin is one of the osteoblast-secreted proteins that can limit BMP signaling through complexing with BMP, thereby preventing receptor binding [31]. In this study, we found that PIN1 inhibition by juglone suppressed Smurf1, but enhanced the expression of BMP-2 protein, and its downstream molecule Smad1/5/8 protein, suggesting that PIN1 inhibition may influence HDPC functions through the BMP pathway. This finding was further supported by the fact that noggin blocked OM-induced BMP-2 and its downstream molecules, and Ad-PIN1 reversed the effects of PIN1 inhibition on BMP pathways. Furthermore, PIN1 negatively regulates Smad signaling, by promoting proteasome-mediated degradation of Smad proteins, in MDA-MB-231, HT1080, and human embryonic kidney 293 cells [32].
Growing evidence demonstrates that Wnt signaling plays a critical role in development and stem cell self-renewal. There are currently 19 identified Wnt family proteins, divided into 2 main categories, canonical and noncanonical, based on their role in cytosolic β-catenin stabilization [33]. The canonical Wnt/β-catenin signaling is another key pathway for regulating bone formation and remodeling and contribute osteoblastic differentiation [34]. In addition, canonical Wnt signaling negatively regulates the odontoblast-like differentiation of DPSCs [6]. In this study, expression level of Wnt ligands and β-catenin were enhanced in the presence of juglone but inhibited in the presence of Ad-PIN1. The application of negative regulator, DKK-1, also caused partial inhibition on the OM-induced Wnt/β-catenin signaling. Therefore, it is postulated that the canonical Wnt/β-catenin and BMP signaling involved in PIN1 inhibits the odontogenesis in DPSCs.
TGF-β superfamily cytokines bind to type I and type II serine/threonine kinase receptors and transduce intracellular signals through Smad proteins [35]. In the present study, expression of TGF-β1 ligands and receptors were upregualted in OM-treated HDPCs, suggesting that endogenous TGF-β signaling is activated in HDPCs. However, PIN1 inhibition and overexpression did not affect OM-induced TGF-β pathway. Moreover, the inhibition of BMP (noggin), Wnt (DKK-1), and TGF-β (SB431542) pathway does not significantly alter the expression of PIN1 in mRNA or protein levels. Therefore, although PIN1 overexpression exerts antiodontogenic effects in part by suppression of BMP and Wnt induction, these odontogenic pathways (including TGF-β) seem not to be related in PIN1 expression. This observation was in accordance with simultaneous decrease of PIN1 expression and increase of BMP, Wnt, and TGF-β during odontogenesis (Figs. 6 –8).
Master regulatory transcription factors, PPARγ2, C/EBPα, C/EBPβ, and C/EBPδ, are involved in adipose differentiation [36,37]. Several studies have demonstrated that PPARγ2 and C/EBPα co-regulate each other's expression. PPARγ2 is the ultimate key regulator of adipogenesis, and C/EBPα might play more of an accessory role for PPARγ2 by inducing and maintaining PPARγ2 expression [38]. In PIN1 KO mouse adipose tissues, gene expression of PPARγ2, C/EBPα, and C/EBPβ was significantly suppressed [12]. Consistent with these results, our study showed that PIN1 inhibitor blocked the AM-induced activation of C/EBPα, C/EBPβ, and PPARγ, but PIN1 overexpression has opposite effects. These results suggested that PIN1 overexpresssion induced the differentiation of HDPCs into adipocytes via PPARγ, C/EBPα, and C/EBPβ transcriptional activity.
MAPK is a proline-directed serine/threonine kinase consisting of three-enzyme modules; its targets, inducing ERK, JNK, and p38 kinases, are important in cellular signal transduction pathways. NF-κB signaling is associated with bone metabolism and considered to be the important signal transduction pathway in osteoblasts differentiation [39]. The MAPK and NF-κB pathways are also crucial for cell proliferation and differentiation in HDPCs [20 –40,41]. In the present study, OM-induced activation, ERK, JNK, and NF-κB, was enhanced by PIN1 inhibition but suppressed by PIN1 activation during odontogenic differentiation. These results are similar to the reports that the activation of ERK and p38 MAPK [42,43], as well as NF-κB [17,20,21], is involved in osteoblastic or odontoblastic differentiation. Furthermore, treatment with PIN inhibitor blocked AM-induced ERK, JNK, and NF-κB activation, but PIN1 overxpression reversed the MAPK and NF-κB signaling during adipogenic differentiation.
PIN1 is tightly regulated at multiple levels, and its deregulation has an important role in a growing number of pathological conditions, notably cancer and Alzheimer's disease [44]. PIN1 is expressed in neurons but not glia in the adult brain [8,45]. Significantly more expression of PIN1 was found in the developmental brain stages than in the adult stage of mouse [11]. In the present study, PIN1 was found intensely expressed in odontoblasts and cells of subodontoblast layer. Furthermore, PIN1 expression was higher in the adult mouse tooth stage than in the neonatal and root forming developmental stage. These observations suggest that PIN1 might be an important factor for odontoblastic differentiation in adult tooth development. Further studies will be needed to elucidate the role of PIN1 in neurogenic differentiation of HDPCs because positive PIN1 expression was noted in nerve fiber of adult mouse dental pulp tissue.
In conclusion, the present study is the first to report that PIN1 inhibition can promote odontogenic differentiation of HDPSCs through activating the BMP/Wnt/β-catenin/ERK and JNK/NF-κB pathway but inhibits adipogenesis through inhibition of the PPARγ and C/EBPβ, α/ERK/NF-κB signaling pathway. In contrast, PIN1 overexpression promoted adipogenesis and, concomitantly, suppressed odontogenesis. Thus, PIN1 may be novel important modulator of HDPSCs differentiation, which can useful in dental tissue regeneration and tissue engineering by its reciprocal effect on odontogenesis and adipogenesis. These data indicate that the ERK, JNK, and NF-κB pathways are involved in the PIN1-mediated regulation of odontogenic and adipogenic differentiation in HDPCs.
Footnotes
Acknowledgments
This work was supported by Mid-career Researcher Program through National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (MEST) (no. 2012004117), and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2012R1A5A2051384).
Author Disclosure Statement
The authors have no conflict of interest to disclose.