
Abstract
Europe is ready to deploy its immense capital of knowledge into the development of effective cell-based therapies and delve into the global race for translating stem cell science into regenerative medicine. But what are the challenges and the emerging issues that lay ahead the realization of Europe's enormous potential in this field? Both researchers and industrial stakeholders tend to impute the slow pace of translation to specific suboptimal features of the regulatory environment in Europe. At the same time, a host of new issues are emerging as testified by a recent public controversy regarding the provision of unproven cell therapy in Italy. We will review this topic and suggest some solutions to foster the responsible development of innovative cell-based therapies in Europe.
Introduction
Europe is ready to deploy its immense capital of knowledge for the development of effective cell-based therapies and to delve into the global race for translating stem cell science into regenerative medicine. But what are the challenges and the emerging issues that lie ahead of the realization of Europe's enormous potential in this field?
This is a time of economic crisis, in Europe as well as in other parts of the world. However, while the present economic crisis per se may be conducive to reduced spending on science in Europe, some Member States are starting to realize that biomedical innovation can be an opportunity for economic recovery [101] and thus decide to prioritize at least some domains of biomedicine—such as regenerative medicine— that hold great promise to deliver new cures and, according to some, reduce soaring healthcare expenditures throughout the continent in the long run [102].
To unleash the therapeutic potential of European stem cell science, sustained public and private investment in this sector is certainly required, but also, and equally importantly, a solid and efficient regulatory framework must be put to work. In this respect, the European Union and Member States share their legal competencies both in public health and on the realization of the internal market, in particular with respect to the free movements of goods (including medicinal products).
It has been estimated that between 2004 and 2010, 318 regenerative medicine clinical trials were initiated in Europe, 78% (n = 248) of which are relative to cell-based medicinal products [1]. To date, however, only two regenerative medicine products obtained marketing authorization as advanced therapy medicinal products (ATMPs) by the European Medicines Agency (EMA): Glybera, a gene therapy product by UniQure to treat lipoprotein lipase deficiency (a severe form of pancreatitis), and Chondrocelect, an autologous cell therapy for cartilage regeneration by Tigenix.
Other scientifically advanced countries such as the United States, Canada, and Japan have not approved more new cellular therapies than Europe. However, both researchers and industrial stakeholders tend to impute the slow pace of translation to specific suboptimal features of the regulatory environment in Europe. Expectations are thus growing for the pipeline to be improved. At the same time, a host of new issues are emerging as testified by a recent public controversy regarding the provision of unproven cell therapy in Italy.
Current challenges in the governance of innovative cell therapies
The translational pipeline from basic research to the delivery of innovative stem cell-based therapies is covered by a variety of European legal instruments [2] ranging from regulations on marketing authorization (Regulation (EC) no. 1394/2007) to Directives about clinical trials (Directive 2001/20/EC) and guidelines of good clinical practice (Directive 2001/83/EC, Directive 2009/120/EC) (Fig. 1). In addition, each Member State has to implement these provisions at the national level, thus accounting for a second level of heterogeneity. In this respect, the legal regulation is far from being totally homogeneous in Europe [3,4], and therefore not all the steps in the translational pipeline are equally addressed from a legal point of view [5]. This leads to heterogeneity in the legal requirements to be fulfilled in the various Members States, a factor that can be seen as slowing down the innovation process.
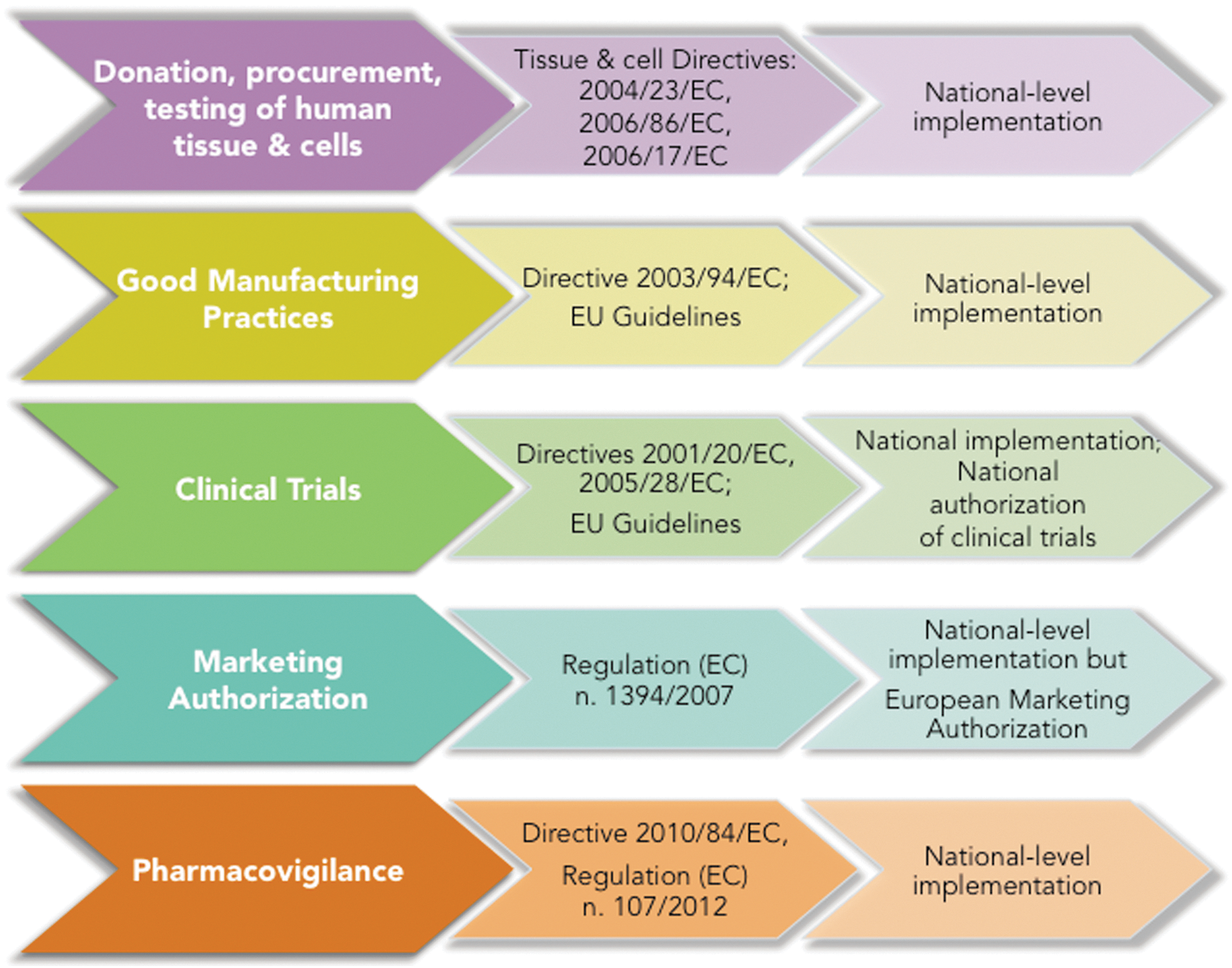
Scheme depicting European legal texts of relevance to cell-based medicine: tissue and cell Directives, clinical trial Directives, and advanced therapy medicinal products, Regulation.
In December 2012, almost 6 years after the approval of the Regulation setting up a centralized procedure for marketing authorization of ATMPs (Fig. 1), the Directorate General for Health and Consumers of the European Commission launched a public consultation on how the requirements laid down by the Regulation fare with respect to the needs and expectations of researchers, clinicians, patients, national regulatory agencies, and the industry. 1
It turned out that the European regulatory pathway is perceived as very demanding, especially for academic institutions and small and medium enterprises that, while constituting the bulk of those attempting the clinical translation of stem cell biology, are less experienced than big pharmaceutical companies in navigating a complex regulatory environment. This is said to account, at least in part, for the current stagnation in new marketing authorizations for ATMPs.
Another aspect of the current regulation that is often highlighted is the lack of clarity in certain important requirements—namely, the notion of substantial manipulation, that of nonhomologous use and, finally, that of hospital exemption. While the former categories create ambiguity in the classification of ATMPs, the latter is seen as a disincentive to engage in the complex, lengthy, and expensive pathway that should lead to marketing approval.
A clearer and better-defined regulatory pathway, many argue, would be more encouraging for investors. To this aim, better coordination between the EMA and national regulatory agencies would be appropriate, so that researchers and their commercial partners can receive improved advice on the complex requirements for ATMP approval.
Some stakeholders in the field also point out that the ATMP Regulation should be more specific, especially as to technical requirements for clinical trials and good manufacturing practices for advanced therapies: cells are inherently more variable than conventional chemical compounds and, since they are viable, it is obviously hard to fully assimilate them to drugs. This calls for more specific safety and efficacy criteria. Moreover, quite a number of promising advanced therapies in an early translational phase target rare diseases [6,7] and may be difficult to validate on large cohorts: this has to be taken into account, and clinical trial design has to be adapted accordingly.
Overall, it is important that the pathway from phase 1 to marketing approval be streamlined, that researchers and companies receive enough support from national and European regulatory agencies in navigating the system, and that the intricate legal landscape constituted by the ATMP Regulation, the clinical trial Directive, and the tissue and cells Directives (Fig. 1) is harmonized and simplified. In this last respect, while many concur that the centralized marketing authorization procedure established by ATMP requires clarification, not enough attention has been given so far to the issue of harmonization of the whole regulatory pipeline from research—thereby including the procurement, storage, and distribution of human material—to clinical use.
To this aim we promoted EUCelLEX 2 (coordinated by Emmanuelle Rial-Sebbag), an FP7 coordination action that brings together a number of European and international experts and stakeholders in the ethical, legal, and social issues that surround cell-based regenerative medicine: the aim of the consortium is to assess the adequacy of the present European regulatory framework for the use of stem cells and to provide recommendations that may facilitate it in all aspects from research to healthcare applications.
A further challenge for the development of cell-based therapies in Europe has to do with the fact that, notwithstanding the existence of a centralized marketing authorization procedure for ATMPs, other crucial aspects of the translational pipeline are still delegated to the individual Member States. For example, whereby the regulatory requirements are set at the European level, the creation of the clinical infrastructure remains largely of national competence. Therefore, since development of innovative cell therapies still requires conspicuous public investments at the national level, chances are that this will create competition instead of collaboration among Member States.
Moreover, national healthcare systems will have to adapt to the specificities of innovative products such as new cell therapies, both in terms of evaluation procedures and in terms of reimbursement policies. This aspect of regenerative medicine in Europe has been evaded for too long. In a not-too-distant future, we may have to cope with a catalog of cell-based products approved for being marketed throughout Europe, but we may lack a harmonized scheme for making them available to all European patients. This may eventually push regenerative medicine into the private market and potentially create inequalities of access to new products among European citizens.
Emerging issues: hope, hype, and innovation
Expectations are high on the part of patients and their families, who see in innovative cell therapies their unique—and sometimes last—hope. However, it takes time for regenerative medicine products to reach the European market (so that in some cases, applications for marketing authorization have been withdrawn by the sponsors, as in the case of Advexin and Cerepro) and to meet those expectations. Probably also as a consequence of this lengthiness, parallel “unorthodox” offers of untested therapies are flourishing in European countries and worldwide, not rarely with deleterious consequences for patients undergoing unproven therapies (Fig. 2). In this context, attention must be paid to the fact that different routes to implementing European Directives and Regulations may be taken by different Member States. This is especially true for the so-called hospital exemption clause [8] and can result in loopholes to be exploited by providers of unproven therapies, thus encouraging intra-European stem cell tourism and the activity of clinics offering yet untested stem cell treatments.

Casualties and controversies with unproven stem cell based therapies in Europe.
A recent case demonstrates that, notwithstanding the existence of a regulatory framework that, as we said, is perceived by some as rigid and restrictive, things can, nonetheless, go wild for regenerative medicine in Europe.
A recent judiciary investigation uncovered that, since 2009, a private group based in northern Italy called Stamina Foundation has been offering mostly allogenic intravenous injections of bone marrow aspirations allegedly containing mesenchymal stem cells to patients affected by a variety of debilitating conditions ranging from Parkinson's disease to metachromatic leukodystrophy, Niemann–Pick disease, and spinal muscular atrophy.
Apparently, Stamina used to charge patients at least until recently, when the organization stipulated official agreements with two public hospitals in the north of Italy, claiming to be acting under the compassionate use framework. The case occasioned an investigation followed by and inspection by the Italian medicines agency (Agenzia Italiana del Farmaco, AIFA). The inspection revealed that Stamina did not comply with a number of regulatory requirements for the compassionate use of cell therapy in Italy; most importantly, AIFA found out that no published preclinical and clinical evidence supports the Stamina procedure.
On May 15, 2012, AIFA issued a formal inhibitory order to stop Stamina's activities. This decision caused a number of patients—most of them very young children—to stop receiving injections. In reaction to this outcome, many families turned to courts and, thanks to surprising judicial decisions based on testimony by children's parents, obtained counter-injunctions to continue with the injections. So far, more than 100 patients were accorded judicial permission to receive Stamina's infusions.
Especially in this early phase, public opinion, after one-sided media reports showing desperate families and their sick children, seemed to be unequivocally supportive of the Stamina method, despite numerous warnings from the scientific community. As a consequence, the regulatory requirements set up to protect patients' safety started to be perceived as unjust obstacles to families and patients claiming access to the injections as their last therapeutic hope. The mounting pressure resulted in late March 2013 in an ad hoc decree by the Ministry of Health that gave Stamina permission to derogate from existing norms and continue the treatment for the patients who had already started it, at the expense of taxpayers' money.
As the decree underwent the usual parliamentary ratification process, a further decision was taken by the House of Representatives: the sum of €3 million was allocated to sponsor a public clinical trial to test for the safety and efficacy of the Stamina method. The starting date of the trial has, however, been postponed because of Stamina's reluctance to provide their protocol to the Italian regulatory agencies. Finally, the protocol was provided, but the Ministry of Health decided to keep it secret and not to share it with the public and the scientific community. The case, in the meantime, trespassed the Italian borders as prominent scientists of international renown forcefully criticized both Stamina and the political decisions taken in this case [9 –11,103]. So did the EMA [104].
The Stamina case is a vivid reminder that the governance of scientific and technological novelties is challenging, especially when the hopes of desperate patients encounter the rush to offer unproven therapies skipping clinical validation. In this climate, claims about therapeutic freedom and patients' right to early access of cell-based therapies can easily gain consensus. They are, however, ethically unjustified and, if translated into a deregulated governance of regenerative medicine, they can indeed hinder its development toward safe and effective cures. Those derogations to the scope of the current legal framework, both at European and national level, call for a deeper appraisal of current practices in Europe and for proposing recommendations for improved coordination and regulation (see EUCelLEX project).
The development of innovative cell therapies should be perceived as a shared responsibility. Although regulatory power is ultimately in the hands of appointed officials, steering clinical translation toward new therapies in an ethically acceptable way depends on a participated effort by all relevant stakeholders: bioethicists, lawyers and social sciences scholars, regulators, scientists, scientific societies, companies, patients' advocates, and the media. Each of these actors can make a difference in how progress in regenerative medicine is communicated to lay publics as well as in the way regulatory jurisdiction by competent authorities is perceived and implemented.
In particular, the Stamina controversy invites more careful ethical and legal analysis of the stakes at play for vulnerable categories of patients such as the very young, those affected by incurable or rare diseases, and the elderly. Their interests do not seem to be enhanced by deregulation of the kind we see in Italy: quite to the contrary, as major advocacy groups have rightly claimed, they need a regulatory environment that guarantees respect for them as persons, patients, and potential research participants (Fig. 3).

Recapitulation of challenges and emerging issues.
Conclusion
To exploit the full therapeutic potential of European stem cell science, the regulatory pathway to centralized marketing authorization for ATMPs in Europe will have to be clarified, streamlined, and simplified. But adjusting the existing framework to avoid unnecessary regulatory burden does not mean deregulation. Emerging issues relative to unproven cell therapies strikingly recall that we must remain vigilant about possible misuses.
The Italian case suggests that in Europe there is room for science policy decisions that contradict the established regulatory framework. The latter, while imperfect and amenable to improvement, at the very least offers a necessary reference for the field. Precipitous revisions of current regulatory norms can represent a concrete impediment to the advancement of regenerative medicine especially when, like we saw, they encourage legislative activities to follow suite emotional waves of public opinion.
In this respect, it is important to close the regulatory loopholes caused by different implementation strategies in different member states. This is crucial to avoid because what is illegal in one European country can be pursued in a neighboring one, thus undermining the efficacy of the European regulatory edifice. Furthermore, Europe should reflect on a good strategy to discourage stem cell tourism to third countries where unproven therapies can be offered to European patients under less strict and less controlled regulatory conditions. Regulatory challenges for the development of regenerative medicine in Europe have to be tackled in a proportionate and responsible way, that is, with the necessary awareness about both regulatory efficacy and possible misuses of innovation.
More work needs to be done to develop solid scientific [11] and ethical standards specifically adapted to the clinical translation of regenerative medicine. This entails acknowledging the uncertainties that lay ahead the realization of the therapeutic potential of stem cells in Europe—be them technical, ethical, legal, or societal. Especially in a time of economic crisis for Europe, promising fields of biomedical innovation represent the occasion for an upturn. Different stakeholders, however, are likely to have different ideas about the future of regenerative medicine—ideas that can be promoted or frustrated depending on the regulatory choices that will be taken in the coming years. Occasions should thus be abundant not only for discussing, but also for renegotiating the values, visions, and interests that different regulatory options embed [109]. It is exactly at this point that Europe needs a good deal of reflexive thinking in shaping the future of cell-based therapies [13].
Footnotes
Acknowledgments
Author disclosure statement
No competing financial interests exist.
1
A summary of the public consultation can be found here:
2
EUCelLEX is funded by the European Commission under the Seventh Framework Programme.