
Abstract
Human and mouse embryonic stem cells (ESCs) exhibit fundamental differences depicting two distinct states of pluripotency: naive and primed. Mouse ESCs (mESCs) are dependent on leukemia inhibitory factor for growth in culture and possess two active X chromosomes in their female cell lines and correspond to the naive state of pluripotency. Human ESCs (hESCs), however, closely resemble mouse epiblast stem cells and correspond to the primed state. Primed stem cells are dependent on basic FGF for growth and show differentiation bias into different cell lineages. Recent studies have revealed that these two pluripotent states can be interconverted by modifying the culture conditions, although unequivocal evidence for obtaining truly naive hESCs has not been found. Accurate identification of the functions of major pluripotency-related signaling pathways and their cross-talk networks should aid in the successful induction of stable naive pluripotency in human cells.
Introduction
P
Naive and Primed Pluripotent Stem Cells Echo Their Embryonic Origin
Although there is limited information in humans compared to mice, new findings suggest that notable differences exist in the early development of these two species, which may be reflected in differences observed in the ESCs derived from them. These differences are in regard to early lineage commitment [8,9], signaling response [10,11], and programs of epigenetic modification [12,13].
During blastocyst formation, by embryonic day 3.5 in the mouse and days 4–5 in humans, two distinct lineages form: the trophectoderm (TE) and ICM, which give rise to the placenta and proper embryo, respectively [9]. The transcription factor Oct4 is considered to be the cardinal marker for ICM cells, whereas Cdx2 is a marker for epithelial TE cells [14]. In the mouse, Oct4 expression is detectable in the 8-cell-stage embryo, while Cdx2 colocalization with Oct4 is found in the 16-cell morula and the mutually exclusive expression of these two transcription factors leads to the segregation of the ICM and TE [15,16]. In humans, while OCT4 expression is detectable at the 8-cell human embryo stage, it persists in the TE until implantation [9]. In addition, CDX2 expression in the TE occurs following cavitation; however, Cdx2 expression is detectable in the morula stage of the mouse embryo [9,16].
The second cell fate decision occurs in the ICM cells, with the segregation of the primitive endoderm (PE; future yolk sac) and the pluripotent epiblast (Fig. 1) [10,17]. The ICM contains distinct subpopulations of cells in terms of the expression of transcription factors Nanog and Gata6, as the key markers for the epiblast and PE, respectively [18]. In the mouse, FGF is needed for the formation of the PE and chemical inhibitors of FGF signaling shift cells from the ICM to the epiblast state, while exogenous FGF mediates the conversion of ICM cells into PE cells [19,20]. Although the exact mechanism that underlies the role of FGF in ICM patterning has yet to be elucidated, individual ICM cells are thought to respond randomly to different levels of FGF4 signaling [17]. Epiblast cells secrete FGF4 and thereby induce the expression of PE-affiliated genes, such as Gata4, Gata6, and Sox17, in the PE cells that highly express FGF receptor 2 (FGFR2) [17,18,20]. As in the mouse, expression of the PE-associated transcription factors GATA4, GATA6, and SOX17 is seen in the cells of human late blastocysts that lack NANOG expression. However, FGF signaling appears to be dispensable for the differentiation of cells into the PE in the human embryo [9,10].
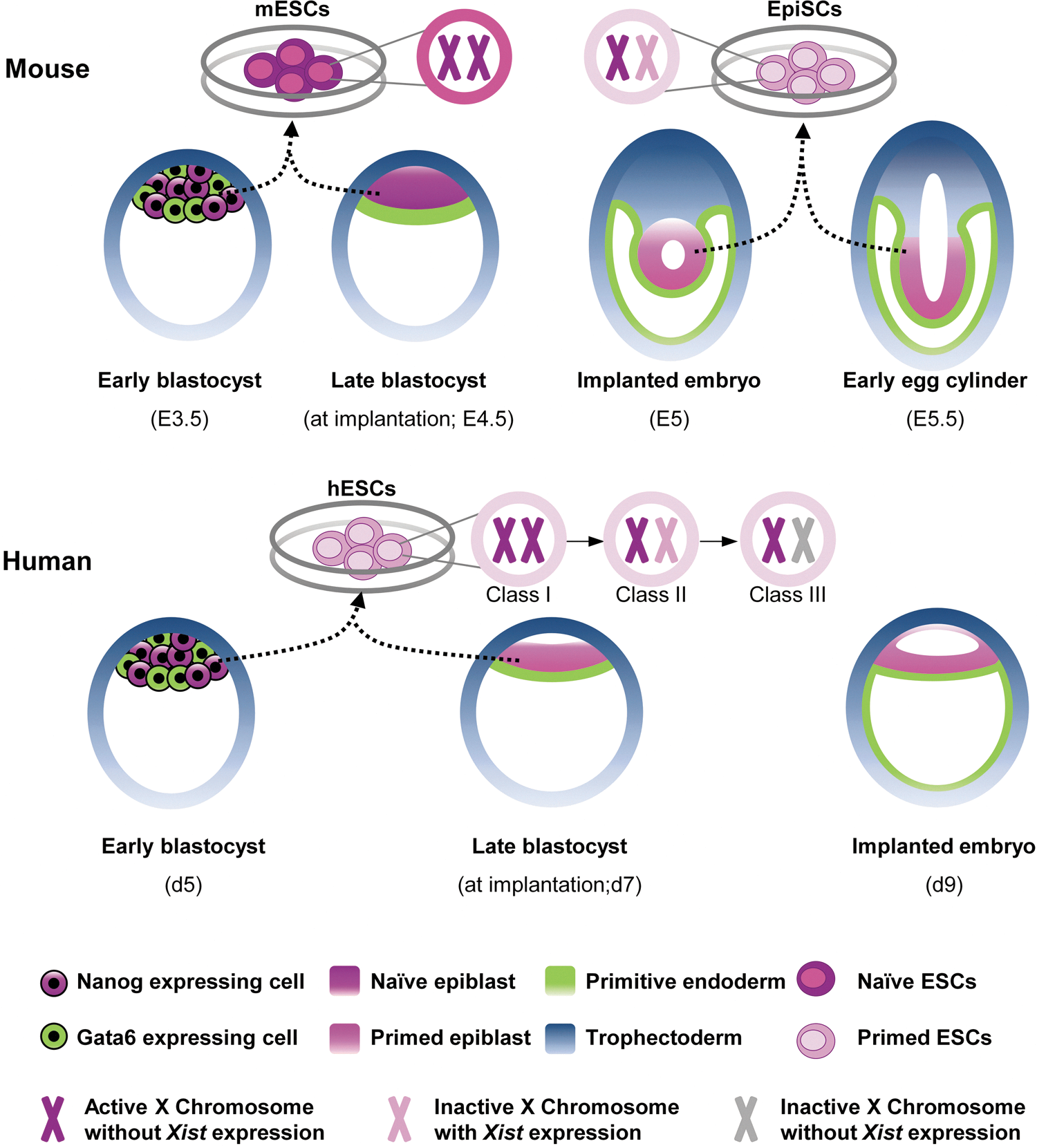
Derivation of pluripotent stem cells from developing mouse and human embryos. Prior to implantation, both mouse and human embryos develop into blastocysts that comprise two different lineages, the ICM and the TE. Pluripotent ICM cells are heterogeneous with respect to Nanog and Gata6 expression, specifying the epiblast and PE lineages, respectively. Both mESCs and hESCs can be generated from the ICM until the late-blastocyst stage; however, mouse EpiSCs are derived from postimplantation epiblasts. mESCs represent naive pluripotency, with two active X chromosomes in female lines, while primed EpiSCs have XCI. In addition, primed hESCs demonstrate three different classes of XCI. ICM, inner cell mass; TE, trophectoderm; PE, primitive endoderm; mESCs, mouse embryonic stem cells; hESCs, human embryonic stem cells; EpiSCs, epiblast stem cells; XCI, X-chromosome inactivation. Color images available online at
At the time of implantation, day 4.5 in the mouse, murine epiblast cells proliferate and form a ball of cells. Subsequently, the internal cells undergo apoptosis and form a cup-shaped epithelium surrounded by the PE; the structure is known as the egg cylinder, which can be seen in the implanted embryo (Fig. 1) [8,21]. In contrast, in humans, the molecular segregation of the epiblast and PE from the ICM leads to the creation of a bilaminar embryonic disc. This event indicates that human pluripotent epiblast cells, unlike their mouse counterparts, promptly progress into primed pluripotent cells [8]. Although hESCs are derived from the ICM of 5–7-day-old blastocysts, the inherent feature of human ICM cells to rapidly create a layer of epiblast cells and also the likely suboptimal culture conditions are the most probable causes to propel the derived hESCs into the murine EpiSC-like cells [8,22]. Another specific feature that distinguishes the early embryonic development of mice and humans is the phenomenon of diapause. In rodents, diapause is a delay, even for several weeks, in blastocyst implantation that occurs in lactating females. In this condition, blastocysts hatch from the zona pelucida and remain in the uterus until estrogen levels are restored [8]. Interestingly, diapause is the first defined way to enhance the efficiency of deriving ESCs from refractory strains in the mouse; however, diapause does not exist in humans [23]. During diapause, epiblast cells can remain in the naive state for a long time, providing another illustration of the feasibility of deriving naive ESCs from mice.
Another feature that outlines the difference in the early development of mice and humans is the status of X-chromosome inactivation (XCI). XCI is a form of dosage compensation that occurs in one of the two X chromosomes in mammals to balance gene expression in female cells. This complex epigenetic process is regulated by long noncoding Xist RNA, which coats the cis X chromosome and triggers gene silencing [24,25]. In the mouse, initially imprinted XCI is found in the 4-cell-stage embryo where the paternal X chromosome (XP) is silenced and can be seen in all blastomeres until the mid-blastula stage [26,27]. XP is then reactivated only in the subpopulation of ICM cells that gives rise to the epiblast; therefore, the cells that form the embryo proper carry two active X chromosomes (XaXa). Subsequently, random XCI (XaXi) occurs upon development of the epiblast in the implanted embryo [12,28,29]. Thus, as with the in vivo status, mESCs represent XaXa, while EpiSCs carry randomly XaXi (Fig. 1) [30,31].
The XCI process differs in humans, compared to mice. Imprinted XCI does not occur in the early embryo, and XIST expression is seen in both active X chromosomes, even in ICM cells [32]. XCI begins later in humans than in mice. XCI occurs downstream of XIST upregulation in humans, while Xist expression is tightly monoallelically regulated during random XCI in mice. Therefore, a substantial diversity in the timing and regulation of XCI initiation exists in the mouse versus the human embryo [32]. This discrepancy in XCI status is also seen between mESCs and hESCs. hESCs are grouped into three classes based on XCI (Fig. 1). Class I (XaXa) expresses low levels of XIST, resembles mESCs, and triggers XCI in one X chromosome upon differentiation. Class I hESCs are unstable in culture and rapidly switch into Class II (XaXi), which contains one inactive X chromosome coated with XIST. Class II often converts into Class III (XaXi), which also has one inactive X chromosome, but it no longer expresses XIST. Class III represents an aberrant epigenetic state [12,13]. These XCI classes of hESCs are thought to arise from the different types of environmental stressors [33]. According to the literature, hESCs derived under conditions of physiological oxygen tension (5%) represent Class I; however, in the presence of 20% oxygen they undergo XCI (Classes II and III) [34]. Moreover, naive-like hESCs, which have been recently derived by the ectopic overexpression of pluripotency transcription factors under culture conditions that support the ground state of pluripotency (see next sections), are classified as Class I [35]. As this situation can also be seen in miPSCs or converted EpiSCs into the naive state by the ectopic overexpression of pluripotency transcription factors, the presence of two Xa could be considered to be the hallmark of naive mouse pluripotency [36]. However, due to the special features of hESCs in regard to XCI described just now, the existence of two Xa could not be undoubtedly considered an acute mark of naive hESCs.
Overall, recent studies have shown fundamental differences in the early embryonic development of mice and humans that certainly translate into differences in these ESCs derived from these two species. However, new breakthroughs in the identification of extrinsic factors and intracellular signaling pathways that control pluripotency in ESCs have created optimism that truly naive hESCs can actually be achieved. This achievement would require the accurate identification of the action of each signal transduction pathway involved in naive and primed pluripotency and would be complemented by useful information on the salient cross-talk between the different pathways.
LIF/Stat3 Signaling: The First-Known Signaling Pathway Involved in Pluripotency
ESCs were first derived in the mouse by plating whole blastocysts or isolated ICM cells of strain 129 on a layer of mouse embryonic fibroblasts (MEFs) as feeder cells in medium that contained fetal calf serum (FCS) [1,2]. The cytokine LIF was subsequently found to act as an effective factor in the secretome of MEFs for sustaining pluripotency in mESC culture, suggesting that it could be used as a substitute for the feeder layer [37,38]. LIF belongs to the interleukin-6 (IL-6) cytokine family and binds to the LIF receptor (LIFR), thereby recruiting the membrane protein gp130 and forming the heterodimer receptor. The heterodimer subsequently phosphorylates and activates a gp130-associated cytoplasmic tyrosine kinase, Janus kinase (Jak), which can trigger three intracellular signaling pathways: the Jak/signal transducer and activator of transcription 3 (Stat3) pathway, the phosphoinositide 3-kinase (PI3K)/protein kinase Akt pathway, and the mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (Erk) pathway (Fig. 2) [39].
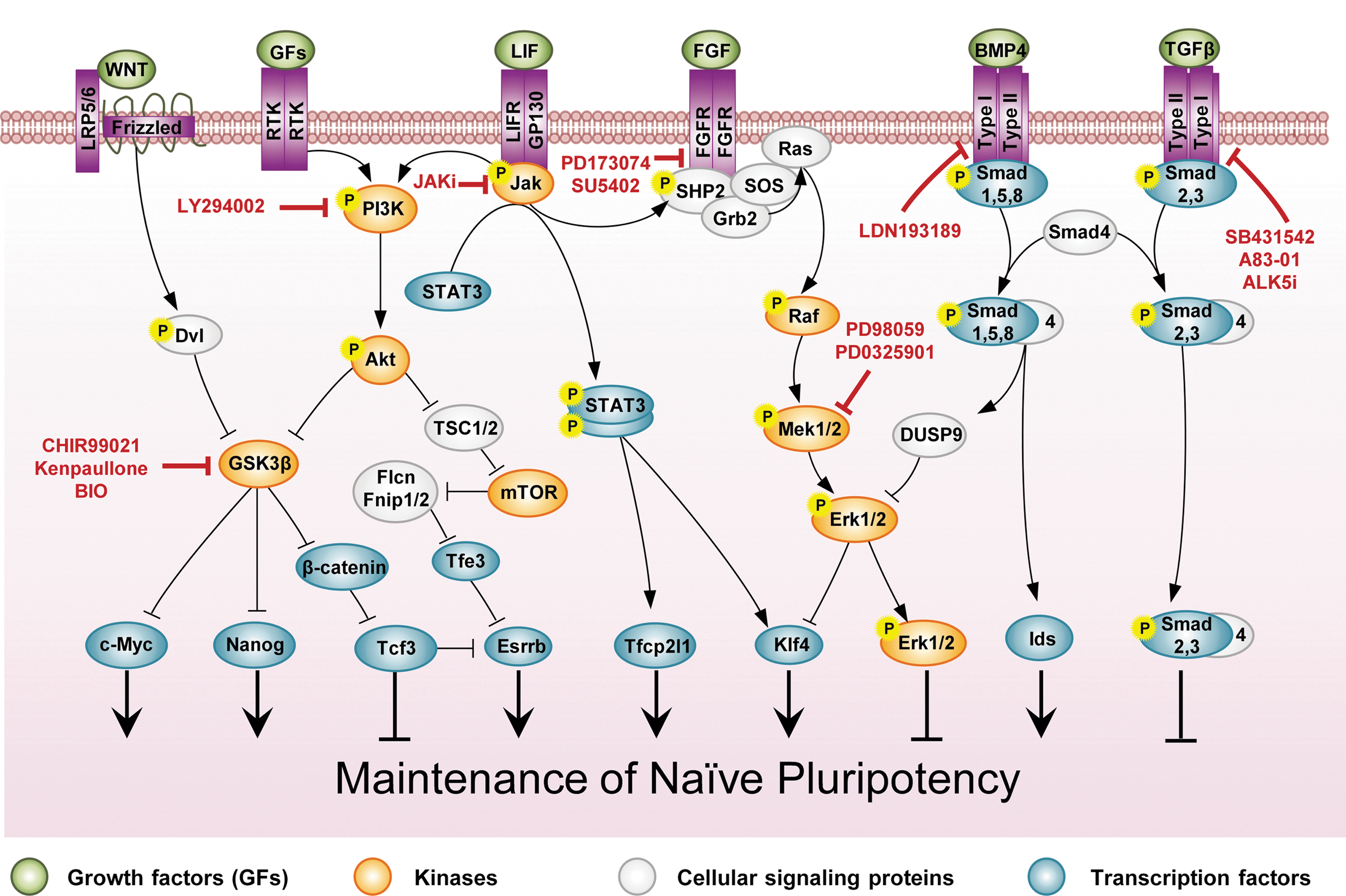
Extrinsic regulation in naive mESCs. LIF, BMP4, PI3K, and Wnt signal transduction is considered to represent the salient pathways that support naive pluripotency. LIF and BMP4 work together to sustain mESC self-renewal. LIF activates the STAT3, PI3K/Akt, and MAPK/Erk signaling pathways in mESCs. While STAT3 and PI3K/Akt signaling pathways support naive pluripotency, the MAPK/Erk signaling pathway induces the differentiation of mESCs. BMP4 activates the Smad1,5,8 signaling pathway, which in turn activates the Id genes and Dusp9. Id proteins suppress mESC differentiation into neural lineages and Dusp9 inactivates Erk, sustaining the pluripotency of mESCs. As STAT3, Id proteins, and Dusp9 maintain ESC self-renewal by primarily restricting multilineage differentiation, the ground-state hypothesis of pluripotency has been postulated and proposes that the small-molecule inhibitors of the FGF4 and GSK3 pathways—PD0325901 and CHIR99021, respectively, known as 2i—support the pluripotency of the mESC culture. GSK3 inhibitors activate Wnt signal transduction, which in turn activates c-Myc, Nanog, β-catenin, and Esrrb expression, thereby promoting mESC self-renewal. Moreover, the combination of PD0325901 and TGFβ inhibitors, such as SB431542, known as R2i, has been recently shown to maintain the ground state of pluripotency. Small molecules that modify the signaling pathways are shown in boxes. BMP4, bone morphogenetic protein 4; Erk, extracellular-signal-regulated kinase; FGF, fibroblast growth factor; FGFR, FGF receptor; Gsk3β, glycogen synthase kinase 3β; Id, inhibitors of differentiation; LIFR, LIF receptor; MAPK, mitogen-activated protein kinase; Mek, MAPK kinase; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; SHP2, SH2-domain-containing tyrosine phosphatase2; TGFβ, transforming growth factor β. Color images available online at
LIF/Jak/Stat3 is the first pluripotency-related signal transduction that has been defined in naive mESCs [40]. Activated Jak can phosphorylate tyrosine residues on gp130 and provide a docking site for the proteins that contain src homology 2 (SH2) domains, such as Stat3. Further, Jak directly phosphorylates Stat3 on tyrosine 705 (Stat3pTyr705), thereby leading to the homodimerization of Stat3. Dimerized Stat3 enters the nucleus and regulates the expression of target genes. Other members of the IL-6 cytokine family, such as oncostatin M, ciliary neurotrophic factor, and cardiotrophin 1, have been shown to activate Stat3 and maintain mESC self-renewal [41]. To date, numerous experiments have been conducted to identify the association between Jak/Stat3 signal transduction and the core circuitry of pluripotency-affiliated genes. Previously high-throughput genome-wide studies had revealed the presence of considerable loci co-occupied by Stat3, Oct4, and Nanog [42,43]. These target genes include both pluripotency-related genes, such as Oct4 and Nanog [42], and lineage-affiliated genes, such as Brachyury, Foxa2, and Eomes [43]. Moreover, it had been shown that the main role of the Jak/Stat3 pathway is activation of the transcription factors c-Myc and Klf4 in mESCs. c-Myc and Klf4 are considered to play important roles in mESCs [39,44], and are two of the four factors involved in the reprogramming of human and mouse somatic cells into a pluripotent state [4,39,45]. Further, genetic manipulation experiments using RNA interference have shown that Stat3 could sustain mESC self-renewal by suppressing the expression of endodermal and mesodermal genes in mESCs [45]. However, the exact role of Stat3 was not determined, as none of these identified Stat3 target genes are neither necessary nor sufficient for the need of LIF [41,46]. A recent study, by taking advantage of Stat3-null mESCs, has revealed that transcription factor Tfcp2l1 (also known as Crtr-1) is the most probable candidate factor among the Stat3 target genes that sustains pluripotency and connects the LIF signaling into the transcription factor core of naive pluripotency. The forced expression of Tfcp2l1 could substitute LIF stimulation in mESCs and the transient expression of Tfcp2l1 was sufficient to reprogram EpiSCs into naive pluripotency without LIF stimulation [46].
LIF also activates PI3K/Akt and MAPK/Erk signaling pathways in mESCs. As will be seen below, PI3K/Akt signaling plays a crucial role in the maintenance of pluripotency in both hESCs and mESCs by increasing the expression of Nanog and c-Myc, genes of the two salient transcription factors of pluripotency [47]. The MAPK/Erk signaling pathway, however, is a well-characterized pathway involved in mESC differentiation [48,49]. According to the activity of each of these pathways, LIF can sustain pluripotency or induce the lineage differentiation of mESCs. In addition, although LIF/Stat3 signaling plays a pivotal role in the self-renewal of mESCs, the knockout mouse embryos for genes of this pathway usually develop normally [41]. This pathway appears to be important in early development during embryonic diapauses, as knockout embryos for gp130 fail during diapause due to apoptosis of the epiblast cells [50].
Moreover, mESCs have recently been shown to be in a ground state of pluripotency and do not need LIF/Stat3 and BMP4 (the main component in serum that sustains pluripotency) when small-molecule inhibitors of the key differentiation pathways are used in culture [51]. This study has shown that by using the chemicals SU5402, PD184352, and CHIR99021 (called 3i), which suppress the FGFR, MAPK kinase (Mek), and glycogen synthase kinase 3β (Gsk3β), respectively, Stat3-null mESCs could maintain self-renewal and a state of pluripotency that is indistinguishable from that of wild-type mESCs. The more potent Mek inhibitor PD0325901 with CHIR99021 (called 2i) is currently used as an appropriate supplement in chemically defined medium for the derivation and long-term maintenance of mESCs from different mouse strains, including nonpermissive NOD [52], as well as rats [53,54]. This supplement (2i) is used instead of the conventional LIF and serum/BMP4-containing medium, although LIF is routinely added to 2i to increase cloning efficiency [55].
LIF can also activate the Jak/Stat3 pathway in primed EpiSCs/hESCs; however, this pathway does not support the self-renewal of these cells [6,7,56]. Although the inefficiency of LIF/Stat3 signaling in maintaining the pluripotency of hESCs has yet to be determined [41], such inefficient signaling in EpiSCs is attributed to the low expression levels of LIFR and Stat3. Interestingly, overexpressed Klf4 [57], LIFR [58], or Tfcp2l1 [46] can activate this pathway in EpiSCs, propelling these cells into a reprogrammed naive state.
BMP4/Smad1,5,8 Signaling: Pluripotency Supporter or TE Inducer?
In addition to MEF/LIF, serum has been shown to play an important role in the traditional cultivation of mESCs. Consistent with the identification of the main factor that supports pluripotency in serum, BMP4 was shown to act as a substitute for FCS, and serum- and feeder-free culture conditions were subsequently established to cultivate mESCs by using LIF/BMP4 [59]. BMPs belong to the TGFβ superfamily proteins, which were originally identified as being involved in bone differentiation, but were subsequently shown to play pivotal roles during the embryogenesis of several organisms [60].
TGFβ superfamily ligands including TGFβ isoforms, Activin, Nodal, growth and differentiation factors, anti-Müllerian hormone, and BMPs bind to type I and II receptor serine/threonine kinases and thereby transduce their signal into cells. By connecting the kinase domains together, type II receptors phosphorylate and activate type I receptors, which in turn phosphorylate the regulatory Smad (R-Smad) proteins. Seven type I receptors, Activin-receptor like kinases (Alk) 1–7, and five type II receptors have been shown to activate R-Smads1,2,3,5,8 in vertebrates. TGFβ/Activin/Nodal signal molecules by activating Alks4/5/7 can phosphorylate R-Smad2,3 and BMP ligands can phosphorylate R-Smad1,5,8 downstream of Alk1/2/3/6. Following phosphorylation, R-Smads bind to co-Smad (Smad4) and translocate into the nucleus, wherein they act as transcription factors regulating the expression of target genes (Figs. 2 and 3) [61]. Moreover, inhibitory Smads (I-Smad6,7) inhibit the activation of R-Smads and thereby negatively regulate TGFβ signaling.
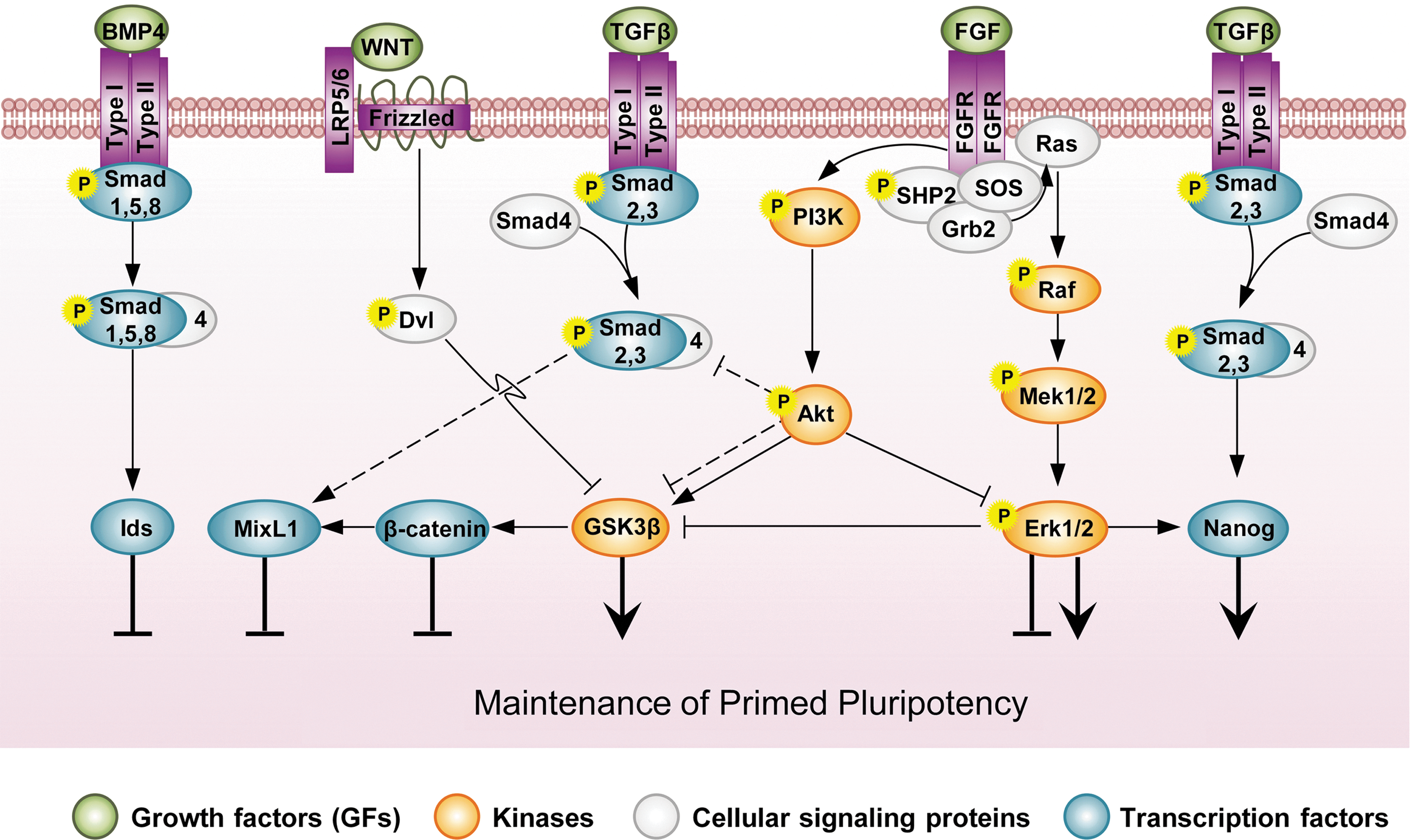
Signaling networks in primed hESCs/EpiSCs. The threshold activity of TGFβ, FGF, and GSK3β is important for sustaining pluripotency, wherein hyperactive PI3K signaling plays crucial roles in modulating these signaling activities. Different growth factors, such as FGF and IGF, trigger the PI3K signaling pathway. Hyperactive PI3K signaling restricts the activation of both Smad2,3 in TGFβ/Activin/Nodal signal transduction and MAPK/Erk in the FGF pathway. Under these conditions, primed ESCs can be maintained in a self-renewing state. Reduced PI3K signaling enhances the phosphorylation of Smad2,3, which, in cooperation with β-catenin, then promotes ESC differentiation into the mesendodermal lineage. PI3k signaling also inhibits GSK3β in primed ESCs, but this pathway also preserves a separate supply of GSK3β in its active form for suppressing Wnt/β-catenin signaling. PI3K mediates the downregulation of the MAPK/Erk pathway, thereby preventing the inhibitory effects of Erk on GSK3β. This active pool of GSK3β alleviates the differentiation stimuli of β-catenin in hESCs. Moreover, in contrast to naive pluripotency, BMP4 triggers the differentiation of primed ESCs. IGF, insulin-like growth factor. Color images available online at
BMP4/Smad1,5,8 signaling proscribes differentiation and thereby sustains pluripotency. BMP4 was primarily shown to induce the expression of inhibitors of differentiation (Id) genes, suppressing neural differentiation in mESCs [59]. Id proteins inhibit neurogenic basic helix-loop-helix transcriptional activators in mESCs, and the forced expression of Id proteins removes the requirement of ESCs for BMP4 [59]. Moreover, a genome-wide-scale experiment has shown that Smad1,5/Smad4 occupy the regulatory regions of several developmental regulator genes in mESCs; importantly, they suppress neural differentiation by inhibiting early neural commitment regulators, such as Dpysl2 and Kdm6b [62]. However, Zhang et al. [63] have claimed that instead of directly inhibiting neural differentiation, BMP4 sustains pluripotency in mESCs by preventing entry into the late epiblast stage. Those authors have shown that during the neural differentiation of mESCs, a BMP4-sensitive window exists when the cells exhibit similar expression characteristics to the epiblast of the egg cylinder. Interestingly, cells at this stage can be maintained as EpiSCs (ESC-derived EpiSCs or ESD-EpiSCs). Accordingly, neural differentiation from mESCs occurs in two phases: first, the conversion of mESCs into ESD-EpiSCs, and then the differentiation of ESD-EpiSCs into neural precursor cells. As BMP4 also suppresses the MAPK/Erk pathway, these researchers have proposed that BMP4 initially inhibits entry into the late epiblast stage by suppressing Erk signaling [63].
Several studies have shown that BMP4 signaling suppresses MAPK/Erk signaling [63,64]. Consistent with this finding, Smith and colleagues have used small-molecule inhibitors of Fgf4/Erk signaling in the 3i or 2i compound, instead of using BMP4, to establish conditions supportive for the ground state of pluripotency [51]. Activated Smad1,5 have been recently shown to upregulate Erk-specific dual specificity phosphatase 9 (DUSP9) and thereby inhibit Erk. DUSP9 inactivates Erk, establishing a link between BMP and MAPK signaling, which sustains pluripotency [65].
In primed EpiSCs/hESCs, however, BMP4 induces differentiation and represses self-renewal [66,67]. BMP4 signaling is considered to be a key regulator of the mesoderm, mesendoderm, extraembryonic endoderm, and a TE inducer in primed ESCs [68]. Treatment of hESCs with BMP4 can cause differentiation into extraembryonic endoderm and TE. However, treatment of hESCs with BMP4, in conjunction with FGF2 or FGF2 and Activin, can induce differentiation into mesoderm or mesendoderm, respectively [66,68]. Controversy surrounds the capability of BMP4 to induce the differentiation of hESCs into the TE cell lineage, as ESCs originate from the ICM [66,69,70]. Specifically, at the blastocyst stage, ICM cells are not capable of generating TE cells, and mESCs have been shown not to form the TE cell lineage during chimera formation [67]. Bernardo et al. have recently questioned this potency by showing that during mesoderm induction of EpiSCs/hESCs mediated by BMP4, TE-affiliated genes such as CDX2 and KRT7, which are common to both the mesoderm and TE lineages, could be expressed in addition to Brachyury (T), a definitive mesoderm marker. Therefore, contrary to previous reports, BMP4 was shown to induce the differentiation of EpiSCs/hESCs into mesoderm, rather than the TE [67]. However, proponents of the TE differentiation capability of hESCs promptly refute the conclusion of Bernardo et al., but rather attribute the results to differences in culture conditions [71]. Overall, BMP4 plays a role in the differentiation of primed ESCs into a wide range of cell types, including extraembryonic lineages, inconsistent with the role of BMP4 in the maintenance of naive pluripotency.
TGFβ, Activin, and Nodal/Smad2,3 Signaling: Balancing Self-Renewal and Differentiation
TGFβ, Activin, and Nodal/Smad2,3 signaling is known to direct differential cell fate decisions in the early embryo. This signal transduction has defined roles in the specification of node [72], mesoderm [73,74], and endoderm [75,76] formation, in addition to promoting the subsequent development of a variety of different tissues and organs [77]. These findings suggest that this signaling could not be involved in pluripotency; however, several studies have identified TGFβ as an active signaling pathway in both naive and primed ESCs, although conflicting views exist about its function in maintaining pluripotency.
TGFβ/Smad2,3 signaling is postulated to be indispensable for the propagation of mESCs [78,79]. Ogawa et al. have indicated that TGFβ signaling is activated in mESCs in an autocrine manner and that its inhibition by chemical inhibitors, such as SB431542, which selectively suppress ALK1/5/7 type I receptors, reduces the propagation of ESCs [78]. However, the present study and previous others have demonstrated that disturbance of TGFβ/Smad2,3 signaling does not affect mESC self-renewal [78,80,81]. Inhibition of this pathway has been shown to significantly enhance the efficiency of miPSC generation [82 –84]. Moreover, we have recently shown that TGFβ suppression by SB431542, in conjunction with inhibition of the Mek inhibitor PD0325901, has allowed for the highly efficient, reproducible generation of mESCs from different refractory and nonpermissive strains [85]. We have demonstrated that in a serum- and feeder-free condition, the combination of SB431542 and PD0325901 (defined as R2i) could support all aspects of the ground state of pluripotency [86]. Consequently, as the main promoter of differentiation in early development, suppression of TGFβ appears to play momentous roles in the maintenance of naive pluripotency. The inhibition of TGFβ/Smad2,3 signaling has been suggested to indirectly activate the BMP4/Id protein pathway and thereby support pluripotency [81,86]. This effect is likely accomplished by repression of I-Smad7 expression. Smad2 has been shown to induce the expression of Smad7, which acts as a negative regulator of both TGFβ and BMP4 in a feedback loop [87]. Therefore, TGFβ inhibition can augment BMP4 signaling, enhancing the self-renewal of mESCs [81].
TGFβ, Activin, and Nodal/Smad2,3 signaling, however, is considered to be a pivotal pathway in primed ESCs. Several studies have identified TGFβ signaling in the secretome of MEFs as the activity responsible for sustaining pluripotency in the hESC culture [88,89]. In a serum- and feeder-free culture condition, Activin was shown to be sufficient for the long-term maintenance of hESCs [90]. In the presence of SB431542, hESCs rapidly exit from their pluripotent state [88,91]. As with mESCs, suppression of TGFβ leads to BMP4 activation, which promotes the differentiation of hESCs into various lineage differentiations, and vice versa, TGFβ has a suppressive effect on BMP4 signaling [92]. However, BMP4 inhibition alone has been shown to be insufficient for the long-term maintenance of hESCs [93]. Activated Smad2,3 directly induce expression of the pluripotency marker gene NANOG [92,94]. Putative Smad binding sites have been identified on the NANOG proximal promoter in hESCs. Mutations in these elements have been shown to reduce NANOG promoter activity [92].
One abstruse problem regarding the role of TGFβ in the maintenance of pluripotency in hESCs is how the well-defined signaling that promotes differentiation could support pluripotency? Several studies have been performed to resolve this conundrum. Chng et al. have demonstrated that activated Smad2,3, in cooperation with NANOG and OCT4, suppresses the Smad-interacting protein 1 (SIP1) and sustains pluripotency. When TGFβ signaling is inhibited, elevated SIP1 levels lead to neuroectodermal differentiation [95]. SB431542, in combination with the BMP4 inhibitor noggin, has been shown to induce the differentiation of both hESCs and hiPSCs into the neural cell lineage [96]. In addition, it has been recently revealed that a limited range of activated Smad2,3 that controlled by PI3K signalling resulted in the maintenance of hESC self-renewability [97]. With reduced PI3K signaling, Smad2,3 phosphorylation is enhanced and differentiation into mesendodermal lineages could be progressed [97]. Another important signaling pathway that balances between self-renewal and differentiation capability of activated Smad2,3 is Wnt signaling, which will be discussed later. Therefore, cross-talk between different pluripotency-associated signaling pathways plays a decisive role in the stability of the undifferentiated state of hESCs.
Similarly to hESCs, TGFβ signaling is considered to be an important pathway that governs the pluripotency of EpiSCs [6,7]. Activated Smad2,3 enhance the pluripotency of EpiSCs through the induction of Nanog expression [98]. Although putative Smad binding sites have been shown not to be conserved between humans and mice, a functional binding site for phosphorylated Smad2,3 has been found on the mouse Nanog promoter several base pairs upstream of the known Sox-Oct bipartite motif that responds to activated Smad2,3 [98].
FGF and MAPK/Erk Signaling: Critical Threshold of Signaling Activity
Similarly to TGFβ signal transduction, FGF ligand binding to FGFR, a family of receptor tyrosine kinases (RTKs), highlights different outcomes in naive and primed pluripotent stem cells. FGF ligand binding to FGFR is considered to be an important stimulus for the proliferation and differentiation of different cell types. This binding leads to receptor dimerization and closing of the two RTKs, followed by cross-phosphorylation of the kinase domains of the two receptor chains at specific tyrosine residues. Phosphorylated RTKs activate important signal transduction pathways, such as the MAPK/Erk and the PI3K/Akt signaling pathways (Fig. 2) [99].
FGF/MAPK/Erk is a well-defined pathway in pluripotent stem cells. In addition to FGF, other growth factors such as LIF could also activate MAPK/Erk signaling. In this pathway, activated receptors phosphorylate SH2-domain-containing tyrosine phosphatase2 (SHP2). SHP2 interacts with the Grb2-SOS complex, resulting in activation of monomeric GTPase Ras. Ras activation then triggers a cascade of phosphorylation and activation events in the kinases of the MAPK pathway consisting of Raf, Mek, and Erk1/2. Activated Erk1/2 then phosphorylates a wide range of proteins, thereby regulating their activity [41,99]. As discussed just now, FGF/Erk signaling underlines the differentiation of mESCs [100,101]. In early embryonic development, FGF signaling is needed for the specification of the PE. A homozygous mutation in FGF signaling components, such as FGF4, FGFR, and Grb2, leads to embryo demise before or during gastrulation and complete lack of PE formation [20]. However, the derivation of FGFR-null mESCs is still possible [102]. Erk1/2 have been shown to phosphorylate the linker region of Smad1, inhibiting BMP4 signaling in mESCs. This phosphorylation causes Smad1 degradation by the proteasome machinery [103]. In addition, MAPK/Erk signaling suppresses the key pluripotency-affiliated factors in mESCs. As predicted from the function of FGF/MAPK/Erk signaling in suppressing Nanog expression during formation of the PE from ICM cells, this pathway has also been shown to inhibit the expression of Nanog in mESCs. Chemical inhibitors of FGFR (SU5402) or Mek (PD98059) prevent Nanog repression as well as PE differentiation in mESC aggregations [100]. Moreover, Kim et al. recently determined that Erk1/2 phosphorylate Klf4 at Ser123, thereby promoting its ubiquitination and degradation [104].
Nonetheless, FGF signaling is essential for the maintenance of hESCs/EpiSCs. Inhibition of this signaling pathway results in hESC differentiation, but the exact mechanism(s) underlying the maintenance of pluripotency has not yet been elucidated. In the presence of MEFs, basic FGF (bFGF) is postulated to act on MEFs to express and release key members of the TGFβ pathway such as Activin A. As mentioned earlier, Activin induces the expression of Nanog in both hESCs/EpiSCs [105]. However, dependence of the hESC culture upon MEFs or MEF-conditioned medium could be obviated by the presence of exogenous FGF2 (bFGF), as culture of hESCs with numerous chemically defined media that contain this factor has been found to maintain hESC self-renewal [106]. As such, bFGF should play more direct roles in hESC pluripotency. Bendall et al. have shown that under serum- and feeder-free conditions, hESC colonies produce differentiated feeder-like cells around themselves that function as an autologous feeder layer. These feeder-like cells express higher levels of FGFRs compared with undifferentiated hESCs, and treatment with bFGF causes these feeder-like cells to release insulin-like growth factor (IGF). IGF acts as a potent activator of PI3K, which plays a crucial role in maintaining hESC self-renewal [107]. Singh et al. have recently shown that bFGF influences pluripotency in a threshold-type manner. At a low concentration (10 ng), bFGF activates MAPK/ERK signaling to a level that is compatible with hESC self-renewal. This low level of ERK could sustain pluripotency, but the underlying mechanism has yet to be elucidated. At a higher concentration (100 ng), bFGF further activates MAPK/ERK signaling but also activates PI3K/Akt signaling, which could suppress ERK signaling. In this scenario, PI3K signaling inhibits the effect of a high level of MAPK/ERK signaling in inducing hESC differentiation into mesendoderm (Fig. 3) [97].
As with hESCs, EpiSCs depend upon bFGF for self-renewal [6,7]. However, the mechanisms that underlie the effect of bFGF on maintaining pluripotency differ between these two primed stem cells. Notwithstanding the effect of bFGF on NANOG expression in hESCs, bFGF appears to be unable to promote the self-renewal of EpiSCs by activating Nanog expression. bFGF could likely enhance EpiSC self-renewal by suppressing EpiSC differentiation into neuroectoderm and by preventing the spontaneous reversion of EpiSCs into the naive state [98].
PI3K/Akt Signaling: The Value of Cross-Talk Between Signaling Pathways
Increasing evidence reveals that PI3K (and the downstream serine/threonine kinase Akt or protein kinase B) is the only signal transduction pathway that plays pivotal roles in the self-renewal of both mouse and human pluripotent stem cells [108,109]. Growth factors, such as LIF, FGF, IGF, heregulin, and epidermal growth factor, activate PI3K/Akt signaling, thereby regulating important downstream effectors such as the mammalian target of rapamycin (mTOR) and Gsk3β [108,110 –112]. Further, there are important cross-talk between PI3K/Akt signaling and different signaling pathways, such as TGFβ/SMAD2,3 and MAPK/Erk (Figs. 2 and 3) [97,113].
While PI3K/Akt signaling is activated mainly by cytokine LIF in mESCs [41], this signaling is activated by insulin, IGF (the components in FCS or serum replacement), heregulin, and bFGF in hESCs [97]. Inhibition of PI3K by the chemical inhibitor LY294002 in mESCs leads to the downregulation of important pluripotency-related transcription factors, such as Klf2, Klf4, Nanog, c-Myc, Esrrb, Tbx3, and Zfp42 [41,114]. Treatment of hESCs with LY294002 causes upregulation of mesendoderm markers, such as Brachyury, Eomes, Goosecoid, and MixL1, and exits hESCs from their pluripotent state [97]. PI3K appears to play a central role in modulating cross-talk between various signaling pathways in pluripotent stem cells. PI3K performs its key functions mainly by regulating mTOR and GSK3β.
PI3K/Akt signaling through activation of mToR supports self-renewal
The serine/threonine protein kinase mTOR is a known major target of PI3K/Akt signaling transduction. Following activation of Akt, it phosphorylates and inactivates TSC2, which acts as an inhibitor of the GTPase Rheb. In its active form, Rheb activates mTOR. In mESCs, mTOR is defined as a key regulator of self-renewal. Inhibition of PI3K by LY294002 also suppresses mTOR and enables mESCs to exit from their pluripotent state [110]. Inducible deletion of mTOR or treatment with rapamycin has been shown to significantly decrease the proliferation of mESCs [115,116]. A recent study has found that knockdown of TSC2 or the molecule that it negatively regulates downstream of mTOR, folliculin (Flcn), prevents the differentiation of mESCs [117]. Flcn, with its partners Fnip1 and Fnip2, restricts the transcription factor Tfe3 to the cytoplasm and impedes access of Tfe3 to pluripotency-related genes such as nuclear receptor Esrrb. Esrrb is considered to be an important regulator of pluripotency that is directly activated by Nanog. During differentiation, TSC2 (and its partner TSC1) causes mESCs to exit from the state of pluripotency by lowering the activity of mTOR (Fig. 2) [118].
The PI3k/AKT/mTOR signaling axis also has an important function in hESCs. mTOR has been found to stabilize the core pluripotency factors—OCT4, NANOG, and SOX2—in hESCs and to support self-renewal by suppressing hESC differentiation into mesodermal and endodermal lineages [113,119]. However, the exact molecular mechanism of mTOR-dependent pluripotency in hESCs remains to be elucidated [111,113].
PI3K/Akt signaling by suppressing GSK3β and Erk affects self-renewal
The serine/threonine protein kinase GSK3β acts as a central switch in numerous intracellular signaling transductions, such as canonical Wnt/β-catenin, PI3K/Akt, and MAPK/Erk [109]. It is well defined that mESCs need to suppress GSK3β [51]. Upon activation by cytokine LIF in mESCs, PI3K/Akt phosphorylates GSK3β on serine 9 (GSK3βpS9) and thereby inactivates it. Phosphorylation of GSK3β regulates the shuttling of GSK3β between the nucleus and cytoplasm through the formation of a complex with the GSK3β-interacting protein Frat. Frat interacts with GSK3βpS9 and carries it out of the nucleus, thereby impeding its access to the pluripotency factor c-Myc. Withdrawal of LIF in mESCs results in decreased PI3K/Akt signaling; therefore, activated GSK3β could enter the nucleus and decrease c-Myc activity by phosphorylating c-Myc on threonine 58 (c-MycpT58) [120]. Use of GSK3β chemical inhibitors, such as BIO, lithium chloride, and CHIR99021, in the absence of LIF, prevents formation of c-MycpT58. Moreover, inhibition of PI3K mediated by chemical inhibitors or genetic tools leads to decreased expression of Nanog via GSK3β activity [121]. Research has shown that the regulation of Nanog function by GSK3β does not depend on Wnt/β-catenin pathways. GSK3β directly downregulates Nanog [121], possibly by a mechanism that is similar to that observed in c-Myc regulation (Fig. 2).
The PI3k/Akt/GSK3β signaling axis also has a salient function in hESCs. However, GSK3β appears to play intricate roles in hESCs. Although activation of PI3k/Akt results in inhibition of GSK3β, this pathway also preserves a separate supply of GSK3β in its active form for suppressing Wnt/β-catenin signaling in hESCs (see next section). In the latter role, PI3K/Akt mediates the downregulation of the MAPK/Erk pathway, thereby preventing the inhibitory effects of Erk on GSK3β. This active pool of GSK3β could antagonize the differentiation stimuli of β-catenin in hESCs [109].
PI3k/Akt signaling also regulates pluripotency in hESCs by modulating the level of activated Smad2,3 so that it is compatible with pluripotency [97]. Under self-renewing conditions, Akt first modulates the threshold level of Smad2,3 activity and then inhibits Erk and maintains GSK3β activity, permitting the activation of a specific subset of target genes required for self-renewal. In the absence of PI3K/Akt signaling, however, Activin A/Smad2,3 signaling is enhanced and Erk is activated, and when coupled with the activation of Wnt signaling, Activin A/Smad2,3 and Erk activities promote GSK3β inhibition and β-catenin activation. Therefore, Smad2/3 and Wnt effectors cooperate, thereby enabling the activation of mesendoderm markers [97]. Therefore, although TGFβ/Smad2,3 signal transduction mediates the induction of NANOG expression and supports hESC pluripotency [92,94], the excess phosphorylated Smad2,3 leads to hESC differentiation. However, the mechanism that underlies PI3K modulation of Smad2,3 in hESCs requires further investigation (Fig. 3) [111].
Wnt/Gsk3/β-Catenin Signaling: A Dilemma in Primed Pluripotency
Wnt proteins are signaling molecules that control numerous developmental processes in all animal cells. The Wnt ligands act by binding to the Frizzled receptor and coreceptors and LDL-receptor-related proteins 5 and 6 (LRP5, 6). In the canonical pathway, Wnt signaling through phosphorylation and inactivation of GSK3β suppresses proteolysis of the multifunctional β-catenin molecule. β-Catenin plays roles both in cell-to-cell adhesion, when associated with cadherin, and in gene regulation as a transcription factor. Upon suppression of GSK3β, β-catenin enters the nucleus and promotes gene transcription by interacting with the Tcf family of transcription factors [122].
Substantial evidence points to Wnt/β-catenin signaling as being involved in the self-renewal of the naive pluripotent state. Activation of Wnt signaling by addition of exogenous Wnt3a or pharmacological inhibitors of GSK3β, overexpression of β-catenin, and depletion of Tcf3 enhances mESC self-renewal [122]. However, recent studies have revealed that mESCs could be established from depleted Ctnb1 (coding gene for β-catenin) [123,124]. One possible explanation for this observation is the compensatory pluripotency-affiliated signal transductions that begin following the culture of mESC under specific conditions. Wray et al. have shown that in a serum-free medium, β-catenin is required only when mESCs are cultured without LIF. In addition, the pluripotency of Stat3-null ESCs relies on CHIR99021 as a GSK3β inhibitor. Of note, long-term maintenance of pluripotency requires PD0325901 with LIF or CHIR99021, or the combination of LIF and CHIR99021. This requirement indicates that these two pathways cannot completely be substituted for one another [124]. New evidence suggests that Wnt/β-catenin could repress differentiation in naive cells by inactivating the transcriptional repressor Tcf3 [123 –125]. Tcf3 co-occupies many target promoters with the core pluripotency transcription factors—Oct4, Nanog, and Sox2 [126]. Genetic depletion of Tcf3 eliminates the need for Wnt3a or GSK3β inhibitors for maintaining pluripotency [125]. The nuclear receptor Esrrb has recently been shown to be a critical target of Tcf3 repression in ESCs. As Esrrb is a direct target of Nanog, its overexpression could maintain the self-renewal of Nanog-null mESCs. Moreover, consistent with the ability of CHIR99021, overexpression of Esrrb could also maintain the LIF-independent self-renewal of mESCs (Fig. 2) [127].
In primed ESCs, rather than maintaining pluripotency, Wnt signaling appears to promote the differentiation of hESCs/EpiSCs [97,128,129]. As previously mentioned, in hESCs, a pool of GSK3β needs to be maintained in the active form to inhibit the activation of β-catenin. β-Catenin in collaboration with phosphorylated SMAD2,3 was found to induce the differentiation of hESCs into mesoderm [97]. Similarly, pharmacological and genetic inhibition of β-catenin was found to enhance the pluripotency of EpiSCs by blocking their spontaneous differentiation toward the mesodermal cell fate [129]. Moreover, suppression of Wnt/β-catenin signaling was found to be necessary for establishing EpiSCs and for preventing reprogramming into the naive state (Fig. 3) [130].
Maneuvering the Extrinsic Signals for Converting Primed and Naive States Together in a Dish
Several groups have attempted to convert primed ESCs into the naive state. In the mouse, primed EpiSCs can be converted into the naive state at a low efficiency by continuously culturing cells in serum/LIF, 2i/LIF, and kenpaullone (GSK3β and CDK1/cyclin B inhibitor)/CHIR99021, or by overexpressing Nanog, Klf2, Klf4, Stat3, Nr5a, c-Myc, and Tfcp2l1 at a higher efficiency (Fig. 4) [52,57,58,98,131 –134]. Conversely, naive mESCs can be readily transformed into the primed state by culturing cells in bFGF/Activin (ESD-EpiSCs) [57,63]. The ESD-EpiSCs can reprogram efficiently into the naive state if they have been cultivated on appropriate feeder layers such as human amnion epithelial cells and LIF-contained medium [135]. EpiSCs can be derived from preimplantation mouse embryos by mechanically passaging ICM outgrowths in bFGF-contained medium [136]. Induced EpiSCs can also be directly generated by reprogramming somatic cells under EpiSC culture conditions [137]. These experiments strongly demonstrate the importance of the culture environment in dictating the state of pluripotency [137].
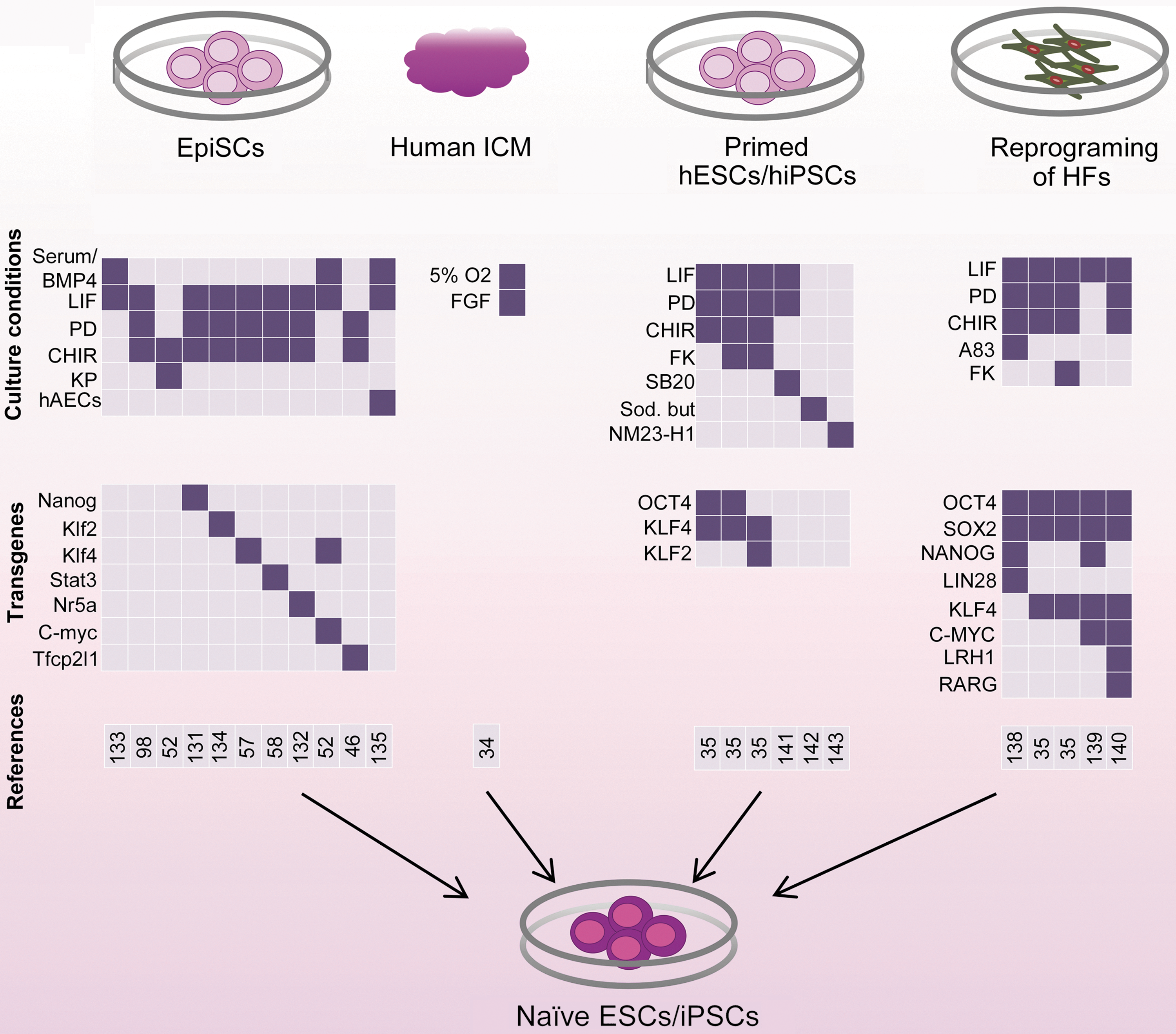
Studies to generate naive pluripotent stem cells from EpiSCs, human ICM cells, and primed hESCs/hiPSCs, and by the direct reprogramming of human fibroblasts. Stem cells of naive pluripotency can be derived from mouse EpiSCs by altering the culture conditions and by overexpressing pluripotency-related transcription factors. In humans, ESCs isolated from preimplantation embryos under physiological oxygen conditions exhibit some features of naive pluripotency, such as representation of two active X chromosomes. However, these lines are bFGF dependent and do not exhibit other features of naive pluripotency. Reversion of conventional primed hESCs/hiPSCs or direct reprograming of human somatic cells into the naive pluripotent state by modifying the culture conditions and overexpressing pluripotency-related transcription factors has yielded more promising results. Although reprogrammed EpiSCs show all features of naive pluripotency, achieving the truly naive pluripotent state of hESCs/hiPSCs requires additional effort. hiPSCs, human induced pluripotent stem cells; HFs, human fibroblasts; PD, PD0325901: Mek inhibitor; CHIR, CHIR99021: GSK3β inhibitor; KP, kenpaullone: GSK3β and CDK1/cyclin B inhibitor; hAECs, human amnion epithelial cells (as feeder cells); FK, forskolin: protein kinase A agonist; SB20, SB203580: p38 inhibitor; sod. but, sodium butyrate: histone deacetylase (HDAC) inhibitor; A83, A-83-01: ALK4/5/7 inhibitor; NM23-H1, MUC1* transmembrane receptor ligand, a tumor suppressor growth factor; LRH1, liver receptor homologue 1 or Nr5a2; RARG, retinoic acid receptor gamma. Color images available online at
More extensive studies have been performed to achieve naive pluripotent stem cells from humans. Some groups have attempted to derive naive hESCs by culturing preimplantation human embryos in 2i/LIF. However, as mentioned earlier, human PE formation is not dependent upon FGF signaling, and 2i/LIF cannot support pluripotency in human embryos [10,11]. Despite this, more promising results have been obtained by directly reprogramming human fibroblasts or reverting hESCs/hiPSCs into the naive state. In such experiments, transfection of human fibroblasts or primed hESCs with common naive and primed pluripotency genes, such as OCT4, SOX2, C-MYC, and LIN28, or with more naive-related pluripotency marker genes, such as KLF2, KLF4, and NANOG, in 2i/LIF, A-83-01 (ALK4/5/7 inhibitor)/2i/LIF, or forskolin (protein kinase A agonist)/2i/LIF, has produced naive hESCs/hiPSCs (Fig. 4) [35,138,139]. In addition, enhancing retinoic acid (RA) signaling by expressing the RA receptor (RAR-γ) and liver receptor homolog 1 (Lrh-1; Nr5a2) with reprogramming factors has been shown to result in the generation of naive hiPSCs [140]. In some studies, reversion of hESCs/hiPSCs has been performed without the forced expression of pluripotency genes. Different small molecules including PD0325901 and SB203580 (p38 inhibitor) [141], sodium butyrate (as histone deacetylase [HDAC] inhibitor) [142], or NM23-H1 (a tumor suppressor growth factor) [143] were used in this regard. However, these studies have been unable to demonstrate definitive evidence for attaining truly naive human cells. Dependence upon exogene expression, deficiency in differentiation, absence of all features of mESCs, and lack of reproducibility are among the obstacles that must be overcome for the successful generation of naive hiPSCs.
Moreover, despite the great attention to the transcription factors and signaling pathways related to naive pluripotency, another important factor that affects the pluripotency pattern of ESCs is the physiological oxygen level. Under reduced oxygen levels (≤5% O2), or hypoxia, which is the natural environment of ICM and epiblast cells, the intracellular signaling pathways have been postulated to exhibit different effects. Cultivation of LIF-dependent mESCs under hypoxic conditions was shown to lead to the downregulation of Stat3 signaling and the attenuation of mESC self-renewal and pluripotency [144,145]. Hypoxia suppresses the expression of LIFR, likely through the induction of hypoxia inducible factor-1α (HIF-1α) [146]. However, under the 2i condition, hypoxia does not induce differentiation [144], likely by enhancing β-catenin activation via HIF-1α [147]. In hESCs, hypoxia has been shown to increase reversion of hESC- or hiPSC-derived differentiated cells back to undifferentiated stem-cell-like state [148]. Hypoxia appears to play an important role in maintaining primed pluripotency. Hypoxia, by inducing HIF-1α, has been shown to be sufficient for deriving naive ESCs into the EpiSC-like stage [149]. Although female hESCs derived from human blastocysts cultured under hypoxic conditions have two active X chromosomes, these lines are bFGF dependent and do not exhibit other features of naive pluripotency (Fig. 4) [34,150]. Overall, these findings indicate that environmental conditions such as physiological oxygen levels increase the complexity of the multifactorial intracellular signaling networks involved in the different states of pluripotency.
Concluding Remarks
Maintenance of the supreme balanced networks of different signaling transductions has been determined to play a major role in the perpetuation of pluripotency in stem cell culture. Interestingly, signaling pathways that support pluripotency in naive stem cells exhibit quite different functions in the primed state of pluripotency, and vice versa. While LIF, BMP4, and Wnt signaling are considered to be salient naive-related signaling pathways, they are dispensable in the maintenance of primed pluripotency or, more specifically, they are responsible for triggering the differentiation of primed stem cells into different lineages. Similarly, FGF, TGFβ, and GSK3 support the pluripotency of primed stem cells and in contrast propel the lineage differentiation of naive stem cells. PI3K appears to be the only signaling pathway that plays crucial roles in the maintenance of both naive and primed pluripotency. But a closer look reveals that it acts in different ways to support pluripotency of the naive and primed pluripotency states. Taken together, in vivo and in vitro evidence suggest that naive mESCs are quite different from primed EpiSCs/hESCs in regard to functional behavior and signal transduction machinery. Although several studies have recently demonstrated the generation of naive human cells, the representation of all features of naive pluripotency under stable long-term cultivation has yet to be achieved. Naive pluripotency appears to be a rodent-specific phenomenon, and the generation of naive hESCs/hiPSCs requires additional attempts to identify the underlying molecular mechanisms and the cross-talk between the different signaling pathways, both in naive and primed states of pluripotency. Obtaining unequivocal evidence of how pluripotency is established in culture is requisite to exploiting pluripotent stem cells for biomedical research purposes.
Footnotes
Acknowledgments
We thank the members of the Department of Stem Cells and Developmental Biology labs for their helpful suggestions and critical reading of the article. We also thank Areti Malapetsas for final proofreading. This study was funded by grants provided from Royan Institute and Iranian Council of Stem Cell Technology and the Iran National Science Foundation (INSF).
Author Disclosure Statement
The authors declare they have no competing financial interests.