
Abstract
Human mesenchymal stem cells (hMSCs) are multipotent cells used in cell therapy research. One of the problems involving hMSCs is the possibility of genetic instability during in vitro expansion required to obtain a suitable number of cells for clinical applications. The cytokinesis-block micronucleus (CBMN) assay measures genetic instability by analyzing the presence of micronucleus (MN), nucleoplasmic bridges (NPBs), and nuclear buds (NBUDs) in binucleated cells. The present study describes modifications in the CBMN assay methodology to analyze genetic instability in hMSCs isolated from the umbilical vein and in vitro expanded. The best protocol to achieve binucleated hMSCs with preserved cytoplasm was as follows: cytochalasin B concentration (4.0 μg/mL), use of hypotonic treatment (3 min), and the fixative solution (9 methanol:1 acetic acid). These adaptations were reproduced in three hMSC primary cell cultures and also in XP4PA and A549 cell lines. The frequency of hMSCs treated with mitomycin-C presenting MN was lower than that with other nuclear alterations, indicating that the hMSCs contain mechanisms to avoid a high level of chromosomal breaks. However, a high frequency of cells with NPBs was detected and spontaneous anaphase bridges under normal hMSC in vitro culture were observed. Considering that anaphase bridges are characteristic alterations in tumor cells, the CBMN assay is indicated as an important tool associated with other genetic analyses in order to ensure the safe clinical use of hMSCs in cell therapy.
Introduction
H
A number of authors have shown that human stem cells remain morphologically and genetically stable after in vitro culture [8 –17]. However, one of the main problems that can arise in stem cell lines isolated from different sources is the possibility of genetic instability during cell expansion [18,19]. Although the occurrence of karyotypic instability in cultured hMSCs has been documented, it remains controversial [19 –29], and only two groups demonstrated the occurrence of in vitro spontaneous transformation of hMSCs isolated from bone marrow [30,31]. Some researchers defend the hypothesis that human stem cells derived from bone marrow do not transform spontaneously in vitro and that aneuploidy can occur without leading to malignant transformation of hMSCs, possibly being only a sign of senescence [32,33].
To draw a firm conclusion regarding the biosecurity of these cells, a larger number of hMSC cultures must be analyzed and more genetic tests need to be included in order to assess genetic integrity before any treatment involving cell therapy. The cytokinesis-block micronucleus (CBMN) assay consists of analyzing nuclear alterations in binucleated cells that were impeded from completing cytokinesis through the use of cytochalasin-B (cyt-B), enabling the accumulation of nearly all the cells that divided in the binucleated stage [34]. Although CBMN analysis is less resolutive and less informative than metaphasic chromosome analysis using the G banding technique, it is much easier and faster [35], since it allows analysis of a larger number of cells and application of statistical tests, and the detection and classification of nuclear alterations do not require as extensive personnel training as human karyotype analysis.
The CBMN assay can assess a number of nuclear alterations, such as micronucleus (MN), nucleoplasmic bridges (NPBs), and nuclear buds (NBUDs). The MN is the result of chromosomal breakage and/or loss caused by errors in DNA repair or in chromosomal segregation. Therefore, MN characterizes whole chromosomes or acentric fragments not included in the main nucleus that are surrounded individually by a nuclear membrane [36]. Analysis of MN in interphasic cells of cell smears has been used to detect chromosomal alterations that arise due to the aneugenic and clastogenic effects caused by a variety of compounds. It is being used in the analysis of exfoliated cells to detect precancerous and cancerous lesions, determining high-risk groups for tumors in the oral cavity, urinary tract (bladder), cervix, and esophagus [37,38]. The frequency of micronucleated cells increases in tissues exposed to carcinogens before any clinical symptom emerges. Therefore, the MN test in exfoliated epithelial cells is a biomarker of occupational exposure to genotoxic chemical agents [39] and a possible indicator of early signs of carcinogenesis. The NPBs indicate the occurrence of rearrangements in which chromatids or chromosomes are pulled to opposite poles during the anaphase: dicentric chromosomes, ring chromosomes, and chromosomes involved in telomeric associations. In CBMN, binucleated cells with NPBs are easily observed because of cytokinesis inhibition by cyt-B, thereby avoiding the breakage of anaphase bridges from which NPBs derive. Therefore, the nuclear membrane forms around the bridge that links the two nuclei, making it possible to detect these chromosomal alterations [40]. The NBUDs are morphologically similar to the MN, except that they remain connected to the nucleus. They are alterations that indicate amplified DNA removed from the cell nucleus and therefore a marker of gene amplification [36]. NBUDs can also form when an NPB between two nuclei breaks and the remnants shrink back toward the nuclei. Further, NBUDs can occur temporarily after the NPB breaks [40]. Recent data have demonstrated that NPBs and NBUDs are also useful biomarkers for monitoring genetic damage, since they are easily recognizable nuclear alterations that allow rapid analysis and counting. MN, NPBs, and NBUDs are nuclear alterations characteristic of chromosomally unstable cells often detected in tumor cells and peripheral blood of patients with cancer [41]. The frequency of nuclear alterations observed by CBMN is a relevant biomarker for the risk of lung cancer and melanoma, and can be used for early detection of the disease, making it a useful tool in clinical and epidemiological studies [42,43]. The CBMN method also allows assessment of the impact of micronutrients in the diet and the combination of micronutrients on DNA damage, revealing the carcinogenic or protective potential of certain diets [44,45].
CBMN is a reliable method for detecting and quantifying DNA damage and chromosomal instability in human cells, initially developed to analyze peripheral blood lymphocyte cultures [34], but adapted for applications in established cell lines, solid-tumor cultures, bone marrow, and even in reconstructed human epidermis [46 –50].
Most genotoxicity tests have been conducted in human cell lines immortalized in vitro; however, they do not represent normal cell types. Some authors have suggested the use of human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) [51] in toxicological studies and drug research as a new strategy for determining the safety of new chemicals [52 –56]. However, there are a number of ethical, religious, and political issues related to the use of hESCs, and the iPSCs derive from somatic cells that need to be genetically handled to return to a state of pluripotency [51]. hMSCs are primary multipotent cells that represent normal cell types; that is, they are isolated and cultured with no genetic handling whatsoever and, therefore, can be used as a cell model in drug research in order to discover the most efficient chemicals and make therapeutic biomonitoring safer. To that end, genotoxicity tests could be carried out with these cells exposed to chemicals, but there are no studies of CBMN using hMSCs for that purpose.
Accordingly, for the first time, the present study describes CBMN assay methodology with the necessary adaptations to analyze hMSCs isolated from the umbilical vein and assess the genetic instability of these hMSCs in vitro expanded.
Materials and Methods
hMSC isolation and cell culture
The procedure used in this study was approved by the UFRN Ethics Committee under protocol No. FR132464, following Brazilian guidelines (Resolution 196/96). Written informed consent was obtained from mothers to collect fresh human umbilical cord specimens from three full-term healthy newborns (two boys, originating primary cell cultures hMSC1 and hMSC2; and one girl, originating primary cell culture hMSC3). The hMSCs for this study were isolated from umbilical veins and expanded in vitro as previously described [57]. Briefly, the umbilical vein was catheterized, cleaned until it was free of excess blood, and filled with a 0.5% collagenase type IV solution (Worthington). After enzymatic disaggregation, the cell suspension was collected after gentle massaging of the cord and by washing with phosphate-buffered saline (PBS). The cell pellet was resuspended in alpha-minimum essential medium (α-MEM; Gibco–Invitrogen) supplemented with 20% fetal bovine serum (FBS; HyClone) and 1% antibiotic solution (prepared with 10,000 U/mL penicillin G sodium and 10,000 μg/mL streptomycin sulfate; Gibco–Invitrogen). The cells were plated onto T25 tissue culture flasks (TPP) and cultures were kept at 37°C in a humidified atmosphere containing 5% CO2. hMSC cultures reaching 60%–70% confluence were harvested for expansion using 0.25% trypsin/1 mM EDTA (Gibco–Invitrogen); cells were counted using the trypan blue (Corning–CellGro) exclusion test and replated at 3,000–4,000 viable cells/cm2 with α-MEM supplemented with 10% FBS and 1% antibiotic solution. This procedure was equivalent to one passage and was repeated every three days approximately. The number of doublings in cell populations (PD) was calculated according to the following formula PD=3.32×(log10Nt−log10No), where No is number of viable cells at seeding and Nt is number of viable cells at harvest. The medium culture was changed and nonadherent cells were removed at least twice a week.
Characterization of hMSCs: determination of the cell-surface antigen profile and multilineage differentiation potential
In accordance with the minimum requirements for defining multipotent mesenchymal stromal cells established by the International Society for Cellular Therapy [1], hMSCs cultured in this study were phenotypically characterized using flow cytometry and induced to differentiate into osteoblasts, adipocytes, and chondroblasts under differentiation conditions and protocols that we previously reported [57]. To phenotypically characterize hMSCs and define their purity, cells were labeled with monoclonal antibodies to analyze the cell-surface expression of typical marker proteins: CD105FITC and CD90PE-Cy5 (purchased from Bioscience), and CD73PE, CD34PE, HLA-DRFITC, CD45FITC, and CD14PE (from Becton Dickinson). For each test, isotype-matched monoclonal antibody was used as a negative control (IgG 1-FITC, PE, and PE-Cy5; Becton Dickinson). Flow cytometer analysis was conducted with a fluorescence activated cell analyzer (FACScan) using Cell Quest Software (Cell QuestTM Software; Becton Dickinson Immunocytometry Systems).
Cells were seeded and cultured into six-well plates (TPP) to induce osteogenic differentiation. When cultures were subconfluent (80%), hMSCs were incubated in osteogenic medium: α-MEM with 10% FBS/1% antibiotic solution supplemented with 50 μM ascorbate 2-phosphate (Sigma-Aldrich), 0.01 μM dexamethasone (Sigma-Aldrich), and 10 mM β-glycerolphosphate (Fluka Sigma-Aldrich). Adipogenic differentiation induction occurred when subconfluent (80%) cultures of hMSCs that were seeded into six-well plates (TPP) were treated with adipogenic medium: Dulbecco's modified Eagle's medium high glucose (DMEM-HG; Gibco–Invitrogen) and 15% FBS/1% antibiotic solution supplemented with 5 μg/mL insulin from bovine pancreas (Sigma-Aldrich), 1 μM dexamethasone (Sigma-Aldrich), and 60 μM indomethacin (Cayman Chemical Company). For chondrogenic differentiation, 2×105 hMSCs were centrifuged to form a cell pellet that was not disturbed and was cultured for 21 days in DMEM-HG and 1% FBS/1% antibiotic solution supplemented with 0.5 μg/mL insulin from bovine pancreas (Sigma-Aldrich), 10 ng/mL transforming growth factor beta 1 (Sigma-Aldrich), and 50 μM ascorbate 2-phosphate (Sigma-Aldrich). After 21 days of treatment in specific differentiation medium, cells treated with osteogenic and adipogenic medium were fixed, and the chondrogenic pellets were fixed with 10% buffered formalin and embedded in paraffin. Osteoblast differentiation was demonstrated by staining the mineralized extracellular matrix with Alizarin Red S (Sigma-Aldrich), adipocyte differentiation was demonstrated by staining lipid-containing vacuoles with Oil Red O (Sigma-Aldrich), and chondroblast differentiation was demonstrated by Alcian Blue (Sigma-Aldrich) staining of the extracellular matrix rich in sulfated glycosaminoglycans.
CBMN assay
CBMN assay was carried out according to methodology described by Fenech [36] with a number of modifications adapted to the primary hMSC culture. The cells were submitted to CBMN assay at passage 10 (a total of 24 PD, considering that the mean cell yield was related to a number of about 2.4 PD per passage). If cultures were healthy—consisting of small, adherent, spindle-shaped fibroblastoid cells—∼60% confluent with frequent refractile doublets of newly dividing cells, and with all cells viable (detected by trypan blue test), then we proceeded with the protocol to obtain binucleated cells. Initially, 5×104 umbilical cord 1 cells (hMSC1) were seeded into each well of the six-well plate (TPP) in order to conduct preliminary experiments to determine cyt-B concentration and incubation time to achieve a significant number of dividing cells blocked at the binucleated stage. The following cyt-B (Sigma Chemical Company) concentrations were checked: 1.5, 2, 2.5, 3, 3.5, 4, 4.5, and 6 μg/mL, after 12 and 24 h of incubation at 37°C in a humidified atmosphere containing 5% CO2. These experiments fared poorly because it was observed that the well area of the six-well plate hindered obtaining an adequate number of binucleated cells for CBMN analysis (see “Results” section).
The experiments were repeated with T25 tissue culture flasks (TPP), resulting in a sufficient number of binucleated cells for CBMN analysis, which was performed in triplicate on hMSCs isolated from three donors (hMSC1, hMSC2, and hMSC3).
In the hMSC culture undergoing active cell division (∼60% confluent), cells were detached with 0.25% trypsin/EDTA solution and replated at 8×104 cells per T25 flask. After cell adhesion (16–24 h), the medium was changed in negative control flasks, and, in the positive control flasks, cells were challenged with fresh medium containing mitomycin-C (mit-C; final concentration 0.2 μg/mL). After 24 h of mit-C treatment, the culture medium in all flasks was changed again and cyt-B was added to each sample, resulting in a final concentration of 4.0 μg/mL. The culture medium was collected 24 h after the addition of cyt-B, and cells were detached from the plastic substratum with 0.25% Trypsin/EDTA solution and transferred to centrifuge tubes, which were centrifuged gently (300 g) for 5 min. The supernatant culture medium was then removed, leaving ∼100 μL of fluid on top of the cell pellets. In this step, the procedure was tested without using a hypotonic solution, according to Fenech [36], but failed. Thus, we tested a new hypotonic treatment in hMSCs. The hMSCs were exposed to 200 μL of hypotonic solution developed in our laboratory: 0.075 M KCl (Merck) mixed to solution 9 distilled water:1 culture medium with FBS at a final proportion of 1:1. The hypotonic solution was slowly added for 3 min and mixed with the cells at room temperature. The hypotonization process was immediately interrupted by adding 100 μL of cold methanol:acetic acid (9:1; Merck) fixative solution freshly made, and the cell suspension was resuspended to prevent clump formation. Before defining the fixative concentration (9 methanol:1 acetic acid), the cytoplasmic area of the cells was evaluated following treatment with fixative containing the proportions of methanol/acetic acid 3/1 [36], 9/1, and 14/1 at the end of hypotonic treatment, the best ratio being 9 methanol:1 acetic acid fixative. The tubes were centrifuged immediately (300 g) for 6 min; the supernatant was removed and replaced slowly with 2 mL of fixative consisting of cold methanol/acetic acid (9/1). It is important to disperse cells gently before adding the fixative. Prior to slide preparation, the tubes were centrifuged, the supernatant was removed, and cells were resuspended in 100–150 μL of fresh fixative depending on cell pellet size; 25–35 μL of cell suspension (depending on cell concentration) was evenly distributed onto a clean glass slide placed above the hot water bath (40–60°C) for 10–20 s and left to air dry. Slides were aged in a 60°C oven for 30 min, and stained using 10% Giemsa stain in potassium phosphate buffer solution for 8 min.
Slides were prepared in duplicate from each triplicate culture and scores were obtained from two different scorers. For each replicate of hMSC cultures, at least 600 binucleated cells were analyzed, for a total of at least 1,800 binucleated cells in each experimental group. The cells were analyzed for the presence of MN (biomarkers of chromosome breakage or loss), NPBs (biomarker of asymmetrical chromosome rearrangement), and NBUDs (biomarkers of gene amplification). Slides were examined at 400 and 1,000 magnification using a bright-field microscope (OLYMPUS CX31). Binucleated cells were evaluated according to the criteria described by Fenech [36] with one exception; in this study, the two main nuclei in a binucleated cell could not touch or overlap each other, and a cell with two overlapping nuclei could not be scored (even if the nuclear boundaries of each nucleus were distinguishable). Since the nuclei of hMSCs are very large and may conceal NPBs that can occur between them, it is important that the nuclei be well spread out in the cytoplasm and separated from one another.
Five hundred viable cells were scored for each replicate of individual hMSC culture in order to obtain the nuclear division index (NDI). The NDI provides an estimated measure of the nuclear division status and cell division kinetics. NDI was calculated as follows: NDI=(M1+2M2+3M3+4M4)/N; where M1–M4 represents the number of viable cells with one to four nuclei and N is the total number of viable cells scored (excluding necrotic and apoptotic cells) [36].
CBMN in other cell lines
CBMN was used for the first time in hMSCs. To determine a possible influence of the methodological procedures used on the results obtained in these cells, the CBMN assay was performed in two cell lines that are routinely cultivated in our laboratory, XP4PA and A549, using the same protocol established for hMSCs, except that in the A549 cell line the hypotonic solution was not used. The XP4PA cell line [58] (kindly supplied by Dr. Alain Sarasin) consists of an immortalized human fibroblast xeroderma pigmentosum (XP), group C. A549 is a lung adenocarcinoma cell line established from culturing cancerous lung tissue from a 58-year-old Caucasian man [59]. Cells were cultivated in DMEM supplemented with 10% FBS and 1% antibiotics in a humidified incubator at 37°C containing 5% CO2. The CBMN assay was performed in triplicate for each cell line, and the slide analysis was done in the same way described for hMSCs.
Detection of natural occurrence of anaphase bridges between hMSCs on in vitro culture
CBMN analysis revealed a large number of NPBs in all hMSC treatments, including the negative control (see description of “Results” section). To observe whether there are anaphase bridges between hMSCs undergoing division in vitro without the addition of cyt-B, proliferative cells were removed from T25 flasks and processed in a 15-mL tube following the same procedure described for CBMN (removing the cyt-B). After the material was fixed, the slides were prepared and stained with Giemsa. hMSC anaphases were observed under 40× and 100× lenses using an Olympus CX31 microscope.
Statistical analysis
The frequencies of binucleated cells with normal nuclei and with nuclear alterations (NPBs, NBUDs, or MN) for each hMSC culture in both standard culture medium without mit-C (SC) group and with mit-C consisting of the positive control group were compared using the t-test. A P value<0.05 was considered significant.
The frequencies of binucleated cells with normal nuclei and with nuclear alterations (NPBs, NBUDs, or MN) for each hMSC culture group, A549 and XP4PA cell lines grown in standard culture medium, were compared using two-way analysis of variance. A P value<0.05 was considered significant and data are shown as mean±SD for all figures.
Results
Isolation, expansion, and characterization of hMSCs
hMSCs were obtained from the umbilical vein of three donors, originating primary cultures hMSC1, hMSC2, and hMSC3. Healthy cultures of hMSCs exhibited a lot of small fusiform cells and a high division rate (Fig. 1A), and, when cells grew in a dispersed manner, morphologically distinguishable cell types could be identified (Supplementary Fig. S1; Supplementary Data are available online at
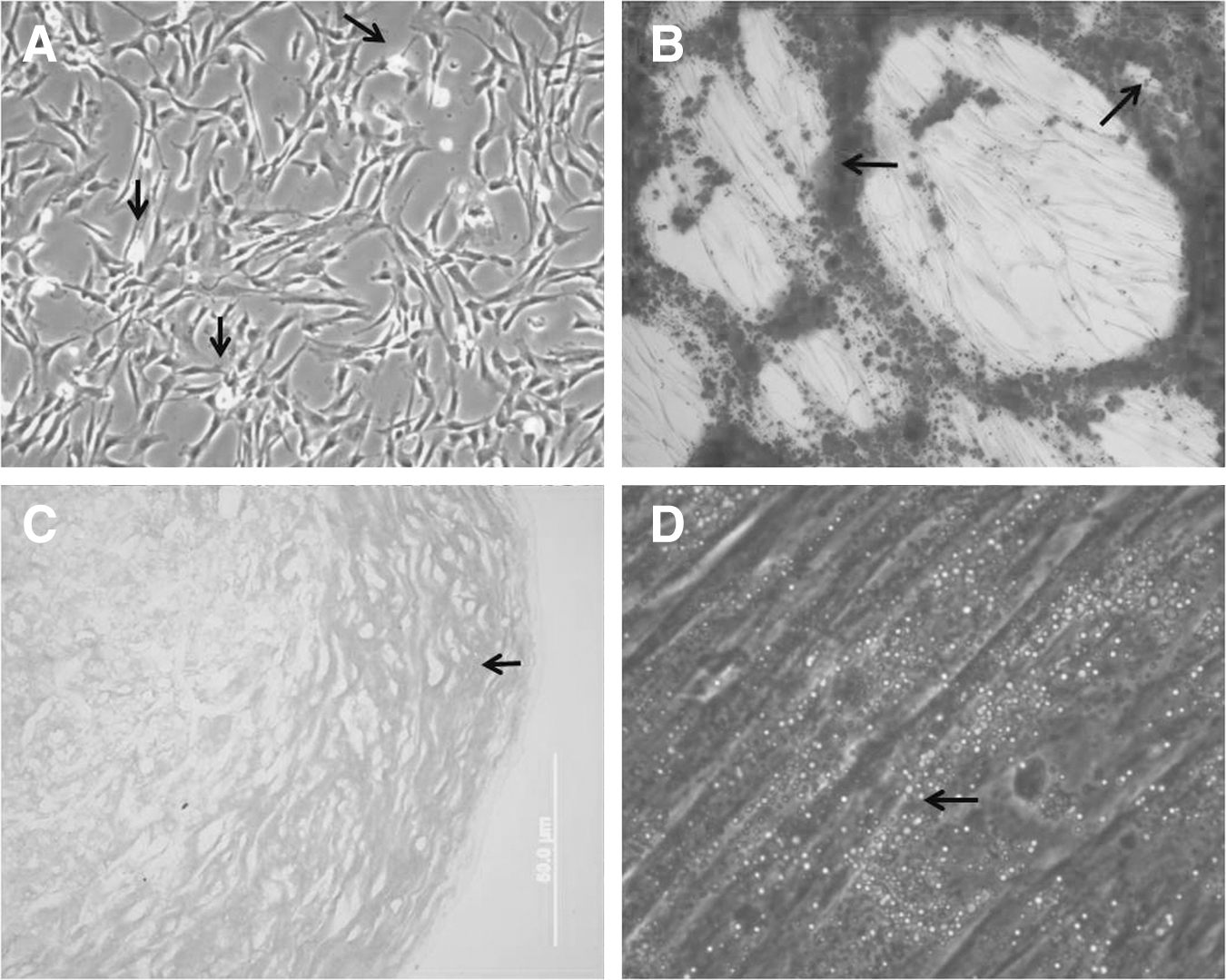
Culture and differentiation of human mesenchymal stem cells (hMSCs) isolated from the umbilical vein.
The capacity for in vitro differentiation of hMSCs in osteoblasts was confirmed by Alizarin Red S staining, showing a calcium-rich extracellular matrix (Fig. 1B). After 21 days of exposition to chondrogenic medium in the micromass culture system, the cartilaginous pellet formed by hMSCs showed extracellular matrix acid-rich polysaccharides stained with Alcian Blue (Fig. 1C). Adipogenic differentiation was detected by the presence of fat vacuoles within cells stained with Oil Red O (Fig. 1D). Cell cultures that were grown in medium without the differentiation supplements did not exhibit spontaneous osteogenic, chondrogenic, or adipogenic differentiation even after 21 days of cultivation.
The cells isolated and expanded in this study were also characterized by immunophenotyping. They were positive for the endoglin receptor CD105, extracelular matrix protein CD90, and surface enzyme CD73. No contamination of hematopoietic cells was detected, since flow cytometry analysis was negative for hematopoietic stem cell markers (CD14, CD34, CD45, and HLA-DR) (Fig. 2).
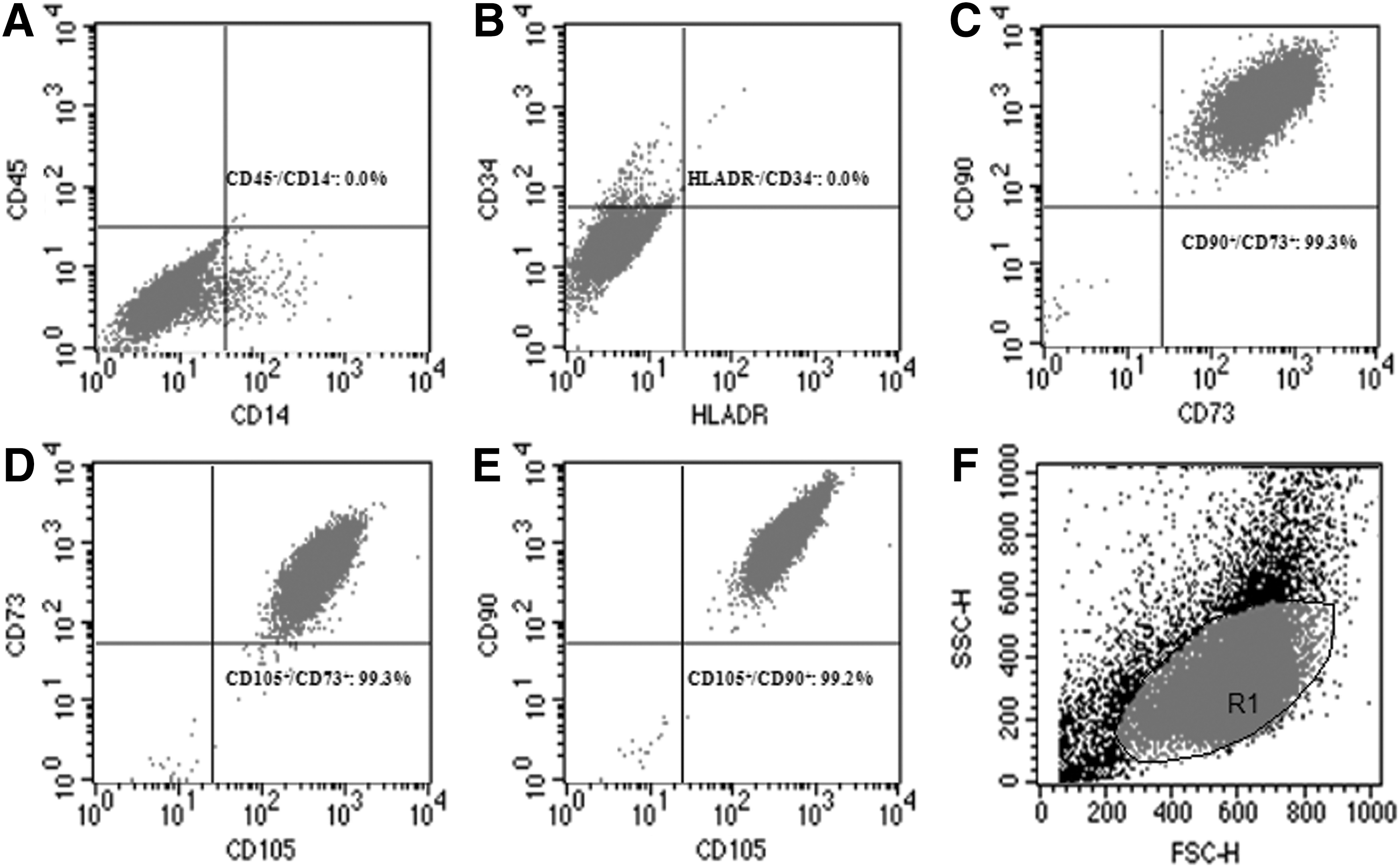
Different expression levels of hMSC markers analyzed by flow cytometry. Panels
Modifications in the CBMN assay for analyzing hMSCs
The well-established protocol for CBMN analysis in peripheral blood cells [36] was modified to measure nuclear alterations in primary hMSC culture. The major adaptations were (1) cell concentration and area of culturing, (2) cyt-B concentration, and (3) hypotonic and fixative solutions.
Concerning the first point, a number of aspects hindered the assay conducted in six-well plates, as follows: the hMSCs adhere and proliferate preferentially in the center of the well, where they form a region of high confluence, thereby increasing cell contact between hMSCs. In this condition, cells presented high sensitivity to cyt-B, considering that 3.5 μg/mL up to 6 μg/mL of cyt-B drastically altered cell morphology, resulting in significant cell death. The cells became branched and arborized (Fig. 3) after only 10 h of treatment with cyt-B. After 24 h of treatment with cyt-B in six-well plate, binucleated cells appeared in the culture, but most detached from the well surface, and there was considerable cellular debris floating in the culture medium. Thus, there were few cells to analyze, and it is not recommended the use of six-well plate for conducting the CBMN assay with hMSCs. Cells were therefore seeded in T25 flasks in order to conduct the CBMN assay. In this growth area, there was no cell aggregation throughout the entire 72-h incubation period, such that the cells proliferated as a monolayer homogeneously spread over the surface of the flask. It was also established that 8×104 cells seeded to perform the CBMN is the ideal initial density for the T25 flask to be 60%–70% confluent at the harvest time, thereby avoiding clot formation during cell processing. According to the replicative potential of each hMSC culture (which can be donor dependent), the initial number of cells to be seeded must be adjusted.
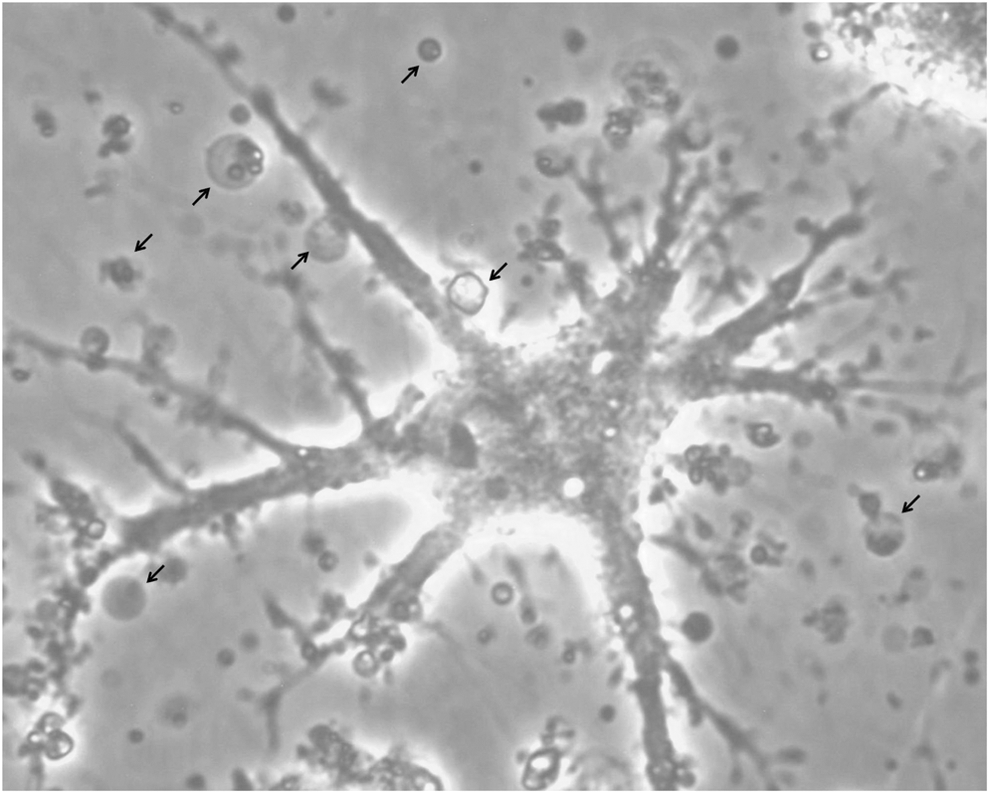
Culture of hMSCs in the six-well plate in the presence of cyt-B (3.5 μg/mL). Cell morphology with altered branching patterns resulting in an arborized aspect. Arrows indicate the presence of cell debris floating in the culture medium.
The second important point in the CBMN protocol was concerning the cyt-B concentration. Among all cyt-B concentrations tested, the 4.0 μg/mL of cyt-B for 24 h in T25 flasks was the best one. It did not cause cell death, did not drastically affect hMSC morphology, and allowed obtaining an adequate number of binucleated cells for CBMN analysis.
The last modification in the original CBMN protocol was the experiment with the hypotonic treatment. In the absence of the hypotonization process, hMSCs remained collapsed, precluding clear visualization of the nuclei. To improve dispersion of the nuclei within the cytoplasm, we used a hypotonic solution developed in our laboratory and routinely employed to eliminate the cytoplasm and enhance chromosome spreading and G banding pattern for cytogenetic analysis in hMSCs. The solution consists of a mixture in equal parts of a solution of 0.075 M KCl with a solution of 9 parts water:1 part FBS-enriched medium, which must be added slowly for 3 min at room temperature for the CBMN assay. The hypotonic solution must not enter in contact with the cells for more than 3 min, the time required to increase cytoplasm swelling, without cell membrane lysis. To preserve the cytoplasm, a requirement for recognizing cell delimitations, the hypotonic action must be interrupted by the fixative solution immediately after the 3 min of hypotonization and before centrifugation. At the end of hypotonic treatment, when cell preparation was fixed at 9 parts methanol:1 part acetic acid, more cells with preserved cytoplasm were observed (Fig. 4). The larger the proportion of acetic acid in the fixative solution (3 methanol:1 acetic acid) the greater the number of cells with cytoplasmic degeneration, and when the proportion of acetic acid was lower (14 methanol:1 acetic acid) the cytoplasm was obscured and the nuclei did not spread correctly within the cell, hindering their visualization (Fig. 4). Further, when using the 9 methanol:1 acetic acid fixative solution, it was not necessary to wash the cells with two additional fixative exchanges, just in that cases with a very obscured cytoplasmic background, which hampered clear visualization of the nuclei.

Preservation differences in the cytoplasm of hMSCs fixed with different concentrations of methanol:acetic acid.
These adaptations for hMSCs could also be used in immortalized cell lines, such as XP4PA and A549.
Characterization of nuclear alterations in hMSCs
To perform the nuclear alteration analysis, it is important that cells are in division; a measure of the cell proliferative status can be obtained by the NDI. The lowest NDI value possible is 1.0 (all of the viable cells have failed to divide during the cytokinesis-block period) and the highest NDI value for CBMN analysis is 2.0, considering that all cells completed one nuclear division and are therefore all binucleated (36). The NDIs for the three hMSCs were 1.56, 1.95, and 1.67 under standard culture condition comparing to 1.43, 1.71, and 1.55 for each positive control, from hMSC1, hMSC2, and hMSC3, respectively.
Binucleated cells with MN, NPBs, and NBUDs were observed in the three primary hMSC cultures as well as in the two immortalized cell lines (XP4PA and A549) (Fig. 5). The mean frequencies of nuclear alterations are shown in Fig. 6. The mean frequency of binucleated hMSCs with MN (3.41±3.94) was low even in the mit-C treatment (12.93±6.37). However, the mean frequency of NPBs (153.59±122.73) was high in all hMSCs analyzed, increasing in the presence of mit-C (379.27±127.08). After the treatment with mit-C, the frequency of binucleated cells with MN was lower than binucleated cells with NPBs (t 34=12.22; P<0.001), and binucleated cells with NBUDs (26.84±16.84) (t 34=3.28; P<0.005). The mean frequency of MN was higher in the XP4PA line (16.88±2.46) than in the A549 line (3.82±4.64) and hMSC cultures (F 4,29=22.53; P<0.05) under normal culture conditions, and very similar to mean MN frequencies found in hMSCs treated with mit-C (F 3,23=2.72; P>0.05).
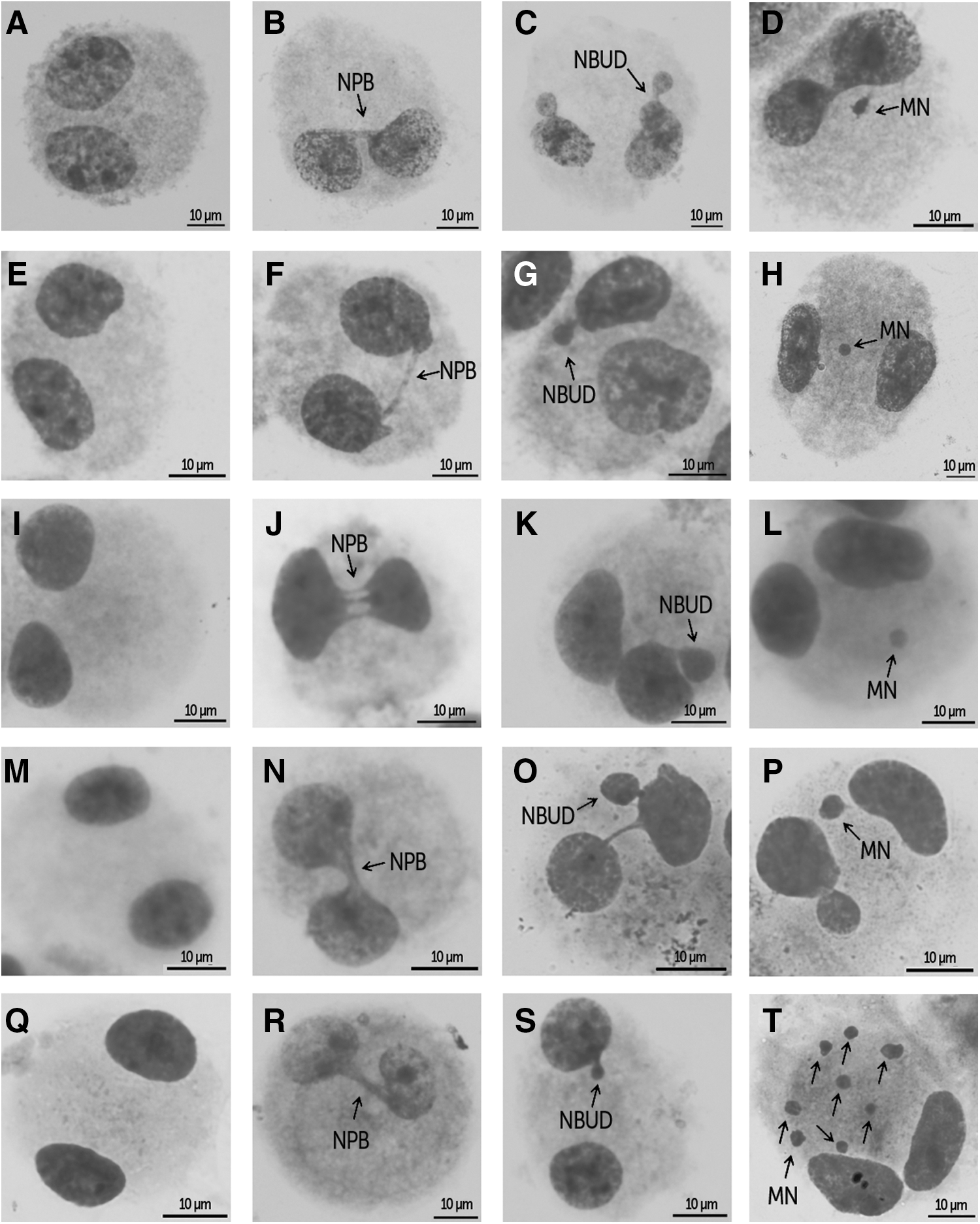
Binucleated cells analyzed by the cytokinesis-block micronucleus (CBMN) assay; micronucleus (MN), nucleoplasmic bridges (NPBs), and nuclear buds (NBUDs) were observed in the three primary hMSC cultures and cell lines A549 and XP4PA.

Cells isolated from cord 1 under normal culture conditions exhibited fewer NPBs when compared with hMSC2 (t=26.57; P<0.0001) and hMSC3 (t=6.34; P<0.0001), but the frequency of hMSC1 with NPBs is higher when compared with A549 (t=8.44; P<0.0001) and XP4PA (t=7.44; P<0.0001) (Fig. 6). The mean frequencies of binucleated cells with NPBs were lower in lines A549 and XP4PA (A549=2.16±1.89 binucleated cells with NPB/1,000 binucleated cells; XP4PA=5.41±4.00 binucleated cells with NPB/1,000 binucleated cells) when compared with mean frequencies observed in hMSC cultures (F 4,29=502.67; P<0.05) (Fig. 6), but there was no difference in the NPB frequency between A549 and XP4PA cells (t=1.8; P=0.1).
To determine whether the emergence of NPBs was due to the use of cyt-B, an experiment was conducted without the addition of cyt-B. The profile of in vitro hMSC division, without the addition of cyt-B, showed the spontaneous occurrence of anaphase bridges and binucleated cells with NPBs during in vitro culture without mit-C (Fig. 7).
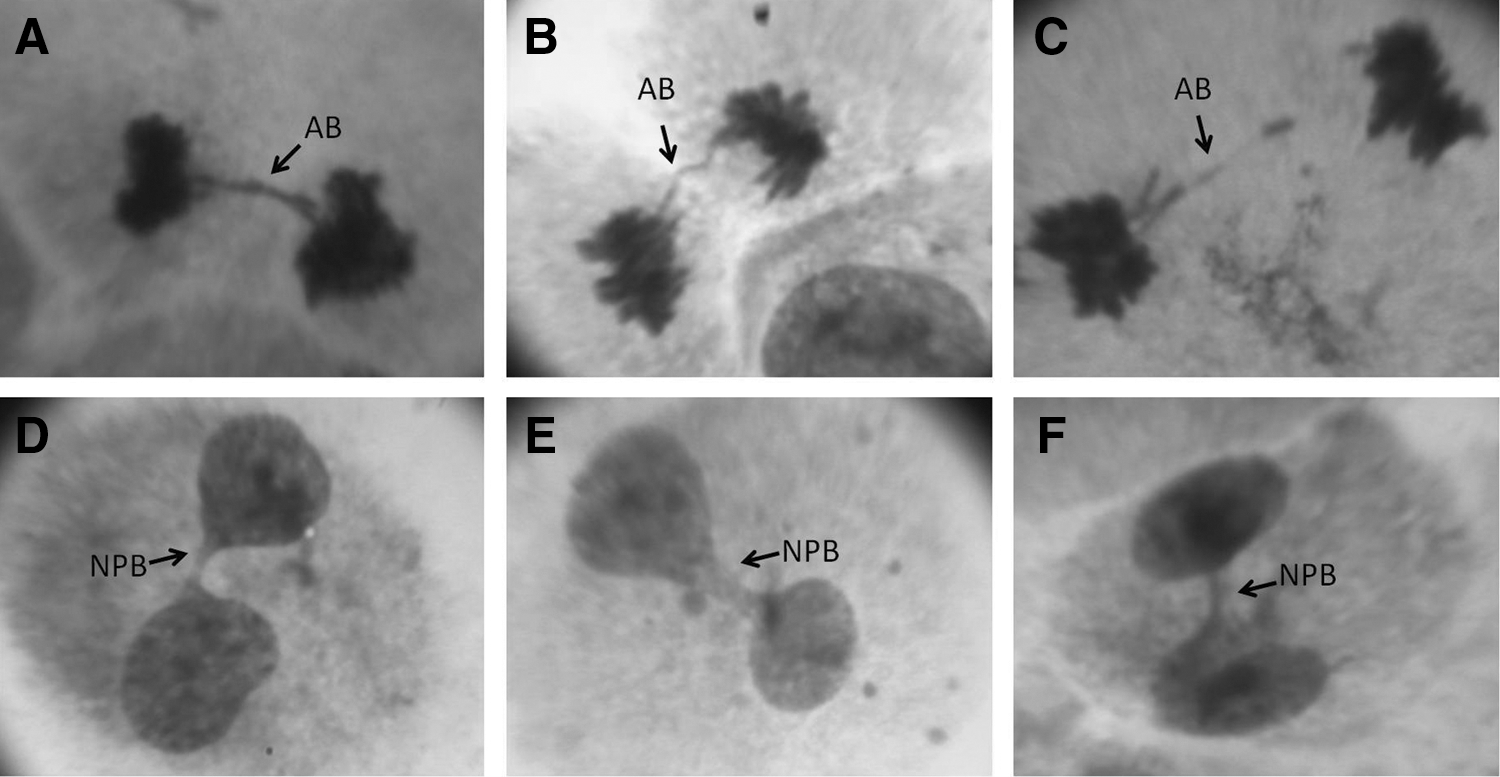
Anaphase bridges
Analysis of nuclear alterations (NPBs and NBUDs) using the CBMN assay shows interindividual differences in hMSC response (Fig. 6 and Table 1) to cell culture under standard conditions (mean frequencies of different NPBs F 2,17=374.11; P<0.05; mean frequencies of different NBUDs F 2,17=16.17; P<0.05) and treatment with mit-C (mean frequencies of different NPBs F 2,17=102.88; P<0.05; mean frequencies of different NBUDs F 2,17=26.35; P<0.05). Cells isolated from cord 1 exhibited fewer alterations, and those from cord 2 showed a large number of cells binucleated with NPBs and NBUDs. However, the mean frequency of cells with MN did not differ among the three different hMSC cultures in the standard cultivation (F 2,17=1.68; P=0.22) nor in the presence of mit-C (F 2,17=2.21; P=0.14).
A P value<0.05 was considered significant.
SC, hMSCs cultivated with standard culture medium without mit-C; PC, positive control consisting of hMSCs treated with mitomycin-C.
hMSCs, human mesenchymal stem cells; NPBs, nucleoplasmic bridges; NBUDs, nuclear buds; MN, micronucleus.
Discussion
The present study describes for the first time the methodological adaptation of CBMN assay for hMSCs isolated from the umbilical vein.
Although CBMN assay is a well-known technology, it must be optimized to hMSC nuclear alteration analysis. It is much easier and faster technique and does not require extensive personnel training as human karyotype analysis [35], allowing counting of a larger number of cells for assessing the genetic instability. So, the modifications described in this methodology will be important for using CBMN assay routinely in biosafety evaluation of the cell therapy.
In the present study, stem cell isolation, expansion, and characterization were conducted successfully as described by our group [57] and other authors who also isolated hMSCs from the umbilical vein [60 –66]. The hMSC morphology was consistent with the guidelines established by Montesinos et al. [67]. The results obtained from the flow cytometry analysis and differentiation studies are in accordance with International Society for Cellular Therapy Guidelines [1], proving that the cells isolated from the umbilical vein are hMSCs.
CBMN analysis must be carried out in healthy cultures of hMSCs with high viability and high proliferation rates. We determined that seeding the cells in T25 flask is better for CBMN assay, avoiding extensive cell confluence (a factor that stresses hMSCs [68]), so the concentration of 4.0 μg/mL of cyt-B was not cytotoxic for hMSCs. Initially, the cyt-B concentration tested in the hMSCs seeded into six-well plate caused drastic alterations in cell morphology. The region of high confluence formed in the center of the well may have increased the cellular stress and cell sensitivity to cyt-B. Previous studies have showed that cytochalasin D alters actin cytoskeleton of MSCs, thereby affecting cell morphology [69,70].
Tests were conducted to determine whether hypotonic treatment of hMSCs was necessary, since, according to Fenech [36], it is not advisable to use the hypotonic treatment in CBMN, because this procedure can destroy necrotic and apoptotic cells, which must be included in analyses of the action mechanism and measure of cell sensitivity to chemical compounds or radiation. However, CBMN assay without the hypotonization step failed in hMSCs because the nuclear boundaries of each nucleus within binucleated cells were not distinguishable. It is believed that this occurred because most hMSCs have thin fusiform bodies with larger nucleus-to-cytoplasm ratio than differentiated cells. Also the cytoplasm of hMSCs is very dense, resulting in intensive synthetic and metabolic activity [71]. Considering the hMSC characteristics described previously, the hypotonic treatment of hMSCs is necessary in a short time of exposure and cannot be skipped. The hMSC cytoplasm has to swell and the two nuclei have to spread, but the cytoplasmic membrane of binucleated cells must be intact and clearly distinct from adjacent cells for optimal CBMN analysis. Thus, it is necessary that any damage to the hMSC cytoplasmic membrane does not occur during the procedure of harvest [72]. Considering these facts, it was determined that hMSC preparation must be fixed at 9 parts methanol:1 part acetic acid for the hMSC membrane preservation. These results corroborate those of other studies that show that alterations in hypotonic treatment and different proportions of methanol–acetic acid fixative solution influence preservation of the cytoplasmic area depending on the cell type [72 –74]. It is therefore important to standardize this step for CBMN analysis in hMSCs.
The methodology described here can be used to obtain binucleated cells originating from other sources; it was successfully tested in hMSCs isolated from three umbilical cord donors and in two cell lines A549 and XP4PA. This standardization favors future studies that will assess the genomic integrity of hMSCs after in vitro culture and determine the response of hMSCs to therapeutically important chemicals, reinforcing the use of hMSCs as an in vitro model of multipotent cells without genetic manipulation in genotoxicity assays.
After CBMN assay methodology was established, nuclear alterations indicative of genetic instability in hMSCs expanded under standard in vitro conditions without mit-C and treated with mit-C were analyzed. NDI determination is always the first step in CBMN analysis, since NDI provides a measure of the proliferative status of cell culture, an indicator of the cytostatic effects of the conditions being tested [36]. All three hMSC cultures showed a decline in proliferative potential in the presence of mit-C (positive control), but the drug was not cytostatic in the concentration used, since it led to the appearance of a sufficient number of binucleated cells for CBMN analysis in the positive control treatment. The fact that hMSC2 exhibited a high replication rate (NDI=1.95), resulting in a large number of trinucleated and tetranucleated cells, in addition to the binucleated, justified the reduced exposure time to cyt-B [72], since it means that several viable cells completed more than one nuclear division in 24 h. However, as we were in the standardization phase of the protocol, we considered it more advisable to use the same incubation time with cyt-B (24 h) in the treatment of hMSCs isolated from the three umbilical cords.
Nuclear alterations in hMSCs were more frequent in the positive control (except NBUDs in hMSC1), where cells were treated with 0.2 μg/mL of mit-C. This indicates that the hMSCs responded to treatment with mit-C, primarily owing to the increased frequency of binucleated cells with NPBs. Mit-C is considered a potent clastogenic agent because it produces several types of DNA lesions, which induce the emergence of double-strand breaks (DSBs) due to the replication fork barrier [75]. Interestingly, the mean frequency of binucleated cells with MN in culture treated with mit-C was very low, indicating that the hMSCs isolated from the umbilical cord likely contain mechanisms to avoid a high level of chromosomal breaks. The XP4PA line exhibits DNA repair deficiency [76], thereby justifying the high MN index, and suggests that the low frequency of MN in hMSCs treated with mit-C is related to its greater repair capacity. Vinoth et al. [52] observed lower levels of chromosomal alterations in hESCs after treatment with mit-C than fibroblasts from the IMR-90 cell line, suggesting that this fact may be related to the presence of more efficient DNA repair mechanisms in hESCs. Increased DNA repair capacity has been frequently detected in stem cells (iPSCs, embryonic, or adult stem cells from humans or mice) when compared with differentiated cells, indicating that stem cells normally protect their genome through an increase in DNA repair machinery [77 –79]. It has been demonstrated that the repair capacity of DSBs is greater in human hematopoietic stem cells isolated from umbilical cord blood and decreases during cell maturation [80]. The authors concluded that stem cells are protected by extensive DNA repair, whereas cells already compromised by cell differentiation, when damaged, can be eliminated by apoptosis. Chen et al. [81] demonstrated that hMSCs isolated from bone marrow have greater antioxidant response and DSB repair capacity, which favor the characteristic resistance to radiation of these stem cells. The role of DNA damage response in the resistance of mouse MSCs to ionizing radiation was assessed by Sugrue et al. [82], who showed that MSCs have efficient DSB repair, expressing high DNA damage response protein (ATM, Chk2, and DNA Ligase IV) and anti-apoptotic protein levels [Bcl-2 and Bcl-(XL)]. A recent study demonstrated that DSB repair is faster in hMSCs than in differentiated osteoblasts after γ-irradiation-induced DNA damage [83]. They also found that hMSCs induced to osteogenic or adipogenic differentiation undergo high levels of apoptosis after γ-irradiation, in contrast to undifferentiated hMSCs, which are highly resistant to treatment and exhibit 95% DSB repair.
The present study detected a high occurrence of binucleated cells with NPBs in all three cords analyzed. NPBs have been validated as biomarkers of DNA damage and chromosomal rearrangement [84]. The formation of NPBs is increased by a wide range of agents, including endogenous oxidative agents, ionizing radiation, polycyclic aromatic hydrocarbons, patulin and folate, and selenium deficiency [40,85,86]. Duan et al. [87] demonstrated that frequencies of NPBs and NBUDs were more sensitive measures than MN for assessing the genotoxicity of polycyclic aromatic hydrocarbons. Interestingly, in the present study, the NPB frequencies were high in hMSCs cultured under standard in vitro culture patterns, without the addition of any genotoxic agent. The cell lines A549 and XP4PA have showed lower frequencies of NPBs, suggesting that these bridges that appear in hMSCs do not result from the methodology used and are characteristic of hMSCs isolated from the umbilical vein and in vitro expanded. In another previous study of CMBN using CHOK1 cells, we detected that high frequencies of NPBs do not occur in all conditions tested [46].
Ebert et al. [88] showed that hMSCs isolated from bone marrow and in vitro cultured with a low amount of selenium exhibit symptoms of oxidative stress and a high frequency of MN, and that selenium supplementation in the culture medium decreases the number of MN by at least 58%. Since the authors did not analyze MN in binucleated cells, there was no description of NPB occurrence. Researchers underscored the importance of preserving the in vitro genomic integrity of hMSCs, concluding that selenium supplementation in the culture medium for hMSC expansion would be an excellent conduct for cell preparations used in tissue engineering, thereby improving the quality of hMSCs ex vivo manipulated. Two other studies described MN analysis in binucleated cells using murine stem cell as a model [89,90]; however, the authors did not analyze the occurrence of other nuclear alterations such as NPBs and NBUDs, precluding comparison with data obtained in the present study.
The occurrence of anaphase bridges and spontaneous binucleated cells with NPBs was detected during expansion under normal conditions of in vitro culture of hMSCs isolated from the umbilical vein. Anaphase bridges and NPBs are signs of genomic instability [84]. This fact is relevant, since the ex vivo expansion process of hMSCs is the strategy selected to produce a large number of cells for use in cell therapy. This procedure, necessary for the clinical application of hMSCs, is associated to the potential risk of cell immunogenicity, with issues of biosecurity regarding components of the culture medium and FBS, as well as the possibility of in vitro transformation of hMSCs [7]. High susceptibility to malignant transformation of MSCs has been described in animal models using stem cells isolated from the bone marrow of mice and monkeys, some becoming highly tumorigenic after in vitro culture, since they caused tumors in mice at sites where these cells were infused [91 –98]. A recent study reported that differentiated hMSCs isolated from human bone marrow became tumorigenic in diabetic mice. The authors believe that this occurred due to spontaneous transformation of hMSCs after prolonged in vitro culture [31]. Genetic alterations have been described in cultured hMSCs [19 –29], but the biological significance of these alterations remains unclear and it is an issue that raises controversy [29,33]. Given this lack of consensus, more studies on the genetic stability of hMSCs are needed to come to a firm conclusion regarding this issue and the CBMN assay can be useful.
In this respect, the present study may contribute to the discussion of this subject, since the anaphase bridges described here in hMSCs in vitro expanded are characteristic alterations in tumor cells [99]. They emerge due to the presence of dicentric chromosomes formed by the fusion of two deficient telomeres and indicate genetic instability within cells, a common characteristic in a wide range of cancers [100 –102]. Anaphase bridges often break [101,103], resulting in chromosomal alterations, such as amplifications, translocations, or deletions, which are biomarkers of genomic instability in malignant cells and used for diagnostic purposes in oncology [104]. After anaphase bridge breakage and fusion of uncapped chromosome ends, in the next interphase, breakage–fusion–bridge cycles initiate and propagate through several divisions, generating more alterations, such as aneuploidy, polyploidy, and genetic mutations [105 –108]. Calado et al. [109] provided direct clinical evidence that telomere shortening in hematopoietic progenitor cells, followed by breakage–fusion–bridge cycles, increases predisposition to malignant transformation in patients with aplastic anemia. Moreover, another study revealed that the mechanism of chromosomal instability in leukemia progenitor cells occurs due to the continuous generation of unstable chromosomal alterations, which undergo repeated breakage–fusion–bridge cycles and subsequent lesion repair [110]. In the present study we detected a high frequency of NPBs in three hMSC cultures; these nuclear alterations can initiate repeated breakage–fusion–bridge cycles during ex vivo expansion of the progenitor cells and cause a high degree of genetic instability in these cells.
On the other hand, a new type of anaphase bridge was recently described: ultrafine anaphase bridges (UFBs) [111,112]. These are ultrafine structures that form bridges between daughter cells in the anaphase (DNA bridges that form between sister chromatids during mitosis); they are BrdU stained, but cannot be evidenced by other DNA stains, such as Giemsa and DAPI [113]. UFBs are common characteristics of cultured cells, and considered physiological structures of normal cells that undergo in vitro replication [114]. Exposure to in vitro expansion causes an increase in decatenated DNA structures and late-replicating intermediates [115]. A number of UFBs occur in the centromeres and may play the important physiological function of uniting DNA structures, acting as a signaling platform for the spindle assembly checkpoint. The spindle assembly checkpoint is activated when sister kinetochores are not bioriented due to errors in microtubule ligation or loss of tension, and it was recently demonstrated that spindle assembly checkpoint persists for longer in cells deficient in resolving the Holliday junction during DSB repair in DNA, with a higher frequency of anaphase bridges and aneuploidy [116]. Another UFB subtype originates in telomeric DNA and its frequency is correlated with defects in telomere replication. UFBs are immunohistochemically stained with the BTR complex [formed of BLM (Bloom syndrome, RecQ helicase-like), topoisomerase IIIα, and RecQ-mediated genome instability 1 and 2 proteins (RMI1 and RMI2)] and PICH (Plk1-interacting checkpoint helicase). BLM has a role in suppressing and/or resolving UFBs, likely contributing to maintaining the telomere acting on late-replicating intermediate resolution [115,117]. There are also UFBs not derived from centromeres characterized by colocalization of PICH with Fanconi anemia proteins FANCD2 and FANCI [118,119]. FANCD2 and FANCI proteins are specifically associated to fragile site loci, marking abnormally intertwined DNA structure induced by replication stress. The association between PICH and the BTR complex and Fanconi anemia proteins suggests that the processing and resolution of UFBs are very important for maintaining genomic stability [120]. The presence of UFBs in anaphases of normal-cultured cells with intact genome shows that the cells are able to resolve late-replicating intermediate sequences or catenane structures, ensuring adequate chromosomal segregation without the need of activating cell cycle checkpoints and avoiding a decline in in vitro proliferation efficiency [115].
Since the number of NPBs is higher in the hMSC2, which exhibited high mitotic index, it can be assumed that the bridges frequently observed in the hMSCs of this study could be in some way related to these ultrafine bridges, although the bridges detected in this study were stained with Giemsa. Therefore, anaphase bridges (characterized as NPBs in CBMN) may be a normal characteristic of in vitro hMSC growth, or on the other hand, they might characterize a pathological condition introduced or enhanced by the manner in which they are cultured, as discussed previously. These issues are important and need to be clarified, since if NPBs are signs of instability generated by the culture condition, then other methodologies for in vitro expansion should be established.
Another factor that must be discussed regarding analysis of the genetic stability of cultured hMSCs is the difference in nuclear alteration frequencies exhibited by distinct donors. Analysis of the gene expression of stem cells isolated from different individuals also exhibited interindividual differences due to genetic variability resulting in a unique gene expression signature [121,122]. The same situation occurs with the gene expression of the DNA repair system, and, in apoptosis induction after cytotoxic treatment in hematopoietic stem cells isolated from umbilical cord blood [80], cells displayed different levels of donor-specific responses. It has also been established that MN formation can be influenced by certain gene polymorphisms involved in DNA repair pathways, in the xenobiotic metabolism, and in folate transport and metabolism [123]. However, the present study found no differences in mean MN frequencies in the three hMSC cultures studied in the absence or presence of mit-C.
Our laboratory is conducting experiments to clarify the genetic significance of a low frequency of binucleated cells with MN in the presence of mit-C and the elevated presence of NPBs in hMSCs even in the absence of genotoxic agents. Spectral karyotyping (SKY) analysis along with CBMN for hMSCs in vitro expanded will help to define whether there is any particular association between NPBs and any specific chromosome. Further, it is important to measure the rate of concurrent gene mutations, chromosomal aberrations, and alterations in gene expression at same cell culture time and conditions in that hMSC CBMN analysis is realized. Afterward, the CBMN could be included in routine analyses of hMSCs as a biomarker in order to improve predictions of risk factors, making it an important tool associated with other genetic analyses to ensure the safe clinical use of hMSCs in cell therapy.
Footnotes
Acknowledgments
The authors would like to thank Mauro Pichorim for the assistance in the statistical analysis, also Gideão Wagner W. Felix da Costa and Carolina Corado for the assistance with the figures. We also would like to thank the CNPq and CAPES for financial support and the scholarship awarded to Déborah Afonso Cornélio, Thais Pimentel, and Silva Regina Batistuzzo de Medeiros.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.