
Abstract
Dental pulp stem cells (DPSCs) remain quiescent until activated in response to severe dental pulp damage. Once activated, they exit quiescence and enter regenerative odontogenesis, producing reparative dentin. The factors and signaling molecules that control the quiescence/activation and commitment to differentiation of human DPSCs are not known. In this study, we determined that the inhibition of insulin-like growth factor 1 receptor (IGF-1R) and p38 mitogen-activated protein kinase (p38 MAPK) signaling commonly activates DPSCs and promotes their exit from the G0 phase of the cell cycle as well as from the pyronin Ylow stem cell compartment. The inhibition of these two pathways, however, inversely determines DPSC fate. In contrast to p38 MAPK inhibitors, IGF-1R inhibitors enhance dental pulp cell sphere-forming capacity and reduce the cells' colony-forming capacity without inducing cell death. The inverse cellular changes initiated by IGF-1R and p38 MAPK inhibitors were accompanied by inverse changes in the levels of active signal transducer and activator of transcription 3 (STAT3) factor, inactive glycogen synthase kinase 3, and matrix extracellular phosphoglycoprotein, a marker of early odontoblast differentiation. Our data suggest that there is cross talk between the IGF-1R and p38 MAPK signaling pathways in DPSCs and that the signals provided by these pathways converge at STAT3 and inversely regulate its activity to maintain quiescence or to promote self-renewal and differentiation of the cells. We propose a working model that explains the possible interactions between IGF-1R and p38 MAPK at the molecular level and describes the cellular consequences of these interactions. This model may inspire further fundamental study and stimulate research on the clinical applications of DPSC in cellular therapy and tissue regeneration.
Introduction
H
DPSCs originate from neural crest cells [9 –11] that acquire dental competence as multipotent stem cells (SCs) [12]. First reported in 2000 [8], the existence of DPSCs has been confirmed by many laboratories, including ours [13]; however, the exact area of the dental pulp in which they are located is still not well established. A recent study by Martens et al. [14] confirmed earlier findings [4,12,15,16] that DPSCs occupy the prevascular niche and, in developing teeth, the cervical niche located near the cementum/dentin zone. A study based on the mRNA expression levels of DPSC markers, including CD166, CD146, and CD105, concluded that in rat molars, “coronal pulp harbors more SCs than the other regions” [17]. A study by Ishikawa et al. [18,19] determined that 5-bromo-2′-deoxyuridine (BrdU)-retaining cells expressing the mesenchymal stem cell marker CD146 were associated with vessels located in the central region of adult rat dental pulp. These label retaining cells (LRCs) possessed proliferative capacity and were responsible for the regeneration of damaged odontoblasts.
Localized in the protective environment of the niche, SCs integrate systemic and local signals that drive them from reversible quiescence into the cell cycle. The asymmetric division of SC produces a SC daughter to maintain the stem cell pool and a transient amplifying (TA) daughter that differentiates after a limited number of divisions. Odontoblasts committed to differentiation express an Msx1 homeobox protein [20] that is downregulated as cells enter the early differentiation stage, which is marked by the appearance of matrix extracellular phosphoglycoprotein (MEPE) [21,22]. MEPE must be downregulated for the late differentiation markers, including dentin sialoprotein (DSP), to be upregulated [1,21 –24]. These markers regulate the mineralization process. The final stage of odontoblast differentiation is indicated by the presence of calcified von Kossa stain-positive nodules.
The homeostasis, growth, and repair of tissues depend on the existence of a balance between cellular quiescence and the proliferation of SCs [25]. The factors that control this balance are currently being identified in hematopoietic, nervous, and epithelial tissues, but the factors that control the quiescence and activation of DPSCs still await discovery. It has been shown, however, that tumor necrosis factor alpha (TNFα) and other inflammatory factors can activate odontoblastic precursors and stimulate their proliferation and differentiation in response to bacterial infection [16,26,27]. Other studies have shown that the downstream targets of TNFα, the p38 mitogen-activated protein kinase (p38 MAPK) [27 –29] and the insulin-like growth factor 1 receptor (IGF-1R) [30 –32], also control odontoblast differentiation [33 –35] and development [36,37]. However, studies on modulation of DPSC are limited. Here, we determined that p38 MAPK and IGF-1R signaling are required to maintain DPSC quiescence and to regulate the differentiation of these cells, although in opposite manners. Importantly, we identified signal transducer and activator of transcription 3 (STAT3) as a converging point that integrates signals from p38 MAPK and IGF-1R pathways and regulates human DPSC responses.
Materials and Methods
Tissue samples and cell cultures
Wisdom or premolar teeth were extracted for orthodontic reasons from young adults at the Dental Clinic of the University Hospital of Lille. Patients agreed to donate their teeth for research by signing a “no opposition” agreement. The storage and use of human biological samples were declared and performed according to the local Person's Protection Committee and the ethical rules approved by the Department of Health of France (File No. DC-2008-642). Dental pulp tissue was snap-frozen for immunohistochemical analysis, sliced and placed in culture for ex vivo study, or enzymatically dissociated for 1 h at 37°C in a solution containing collagenase type I (3 mg/mL) and dispase (4 mg/mL), both purchased from Invitrogen. The digested tissue was filtered through a 70-μm pore strainer (BD Biosciences), and the obtained cell suspension was rinsed and plated at the indicated densities in the standard odontoblast growth medium [Dulbecco's modified Eagle medium (DMEM) Glutamax supplemented with 1 mg/mL glucose, 1% penicillin/streptomycin solution (Invitrogen), 10% fetal calf serum (FCS; Lonza), and 100 μM vitamin C (Sigma-Aldrich)]. The cells were cultured at 37°C under 5% CO2 and re-fed every 3 days with the fresh medium. Inhibitors of IGF-1R (sc204008; 125 nM) and of STAT3 (Stattic5-sc20281; 5 μM; Santa Cruz Biotech.) and of p38 MAPK (PD169316; 250 nM; Calbiochem) were added to the cell culture medium for the indicated times. The solvents in which the inhibitors were dissolved were added at the same concentrations to the control cultures. Colony-forming units (CFUs) and sphere-forming units (SFUs) were calculated as the percentage of the colonies or spheres of, respectively, 200 and 4,000 seeded live cells formed, as described previously [13]. Cell proliferation was monitored on 96-well plates using IncuCyte 2011A Kinetic Imaging System and software (Essen BioScience).
Ex vivo cultures of dental pulp
Freshly isolated pulp tissue was sectioned under sterile conditions using a scalpel. Slices were placed in the upper chambers of transwell plates and covered with the medium placed in the lower chamber. Inhibitors at nontoxic doses (sc204008; 125 nM) for IGF-1R and PD169316 (250 nM for p38) were added to this lower chamber for 5 days. The medium and inhibitors were changed twice during this period, after which the tissue was snap-frozen and used for immunohistochemistry.
Flow cytometry and cell sorting
Cells were trypsinized and resuspended at a density of 1×106 cells/mL in phosphate-buffered saline containing 2% bovine serum albumin. Aliquots of the cell suspension (100 μL) were incubated with primary antibodies (Abs) for 30 min at 4°C in the dark and with secondary fluorochrome-conjugated Abs for another 30 min if needed. A minimum of 1×105 cells were analyzed with a Cyan-ADP (Beckman Coulter, Inc.) instrument; the data were processed using Summit 4.3 software (Beckman Coulter, Inc.). PE-conjugated monoclonal Abs against CD271 were from Mitenyi Biotech. CD166-FITC conjugate was from BD Pharmingen, CD105-FITC from Immunotools, and Ki67-FITC conjugate was from Santa Cruz Biotech. CD57 (HNK1) Ab was a gift from Dr. Elisabeth Dupin, Institut de la Vision, Paris, France.
Propidium iodide/Ki67 assay
Cells were adjusted to a concentration of 106 cells/mL in DMEM with 10% FCS, and the assay was performed as previously described [38]. Propidium iodide (PI)- and Ki67-negative cells within the G0/G1 fraction were considered to represent quiescent cells in the G0 phase of the cell cycle.
DiI and BrdU staining and cell sorting
Cells were resuspended in serum-free DMEM and stained with 3 μL of Vybrant DiI cell-labeling solution (Invitrogen) per 106 cells for 25 min at 37°C. After rinsing, the cells were re-plated in the standard medium. The 5% of cells with the highest and lowest DiI staining indices were sorted by fluorescence-activated cell sorting using a Beckman Coulter Altra instrument. The sorted cells were used for further analysis. For BrdU incorporation, DiI-labeled cells were pulsed with 0.5 μM BrdU (Roche®) 24 h later and allowed to incorporate BrdU for 6 days. The label was chased for another 6 days by growing the labeled cells in a label-free medium. Staining with anti-BrdU Ab was performed according to the supplier's recommendations. The pyronin Y (PY)/Hoechst 33342 assay was performed with 5×105 cells, as described previously [38]. Flow cytometry analysis of the total population before and after PY staining permitted the gating and identification of cells with low and high PY (FL2) fluorescence intensities.
Immunohistochemistry
Dental pulp samples were embedded in Tissue-Tek OCT (Sakura Finetechnical) and cryosectioned. Sections (8 μm) were placed on Superfrost plus (Thermo Scientific) glass slides, air-dried, and fixed in the paraformaldehyde (PAF) solution (Sigma-Aldrich), and immunohistochemistry was performed according to the standard procedures. Abs against CD271 (p75NTR) and p38 MAPK were from Abcam and Cell Signaling Tech., respectively. Negative controls were performed by replacing the primary Abs with irrelevant Abs of the same isotype. Hoechst 33342 (1 μg/mL) was used for nuclear counterstaining. All slides were mounted under coverslips with the Vectashield Mounting Medium (Vector Laboratories) and photographed using a Leica® fluorescence microscope (Leica). To quantify the proportion of Ki67-positive cells, we manually counted the total number of Ki67-positive and Hoechst 33342-positive nuclei present on 5–7 images obtained in three independent experiments per condition. The percentage of Ki67-positive nuclei was calculated using the following formula: number of Ki67 − positive/Hoechst 33342 − positive nuclei×100.
von Kossa staining
To detect mineralization, cells were incubated in the odontoblast growth medium supplemented with 10 mM β-glycerol phosphate (βGP) for the indicated times, fixed in PAF for 10 min, and stained as described previously [13].
Western blot analysis
Western blot analysis was performed using ready-to-use NuPAGE® 4–12% Bis–Tris polyacrylamide gels (Invitrogen) according to the manufacturer's instructions. Blots were probed with primary Abs to actin (Sigma-Aldrich), cyclin D1, phosphorylation of cAMP-responsive-element-binding protein 1 (pCREB), c-Myc, Bcl-2, MEPE, DSP (Santa Cruz Biotech.), p38, pp38, pSTAT3, phosphorylation of ATF2 (pATF2), and phosphorylation of glycogen synthase kinase 3 (pGSK3; Cell Signaling Technology) followed by horseradish peroxidase-conjugated secondary Ab (Bio-Rad). Immunodetection was performed using an ECL-Prim chemiluminescence kit from Amersham and a LAS4000 apparatus. The results were integrated using Gel Analyst software® (Claravision).
Statistical analysis
Results are expressed as the mean±standard error of the mean of at least two independent experiments. Comparisons between means were assessed using Student's t test for unpaired data. If unequal variance was observed, Welch's correction was applied. Comparisons between several groups were assessed using a one-way analysis of variance followed by Dunnett's multiple comparison test, using an appropriate control group as the reference. The statistical analyses were performed using the GraphPad Prism 4.0 software. A P value of<0.05 was considered significant.
Results
Inactivation of IGF-1R and p38 MAPK induces cycling of quiescent human dental pulp cells ex vivo
IGF-1R is known to activate various downstream targets, including AKT/mTOR, extracellular signal-regulated kinases (ERK1/2), p38 MAPK pathways, and the Janus kinase (JAK)/STAT3 pathway [32,39 –41], all of which have been shown to activate quiescent SCs [38,42 –44]. Interestingly, p38 MAPK, a serine/threonine kinase that is activated in response to stress and inflammatory cytokines [45], controls the quiescence of SC via several mechanisms [46 –49], including the inactivation of GSK3, an important effector of the Wnt self-renewing pathway [50]. To investigate the role of IGF-1R and p38 MAPK in dental pulp cell (DPC) biology, we first performed immunohistochemical analysis of snap-frozen human dental pulp tissue to localize the expression of p38 MAPK and IGF-1R in this tissue (Fig. 1A). p38 MAPK was detected in the nuclei of the majority of DPCs. To the best of our knowledge, this is the first study to localize p38 MAPK in human dental pulp. In agreement with an earlier study by Götz et al. [51], IGF-1R was detected in undifferentiated odontoblasts of the odontogenic zone at the dental pulp periphery. The observed nuclear localization of p38 MAPK represents the active form of this kinase [52,53] and suggests that p38 MAPK is constitutively active in DPCs and, as such, may contribute to their quiescence. IGF-1R expression by the mitotically quiescent cells suggests that in addition to its known function in controlling the cell cycle, cell survival, motility, and attachment [54], IGF-1R may also function to maintain odontoblasts quiescence and preserve their undifferentiated phenotype.
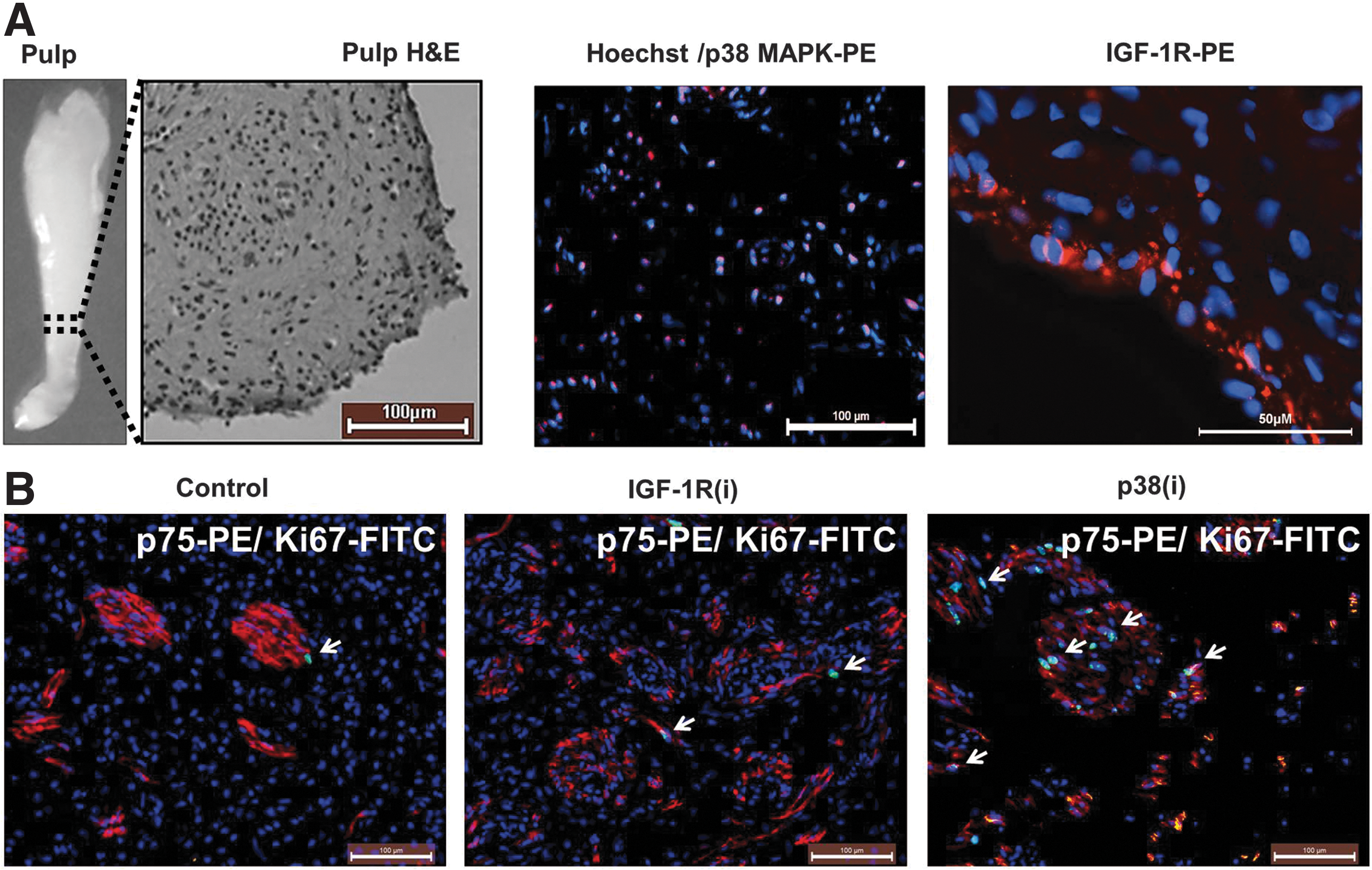
p38 mitogen-activated protein kinase (p38 MAPK) and insulin-like growth factor 1 receptor (IGF-1R) inhibitors induce human dental pulp proliferation ex vivo. Dental pulps were removed from extracted premolar teeth, immediately dissected, and placed on transwell plates in the medium supplemented with the IGF-1R or p38 MAPK inhibitors (i) sc204008 or PD169316 (125 and 250 nM, respectively; see “Materials and Methods” for details) for 5 days.
If p38 MAPK and IGF-1R, which functions upstream of p38 MAPK, maintain the quiescence of DPCs, their inactivation should provoke the exit of the cells from quiescence and their entry into the cell cycle. Cycling cells are detectable by positive staining with Ki67 Ab; this Ab does not recognize resting G0 cells. We incubated slices of freshly extracted dental pulp in the odontoblast growth medium supplemented with 125 nM sc204008 and 250 nM PD169316, inhibitors of IGF-1R and p38 MAPK, respectively, for 5 days. Representative images from these ex vivo studies (Fig. 1B) show that IGF-1R inhibitor [IGF-1R(i)] and p38 MAPK inhibitor [p38(i)] induced the appearance of Ki67-positive cells. The quantitation by manual counting of the Ki67-positive cells per total number of cells showed that, compared to control, 3.2-fold more cells began cycling in response to IGF-1R(i) and 14.8-fold more cells entered the cell cycle in response to p38(i) (Supplementary Table S1; Supplementary Data are available online at
IGF-1R and p38 MAPK inhibitors decrease the pool of quiescent DPSCs in vitro
To substantiate the ex vivo data, we performed several functional tests to characterize the Ki67-positive cells in vitro. The findings obtained using a PI/Ki67 assay (example shown in Fig. 2A), which distinguishes cycling cells from quiescent SCs resting in the G0 phase of the cell cycle, are summarized in Table 1. As seen, IGF-1R(i) and p38(i) diminished the proportion of G0 cells by 57% and 53%, respectively, compared to control cells and stimulated the transition of cells from the G0/G1 to the S/G2/M phase of the cell cycle. Interestingly, neither IGF-1R(i) nor p38(i) increased the subG1 fraction of dead cells, suggesting that under our experimental conditions, these inhibitors do not significantly induce the death of DPCs relative to the control. This was confirmed by AnnexinV/7-AAD and trypan blue assays (Supplementary Fig. S1A, B). Thus, it appears that the inactivation of either the IGF-1R or the p38 MAPK signaling pathway stimulates the cycling of quiescent DPSCs in vitro.
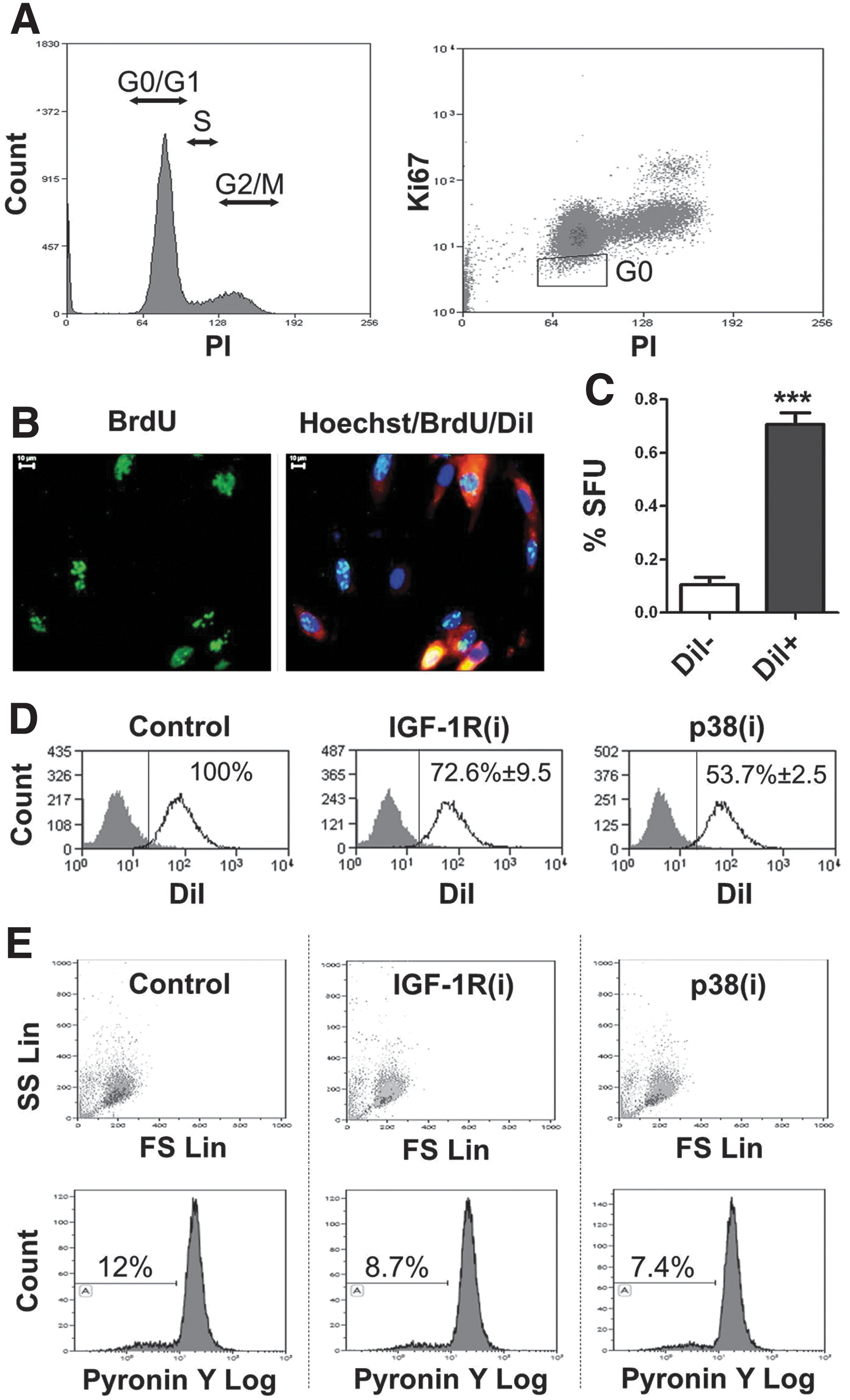
Dental pulp contains a small fraction of quiescent cells.
Human dental pulp propidium iodide Ki67 cell cycle analysis was used to identify the G0 fraction of quiescent cells. Please note that neither IGF-1R inhibitor [IGF-1R(i)] nor p38 inhibitor [p38(i)] (125 nM sc204008 and 250 nM PD169316, respectively) induced cell death (subG1 fraction) during 24 h of treatment.
IGF-1R, insulin-like growth factor 1 receptor.
To confirm that the inhibition of IGF-1R and p38 MAPK signaling affects the stem cell compartment in dental pulp cultures, we determine the expression of DPSC and neural crest stem cell surface markers. The primary DPC cultures used in our experiments expressed 86.4%±6.02 and 81.03%±8.11 of CD166 and CD105 DPSC markers, respectively, but consistent with our previous study [13], only a very few cells expressed CD271 (0.23±0.2) and CD57 (1.39±1.5) neural crest stem cell markers. Since the expression of surface markers is condition dependent, we did not relay on them in defining SCs and used a vital DiI dye-based functional assay to distinguish quiescent LRCs from the cycling population. Cycling cells dilute this dye by half with each cell division and thus rapidly shed the label. Figure 2B shows a representative image of DPCs labeled simultaneously with DiI and BrdU. BrdU incorporation is another method of LRC assay [19]. This double labeling ensured the accurate identification of quiescent LRCs and/or of slowly cycling cells possessing SC attributes. To further confirm the stemness of LRCs, we sorted DiIhigh LRC and DiIlow cycling cells and determined their sphere-forming capacity, another trait that defines SCs. In agreement with our previous results [13], DiIhigh cells showed a greater ability to form spheres than DiIlow cells (Fig. 2C), indicating that the DiIhigh cell population is enriched for DPSCs. Flow cytometric analysis of DPCs consistently showed that 24 h treatment with IGF-1R or p38 MAPK inhibitors significantly decreased the number of DiIhigh LRCs in relation to the control (Fig. 2D). This finding corroborates our G0 results and demonstrates that both inhibitors stimulate the division of quiescent/slowly cycling LRC DPSCs.
SCs that exit the quiescent state divide to reproduce themselves and to generate TA progeny that are committed to differentiating. Quiescent SCs were shown to have low metabolic activity, as reflected, among other properties, by their low content of cellular RNA resulting in poor labeling by PY, an RNA-binding vital dye [38,56,57]. The results of PY assays provided additional evidence that IGF-1R(i) and p38(i) impinge on the DPSC compartment. Exponentially growing dental pulp primary cultures contained 3.81–11.96% PYlow cells. These cells are small and of low granularity, as determined by their low forward and side scatter profiles, respectively (Fig. 2E). IGF-1R and p38 MAPK inhibitors decreased persistently the size of the PYlow fraction to 72% and 62% of controls, respectively (example in Fig. 2E), demonstrating that during IGF-1R or p38 MAPK inhibition, a small number of cells exit the PYlow pool and enter the PYhigh pool of metabolically active cells. Again, these data confirm that IGF-1R and p38 MAPK inhibitors act on quiescent DPSCs and stimulate their metabolic activity, a characteristic of cycling TA cells.
Inactivation of IGF-1R and p38 MAPK stimulates DPC proliferation with different kinetics and inversely and differentially regulates stem and TA cell compartments
The proliferation profile of the DPC total population in response to IGF-1R and p38 MAPK inhibitors confirmed their pro-proliferative action but also revealed differences between them. Figure 3A shows that short 24–48 h treatment with IGF-1R(i) did not inhibit, and p38(i) increased, the proliferation of DPCs compared to that of untreated cultures. Prolonged exposure (144 h) to IGF-1R(i), however, reduced proliferation, whereas prolonged exposure to p38(i) continued to stimulate the DPC proliferative activity (Fig. 3A). The stimulation of proliferation following both short and prolonged inhibition of p38 MAPK is consistent with the known ability of p38 MAPK to induce quiescence [46]. It is likely that the short-term effect is reversible because prolonged inhibition sustained the effect. In contrast, the inhibition of proliferation by short and prolonged treatment with IGF-1R(i) reveals a paradoxical ability of IGF-1R to support quiescence and proliferation.

IGF-1R and 38 MAPK inhibitors stimulate dental pulp stem cell propagation but instigate different fates.
To resolve the paradoxical effect of IGF-1R and p38 MAPK inhibitors, we assumed that they act differently on the SC and TA cell compartments. Whether the SCs self-renew or proceed to their differentiation fate can be estimated by the measurement of SFU values, which reflect the size of the SC compartment. The data in Fig. 3B and C demonstrate that exposure of DPCs to IGF-1R and p38 MAPK inhibitors at the time of seeding affects the sphere-forming capacity of the cells in opposite ways. Surprisingly, IGF-1R(i) augmented and p38(i) decreased the number of spheres formed, implying that IGF-1R(i) expands the SC pool, whereas p38(i) reduces it. Thus, the observed differences in the mitotic activity of DPSC in response to each of the inhibitors have distinct biological consequences.
The exit of SCs from their compartment and their entry into the destined-to-differentiate TA cell compartment reduces their pool and self-renewing activity. The number of cells capable to initiate a colony is a measure of the proliferative potential of SCs and their TA daughters together. The CFU values shown in Fig. 3D indicate that, in contrast to their effects on the sphere-forming ability, IGF-1R(i) significantly decreased (P<0.001) and p38(i) significantly augmented (P<0.01) the number of colonies. The increased CFUs and decreased SFUs observed after p38 MAPK inactivation suggest that such inactivation stimulates cycling and at the same time increases the exit of sphere-forming DPSCs from the SC compartment, thus depleting this compartment. In contrast, the inactivation of IGF-1R seems to prevent the exit of sphere-forming DPSCs from the SC compartment and, consequently, to prevent the exhaustion of DPSCs.
This conclusion was strongly supported by the results of CFU assays performed with sorted PYlow DPSCs and their PYhigh progeny. The differences in the growth rates of untreated and IGF-1R(i)- and p38(i)-treated PYlow cultures were easily noticeable. Fig. 4A shows how strongly PYlow DPSC responded to p38(i). IGF-1R(i) was less powerful, but it also stimulated proliferation, whereas untreated PYlow cells grew very slowly. Sorted PYlow control cells remained quiescent for 3 weeks and did not form colonies at the time of scoring (after 2 weeks). In continuously IGF-1R(i) and p38(i) treated cultures, colonies were visible 1 week earlier. IGF-1R(i) stimulated colony formation to a lesser extent than p38(i) (Fig. 4B). In contrast, untreated and p38(i)-treated PYhigh cells grew much faster. However, IGF-1R(i) treatment of cycling PYhigh cells limited the growth to spindle-like, stem-like cells with a high nuclear/cytoplasmic ratio, clearly restricting the growth of the fibroblast-like TA cells that were visible in the control and p38(i)-treated cultures. Importantly, PYhigh cells treated with p38(i) produced more colonies than control PYhigh cells, and those treated with IGF-1R(i) produced fewer colonies than controls. This finding indicates that the SC and the TA cell compartments, as a whole, are enlarged in response to p38(i). IGF-1R(i), however, appears to stimulate only the SC pool because colonies in PYhigh cultures treated with IGF-1R(i) exhibited an SC-like morphology.
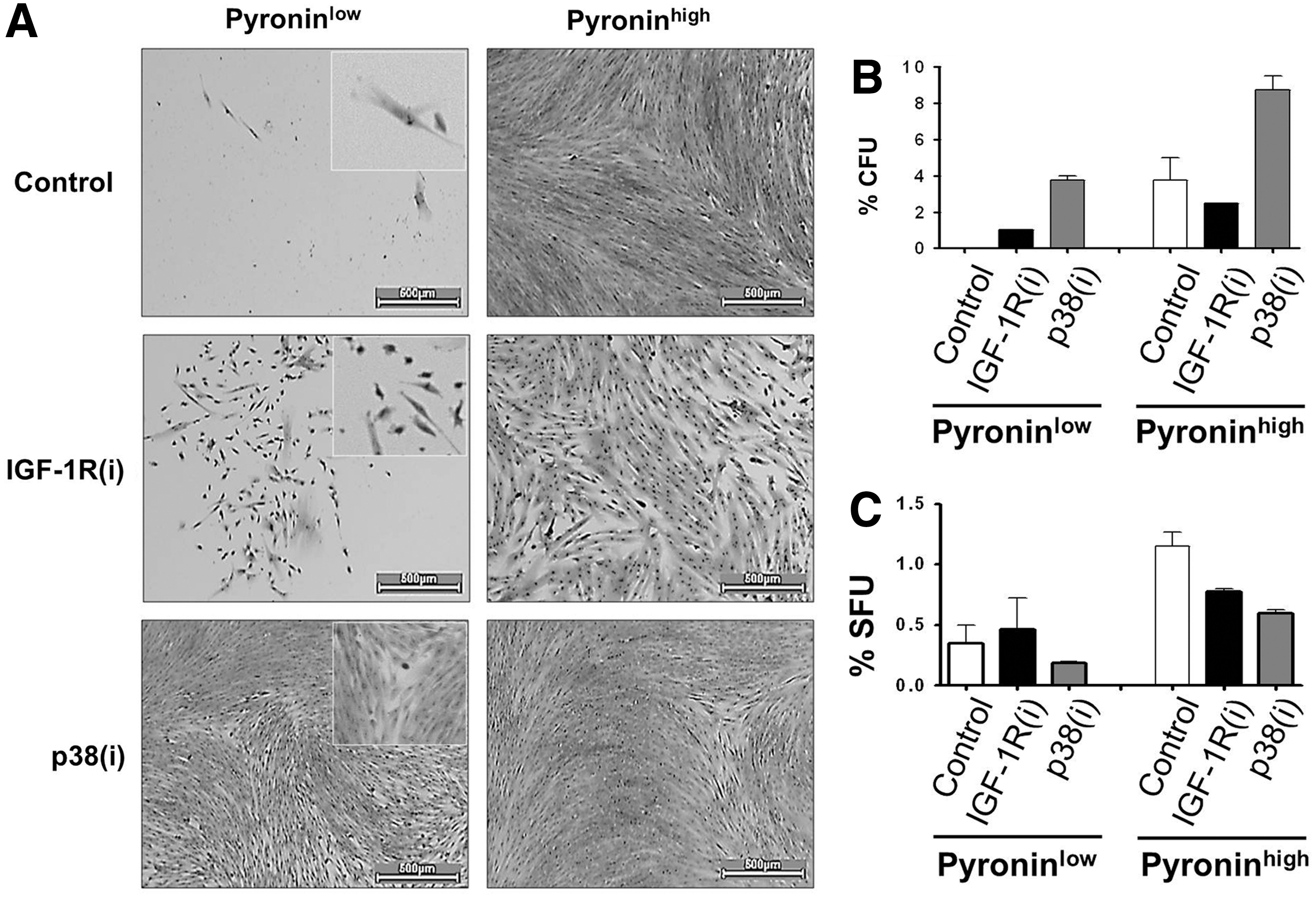
IGF-1R and p38 MAPK inhibitors diminish the proportion of PYlow human dental pulp stem cells.
This conclusion was substantiated by the results of SFU assays, which indicated that PYlow cells produced more first-generation spheres in the presence of IGF-1R(i) than in the presence of p38(i) (Fig. 4C) and had a 10.8-fold higher self-renewing capacity than PYhigh cells, as judged by the percentage of spheres in the second generation (1.3%PYlow/0.12%PYhigh). The delayed growth of PYlow cells is consistent with their properties of quiescent cells and suggests that they represent a pool of long-term quiescent SCs as opposed to the short-term poised SCs [25,58]. The PYhigh population apparently contained a subpopulation of cycling SCs that diminishes upon IGF-1R and p38 inhibitor treatment. Taken together, these data demonstrate that both IGF-1R(i) and p38(i) activate the slow-growing PYlow cells of the SC compartment. In addition, p38(i) may specifically accelerate division of SCs in this compartment while simultaneously feeding them into the PYhigh fast-growing, destined-to-differentiate TA cell subset. In contrast, by repressing the well-known pro-proliferation activity of IGF-1R [40], IGF-1R(i) clearly blocks the development of the PYhigh cell compartment.
IGF-1R and p38 MAPK inhibitors activate DPSCs by targeting different signaling pathways
Thus far, we have established that IGF-1R and p38 MAPK signals can maintain DPSC quiescence and induce cell cycling, two antagonistic cellular functions. How do these two pathways interact to regulate these opposite cellular activities? p38 MAPK is a downstream target of the IGF-1R. This suggests that IGF-1R(i) affects odontoblast quiescence through p38 MAPK signaling. To investigate this possibility, we cultured DPCs simultaneously with both IGF-1R and p38 MAPK inhibitors and measured the effects of such treatment on the colony-forming capacity of the cells. We found that the treatment with p38(i) enhanced the CFU values of IGF-1R(i)-treated cultures to the same level as treatment with p38(i) alone (Fig. 5A, B), suggesting that, in fact, p38(i) overcomes the inhibitory effect of IGF-1R(i). The well-established pro-proliferative activity of IGF-1R is mediated by its intracellular downstream targets, which include the PI3K/AKTmTOR signaling pathway [40,42]. To determine which, if any, of the intracellular downstream targets of IGF-1R may be affected by the p38 MAPK signals, we double inhibited this pathway and the PI3K/AKT pathway with LY294002 or mTOR with rapamycin. We found that mTOR inhibitors, but not PI3K/AKT inhibitors, reduced CFU values. Intriguingly, p38(i) alone or in combination with any of the other inhibitors always resulted in a similar increase in CFUs (Fig. 5A, B), demonstrating a prevailing action of p38 MAPK. The colonies formed in the presence of PI3K/AKT or mTOR inhibitors were smaller, however, than those formed in the presence of p38(i) alone, indicating that, conversely to p38 MAPK, PI3K/AKT/mTOR signaling may regulate the proliferative potential of DPCs. mTOR in particular seemed to contribute to the colony growth because double inhibition with LY294002 and rapamycin stopped cell division before colonies developed to the 50-cell stage. Taken together, these data suggest that IGF-1R may control stem cell quiescence via p38 MAPK signaling and proliferation potential via PI3K/AKT/mTOR signaling. The inactivation of p38 MAPK appears to have a regulatory effect on both of these cellular processes.
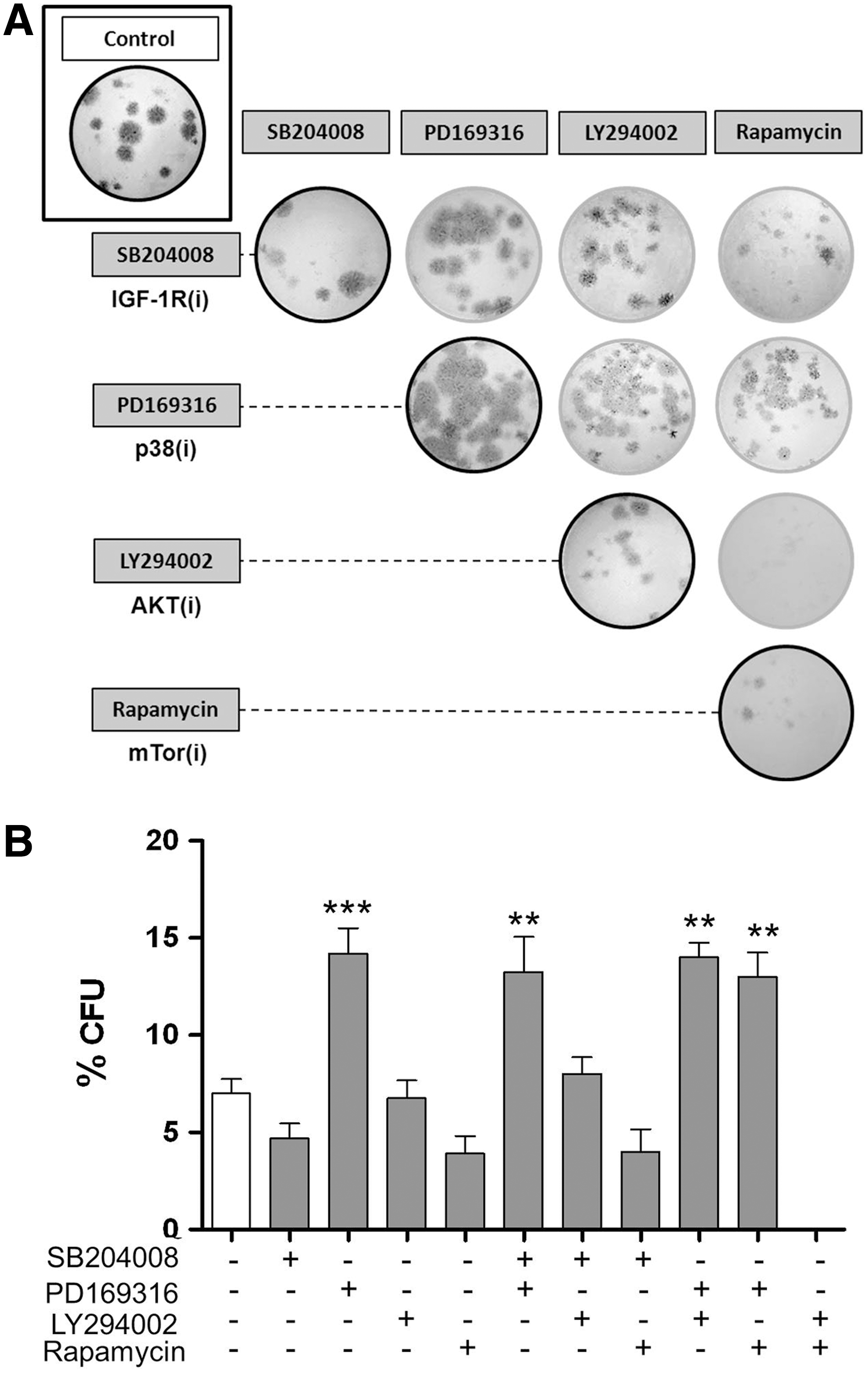
Inhibition of p38 MAPK has a dominant effect on colony-forming capacity and colony size.
Inactivation of IGF-1R and p38 MAPK inversely regulate STAT3 and MEPE-related DPC differentiation and mineralization
To gain insight into the mechanistic links between cellular responses and molecular events, we generated profiles of the phosphorylated proteins that regulate cell growth and cycling activity in control, IGF-1R(i)- and p38(i)-treated dental pulp cultures. Western blotting (Fig. 6A) of IGF-1R(i)- and p38(i)-treated cells shows a set of proteins that were affected by the treatment. We found that IGF-1R(i) mainly stimulated the pCREB and pGSK3β, suggesting that in the absence of active IGF-1R that normally phosphorylates CREB and GSK3, other signaling pathways upregulate pCREB and pGSK3, perhaps the active p38 MAPK pathway, which is known to phosphorylate CREB [59,60] and inactivate GSK3 [50,61]. Consistently, the inactivation of p38 MAPK had no effect on these targets (Fig. 6A). In accord with the capability of IGF-1R(i) and p38(i) to drive cells out of cellular quiescence, these two inhibitors upregulated the expression of c-Myc and cyclin D1, which drive the cell cycle, and of Bcl-2, a pro-survival factor, to comparable levels, suggesting that the IGF-1R and p38 MAPK signaling pathways converge at this level of regulation.
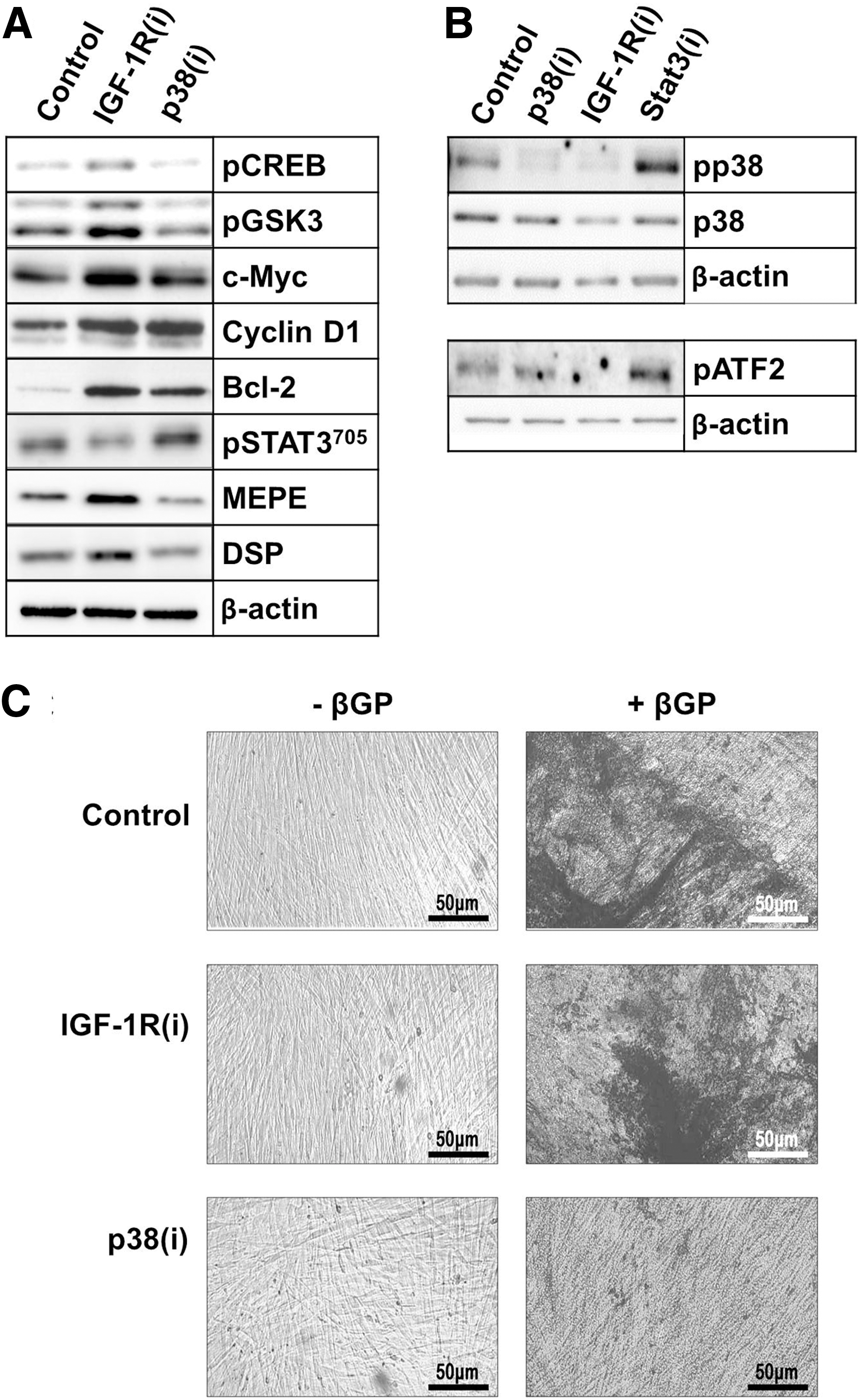
IGF-1R and p38 MAPK inhibitors inversely affect signal transducer and activator of transcription 3 (STAT3) and matrix extracellular phosphoglycoprotein (MEPE) expression and dental pulp cell differentiation.
Interestingly, p38(i) primarily upregulated STAT3 phosphorylation at the Y705 residue (pSTAT3705) (Fig. 6A). The increased phosphorylation of STAT3 was blocked by IGF-1R(i), confirming that STAT3 is a downstream substrate of IGF-1R signaling [32] and suggesting that active p38 MAPK interferes with this signaling. As shown in Fig. 6A, IGF-1R(i) and p38(i) differentially regulated also the expression of MEPE, a marker of early odontoblast differentiation [22,62]. Treatment of cells with p38(i), which induced pSTAT3705, reduced MEPE levels; conversely, treatment with IGF-1R(i) repressed pSTAT3705 and upregulated MEPE expression. The inverse relationship between pSTAT3 and MEPE was evident in all the experiments in which we were able to pharmacologically manipulate the activity of p38 MAPK (Supplementary Fig. S2A) or to knock down its expression using siRNA (Supplementary Fig. S2B). Moreover, inhibition of pSTAT3705 by STAT3 inhibitor induced MEPE (Supplementary Fig. S2C). Taken together, these data provide strong evidence that STAT3 inhibits MEPE expression. Because pSTAT3705 has been shown to control stem cell self-renewal [63 –65], its presence at higher levels in p38(i)-treated cells may provide a link between p38 MAPK, IGF-1R and the quiescence of DPSCs. Figure 6B shows that both inhibitors rapidly repressed phosphorylation of p38 MAPK (pp38 MAPK). Inactivation of STAT3 by its specific inhibitor sc202818 reversed this repression and, consequently, upregulated pATF2, a p38 MAPK downstream substrate. This confirms, to some extent, that pSTAT3705 negatively feedback-regulates p38 MAPK.
The SC-derived TA cells have a limited proliferative capacity and stop cycling after several rounds of replication to enter the differentiation process [66,67]. MEPE is a prerequisite for late odontoblast differentiation [22,62]. IGF-1R(i), which stimulated MEPE production, also consistently stimulated the production of DSP, a marker of late odontoblast differentiation. As evidenced by the appearance of von Kossa-positive mineralization nodules (Fig. 6C) and the expression of alkaline phosphatase (Supplementary Fig. S3), a marker of mineralization [68,69], IGF-1R(i)-treated DPCs entered terminal differentiation in the presence of βGP, an odontoblast differentiation-inducing factor [23,70]. In contrast, p38(i), which stimulated proliferation and reduced MEPE levels, did not affect the levels of DSP, and cells treated with p38(i) did not initiate the mineralization process (Fig. 6C and Supplementary Fig. S3). These results suggest that odontoblast proliferation requires the constant inactivation of p38 MAPK and only a temporal inactivation of IGF-1R. The repression of IGF-1R-signaling and the activation of p38 MAPK, however, appear to be necessary for odontoblast differentiation. Taken together, these data provide strong evidence that STAT3 integrates IGF-1R- and p38-mediated signals to determine DPSC fate.
Altogether, our data demonstrate that although IGF-1R and p38 MAPK inactivation impinge on different downstream targets, their signals are integrated within the cell at STAT3 to activate the cycling of quiescent DPSC and to determine their fate.
Discussion
DPSCs, which are normally quiescent, enter the cell cycle in response to odontoblast injury in vivo [4] and can proliferate and differentiate in vitro [12,13,71]. The actual factors that regulate the transition of DPSCs from the quiescent to the active state, however, are not well defined, and we have a limited understanding of how the self-renewal and differentiation of these cells is regulated. Given the well-established role of p38 MAPK in mediating inflammatory responses [72] and its roles in dentinogenesis [27,35] and in the control of cellular quiescence [46], we investigated whether p38 MAPK regulates the fate of human DPSCs and whether this fate also depends on IGF-1R signaling pathways upstream of p38 MAPK that control the cellular reprogramming and pluripotency of SCs [40].
We determined that integrated p38 MAPK and IGF-1R signals inversely control the STAT3 activity to maintain human DPSC quiescence and activation. Consistent with the proposed roles of p38 MAPK and IGF-1R in the maintenance of cellular quiescence, we determined that the majority of mitotically arrested DPCs in vivo express the nuclear active [52,53] form of p38 MAPK in all zones of dental pulp. In agreement with the results of an earlier study [51], however, IGF-1R was detected mainly at the pulp periphery, implying that it may have a specific function in the committed odontoblasts that are present in this region [12,73]. As expected of G0-arrested cells, DPCs were almost exclusively Ki67-negative. Inactivation of IGF-1R and p38 MAPK by their respective inhibitors, sc204008 and PD169316, induced Ki67 in DPCs ex vivo and decreased the quiescence of PYlow and G0-arrested human DPSCs cells in vitro, demonstrating that p38 MAPK and IGF-1R are responsible for the mitotic quiescence of these cells. The identity of DPSCs was corroborated by their phenotype, stem cell activity, and localization of Ki67-positive cells perfectly matching the position of BrdU-positive DPSCs reported by Téclès et al. [4] and Ishikawa et al. [19] and the position of Oct4-positive pluripotent DPSCs reported in another study [74]. Taken together, our data establish that IGF-1R and p38 MAPK signaling pathways control human DPSC quiescence and demonstrate that these pathways must be inactivated for mitotic division to occur in DPSCs.
Several studies have identified other factors that promote DPSC expansion in vitro, including leukemia inhibitory factor, which activates JAK/STAT3 signaling [75]. Interestingly, STAT3 is a central molecule on which the signals from many cytokine and growth factor receptors, including IGF-1R and p38 MAPK, converge [32,40,76,77]. STAT3 stimulates embryonic and somatic stem cell self-renewal [32,63,64,78] by promoting Klf4 expression, which affects Oct4, Sox 2, and c-Myc, Yamanaka pluripotency and cellular reprogramming factors (see refs. [79 –81]). STAT3 also cooperates with GSK3 inhibitors to maintain pluripotency and self-renewal by blocking embryonic stem cell (ESC) differentiation [80,81]. We observed that p38 MAPK inhibitors upregulated STAT3Y705 and propagated DPCs without affecting the levels of pGSK3, an inactive form of GSK3. Together with the size of the stem cell pool, arrested differentiation, and the increase in the clonogenic capacity of the cells, this finding implies that p38 MAPK maintains DPSC quiescence and prevents the exhaustion of the stem cell pool by inhibiting STAT3 activity. The inactivation of STAT3 permits MEPE expression and DPC differentiation. In contrast to p38(i), IGF-1R(i) downregulated STAT3Y705 but increased pGSK3 levels and the size of the stem cell compartment while reducing the clonogenic potential of DPCs. This activity suggests that STAT3 stimulates DPSC propagation and pGSK3, as in ESC [80], represses DPSC commitment to differentiation. Because both IGF-1R(i) and p38(i) induced DPSC proliferation but p38(i) overcomes the inhibitory effect of IGF-1R(i) on the clonogenic potential of DPSCs as of endothelial progenitors [82], we propose that p38 MAPK constitutively inhibits IGF-1R-mediated STAT3 activity and inactivates GSK3 directly ([61] and our data) to maintain DPSCs as undifferentiated quiescent cells. IGF-1R, even if present, would be unable to activate STAT3 and cause DPSCs to exit from quiescence; hence, IGF-1R also maintains DPSC quiescence.
The observed transient stimulation of DPC proliferation by IGF-1R(i) implies that active STAT3 may repress p38 MAPK via a feedback effect. Indeed, inhibition of the STAT3 activity increased the level of pp38 MAPK and its substrate, ATF2 as well as MEPE early marker of odontoblast differentiation. How, then, can IGF-1R simultaneously stimulate cell proliferation and maintain the quiescence of DPSCs, and how do p38 MAPK and IGF-1R exert their opposite effects on DPSC proliferation and differentiation? To consolidate our results, we propose a working model (Fig. 7) that explains how IGF-1R and p38 MAPK signaling pathways may interact to regulate DPSC. In building the model, we assumed that adult human DPSCs, like ESCs [80,83], self-renew and generate undifferentiated progeny due to the inactivation of GSK3 and the increased activity of STAT3 transcription factor. We also assumed that the p38 MAPK signaling pathway represses the STAT3 activity not by repressing all the functions of IGF-1R but, rather, by specifically repressing the function of its downstream STAT3-activating substrate, most likely JAK [32,41,42], which has recently been shown to stimulate odontoblasts proliferation and to inhibit odontoblasts differentiation [75].
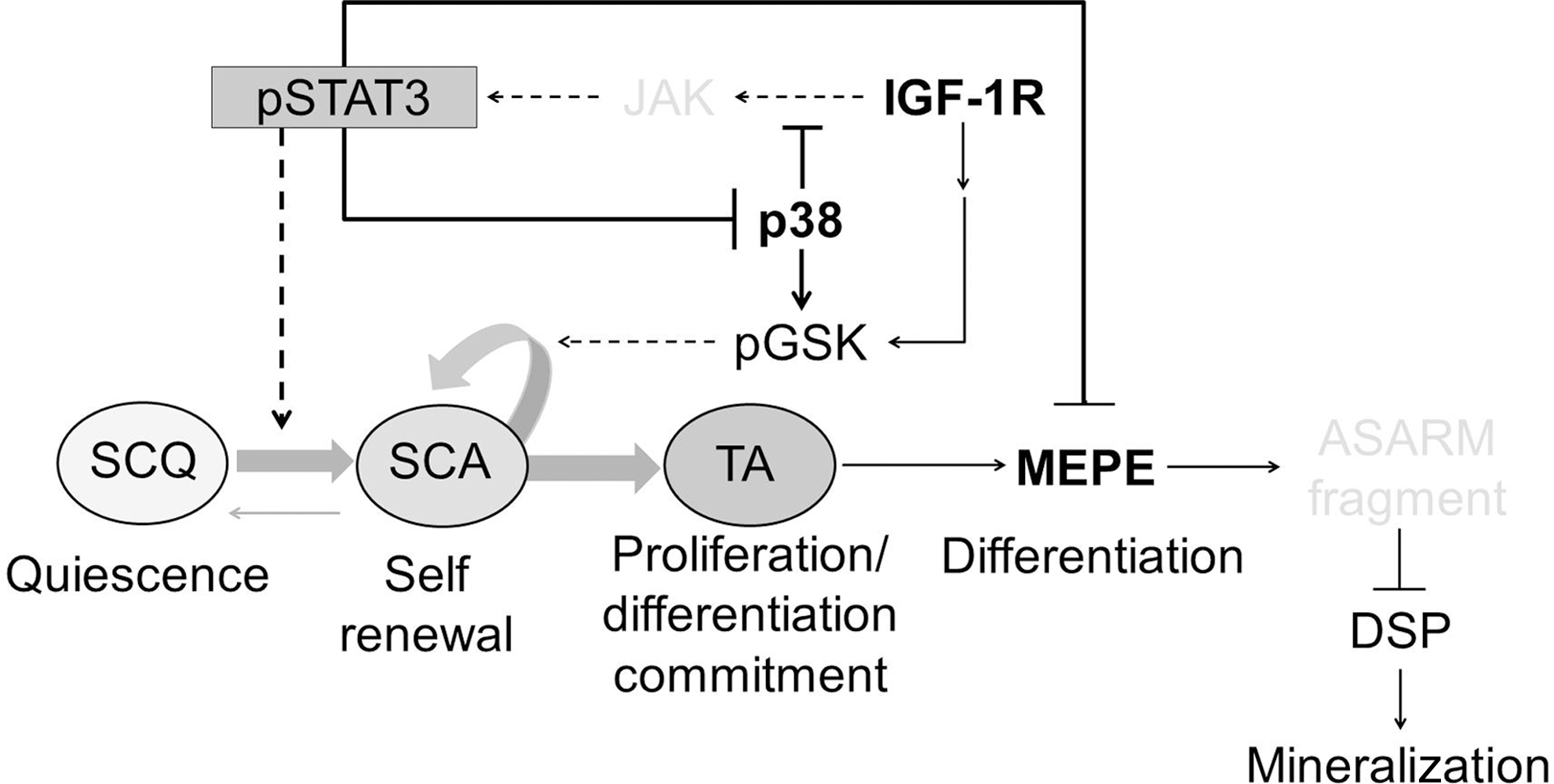
A working model of cross talk between IGF-1R and p38 MAPK signaling pathways integrating cellular and molecular events that control the self-renewal and differentiation of dental pulp stem cells. Please see the text for details. SCQ, quiescent stem cell; SCA, activated stem cell; TA, transient amplifying committed progenitor; MEPE, matrix extracellular phosphoglycoprotein, a marker of early odontoblast differentiation; ASARM, proteolytic fragment of MEPE that inhibits DSPP (dentin sialophosphoprotein) expression [22]; DSP, dentin sialoprotein, a DSPP processing product associated with late odontoblast differentiation and mineralization; pSTAT3, phosphorylated signal transducer and activator of transcription 3; pGSK3, phosphorylated glycogen synthase kinase 3; JAK, Janus kinase; IGF-1R, insulin growth factor 1 receptor. Arrows indicate activation, and perpendicular lines indicate inhibition. The bent arrow indicates a self-renewing function. The model is based on results reported in this study (black fonts and solid lines) and in the literature (gray fonts and broken lines).
The importance of p38 MAPK in controlling quiescence has been recognized previously [46,49]. Interestingly, the inhibition of p38 stimulates the expansion of hematopoietic stem cells (HSC) in vitro and increases their repopulation potential in vivo [84]. In contrast, reactive oxygen species-induced p38 MAPK signaling impairs HSC self-renewal [85,86], demonstrating a dual activity of p38 MAPK through which it may either promote or inhibit cellular quiescence [59,61]. The mechanism for the observed duality of action of p38 MAPK is not clear, but its control of cell quiescence may involve the inhibition of GSK3 [50,61,87] and STAT3, as work of others [50,59,77] and our data imply. Inactive GSK3 stimulates ESC propagation and inhibits ESC differentiation [80]. This previous finding is consistent with our results demonstrating the proliferative activity of DPSCs in the presence of an IGF-1R inhibitor. However, in the absence of STAT3, which promotes stem cell pluripotency, cycling DPSCs, like ESCs [80], could not maintain their propagation by the inactivation of GSK3 alone and would eventually succumb to differentiation. Indeed, the inhibition of IGF-1R stimulated MEPE expression, suggesting that STAT3 negatively regulates MEPE and odontoblast differentiation.
In summary, we show that IGF-1R and p38 MAPK interact to balance DPSC quiescence and activation and that their signals are integrated to inversely regulate STAT3 and perhaps the GSK3 activity. We consider that p38 MAPK has a dominant effect and that it inactivates both STAT3 and GSK3 to maintain DPSC quiescence and sustain their self-renewal while inhibiting their commitment to differentiation. By inhibiting STAT3, however, p38 MAPK can stimulate MEPE expression and the differentiation of committed DPSCs. The IGF-1R signaling dominated by p38 MAPK also maintains DPSC quiescence, but when STAT3 is activated in a feedback reaction, p38 MAPK is inhibited. The presence of IGF-1R at the pulp periphery suggests that IGF-1R blocks odontoblast proliferation and differentiation. Due to their location, cells at the periphery are predisposed to respond rapidly to factors released by infected or damaged pulp. A precedent for a mechanism linking IGF-1R expression to cellular quiescence has been described in pancreatic tumor cells, which exhibit a growth factor-independent phenotype that enables them to rapidly re-enter the cell cycle [88]. We believe that our study opens an avenue for further investigation of DPSC expansion and differentiation in vitro and that it may also advance our understanding of the mechanisms controlling reparative dentinogenesis in inflamed and damaged teeth in vivo.
Footnotes
Acknowledgments
This work was supported by La Fondation pour la Recherche Médicale (FRM), which provided a PhD fellowship to J.V. for 2 years, and by the Institut Nationale de la Santé et de la Recherche Médicale (INSERM). We acknowledge Marie-Helene Gevaert, Histology Laboratory, University Lille 2, and Nathalie Jouy, BiCell-IFR 114, for their greatly appreciated help with frozen sections and hematoxylin–eosin (H&E) staining and with flow cytometry analyses, respectively, and Dr. Gilbert Nafash from the Department of Dentistry at the University Lille 2 for providing us with extracted teeth.
Author Disclosure Statement
The authors have no conflicts of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.