
Abstract
Primordial germ cells (PGCs) have the ability to be reprogrammed into a pluripotent state and are defined as embryonic germ cells (EGCs) in vitro. EGC formation is more efficient, has a shorter culture period than somatic cell reprogramming, and does not require exogenous genetic manipulation. Therefore, EGCs are a good model to analyze mechanisms by which committed cells acquire a pluripotent state. In the present study we have attempted to elucidate a more defined and robust protocol that promotes EGC generation through the suppression of transforming growth factor-β (TGF-β) and extracellular signal-regulated kinase (ERK) signaling pathways by SB431542 (SB) and PD0325901 (PD), respectively. Under this condition the efficiency of transformation of PGCs into EGCs was more than the inhibition of glycogen synthase kinase 3 and ERK signaling pathways. Pluripotency of the resultant-derived EGC lines were further confirmed by gene expression, immunofluorescent staining, directed differentiation ability, teratoma formation, and their contribution to chimeric mice and germ-line transmission. These results showed that PGCs from different embryonic stages (E8.5 and E12.5) could be reprogrammed, maintained, and expanded efficiently under feeder- and serum-free chemically defined conditions by dual inhibition of TGF-β and ERK signaling pathways, regardless of the animal's genetic background.
Introduction
P
In the conventional EGC derivation method, media that contain serum supplemented with leukemia inhibitory factor (LIF), basic fibroblast growth factor (bFGF), and stem cell factor (SCF) are commonly used [12,14]. Because of the undefined and less-efficient condition of the conventional method, researchers have searched for a more defined, efficient protocol [15]. Leitch et al., in their research, have used a chemically defined medium for efficient EGC induction from 8.5 dpc embryos [15]. They used chemical inhibitors of glycogen synthase kinase-3 (GSK3) and extracellular signal-regulated kinase (ERK) pathways in combination with LIF (referred to as the CHIR+PD condition), which had been previously used for the derivation and expansion of ESCs from mice in a serum- and feeder-free condition [16]. Because GSK3 is a multifunctional molecule that plays central roles in several intracellular signaling pathways [17], its inhibition could complicate understanding of the reprogramming mechanism. Recently, we have shown that inhibition of transforming growth factor-β (TGF-β) by SB431542 (SB) and the ERK pathway by PD0325901 (PD) could have an impressive effect on the derivation and maintenance of ESCs derived from mouse blastocysts [18]. Very recently, we also reported this combination of two inhibitors which hereafter referred to as SB+PD could provide the ground state pluripotency in generation and culture of naive ESCs from different mouse strains in a chemically defined medium [19]. It has been shown that inhibition of the TGF-β receptor by small molecules spurred reprogramming of mouse and rat somatic cells into iPS cells [20,21]. Therefore, we aimed to apply the SB+PD condition in the derivation of EGCs from 8.5 and 12.5 dpc PGCs. Of note, we have eliminated the use of GSK3 inhibitor, which consequently would direct us to locate the reprogramming mechanism with fewer complications. In the present study we introduced SB+PD as a fully chemically defined condition that led to more efficiently derived EGCs from embryos of various ages compared with the previously reported defined media.
Materials and Methods
Mice and embryos
We used outbred (NMRI) and inbred (DBA2; Pasteur Institute, Tehran, Iran) mice strains to derive EGCs. Animals were maintained under 12-h light/dark conditions. All embryos were obtained by natural mating. The copulation plug was verified the morning following coupling of females with males; in cases of plug formation, we considered the embryos to be 0.5 day dpc. Mouse embryonic fibroblasts (MEFs) were obtained from 12.5 dpc embryos of the NMRI strain and used as feeder cells. All animal studies received approval from the Institutional Review Board and Ethics Committee.
Isolation of PGCs, and establishment and culture of EGC lines
We isolated PGCs from 8.5 and 12.5 dpc mouse embryos, as previously described [22]. PGCs were isolated from 8.5 dpc embryos by removal of the decidua and all extraembryonic membranes, after which the posterior part of each embryo at the base of the allantois was dissected and collected in 1.5-mL tubes. For 12.5 dpc embryos, we cut off the head of each embryo, followed by the removal of urogenital ridges (genital ridges with mesonephros) from the dorsal wall of each embryo body. We used a fine needle to separate the genital ridges from the mesonephros, which were collected in a tub that contained dissecting medium. After trypsinization and centrifugation of the part that contained the PGCs, we cultured single cells from each embryo on 2-cm2 gelatin-coated dishes in a medium designated as day 0 medium. This medium contained knockout Dulbecco's modified Eagle's medium (KO-DMEM; Invitrogen), 2% fetal calf serum (FCS; HyClone) or knockout serum replacement (KSR; Gibco), 0.1% MEM nonessential amino acids (Invitrogen), 2 mM L-glutamine (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 100 U/mL penicillin and 100 mg/mL streptomycin (Invitrogen) supplemented with recombinant mouse LIF (1000 U/mL; ESGRO, Chemicon), homemade bFGF (25 ng/mL; Royan Institute), and SCF (100 ng/mL; R&D). The following day we replaced the medium with serum-free N2B27-supplemented medium that consisted of DMEM/F12 (Invitrogen) and neurobasal (Invitrogen) at a 1:1 ratio, 1% N2 supplement (Invitrogen), 1% B27 supplement (Invitrogen), 2 mM L-glutamine, 1% MEM nonessential amino acids, 100 U/mL penicillin, 100 mg/mL streptomycin, 0.1 mM β-mercaptoethanol and 5 mg/mL BSA (Sigma-Aldrich), LIF (1000 U/mL), small molecules (see Chemical compounds section), and lacking bFGF and SCF. We renewed the medium every other day. EGC colonies were observed after 7–10 days. The resultant colonies were selected and transferred to gelatin-coated dishes in the medium from which they were derived. Subsequently, the plated cells were allowed to expand into EGC lines by dissociation with trypsin and replated every 2–4 days.
Chemical compounds
We used the following small molecules and their concentrations in this study: DMSO (Sigma-Aldrich) as the solvent, PD0325901 (PD, 1 μM, ERK1/2 inhibitor; Stemgent), CHIR99021 (CHIR, 3 μM, GSK3 inhibitor; Stemgent), SB431542 (SB, 10 μM, TGF-β inhibitor; Sigma-Aldrich), A-83-01 (A83, 250 nM, TGF-β inhibitor; Stemgent), and ALK5 inhibitor (ALK5i, 1 μM, TGF-β inhibitor; Stemgent).
Antibodies
For immunofluorescence staining, EGCs were fixed in 4% paraformaldehyde prepared in phosphate-buffered saline (PBS), at pH 7.4 (Invitrogen) for 20 min. To remove the remaining fixative, cells were washed twice with 0.1% Tween-20 in PBS, followed by permeabilization with 0.2% Triton X-100 in PBS for 20 min prior to blocking with 10% goat serum in PBS for 60 min. Subsequently, EGCs were allowed to incubate overnight in primary antibody solution at 4°C. The primary antibodies utilized in this study were as follows: Oct4 (1:100; Santa Cruz Biotechnology, SC-5279), Nanog (1:100; Santa Cruz Biotechnology, SC-30329), Sox2 (1:100; Santa Cruz Biotechnology, SC-365823), and stage-specific embryonic antigen-1 (SSEA1, 1:50; R&D, MAB2155) for detection of the pluripotency state. Connexin 43 (1:200; Abcam, ab11370), Mef2c (1:200; Abcam, ab64644), α-Mhc (1:200; Abcam, ab15), Tuj1 (1:200; Sigma-Aldrich, T-8660), Map2 (1:200; Sigma-Aldrich, M1406), and Gfap (1:200; Sigma, G3893) were used to detect differentiated cardiac and neural cells. After washing twice with 0.1% Tween-20 in PBS for 10 min, cells were incubated with the secondary antibody dissolved in PBS. We used the following fluorescence-conjugated secondary antibodies in this study: goat anti-mouse IgM-PE (1:500; Santa Cruz Biotechnology, SC-3768), goat anti-mouse immunoglobulin IgG fluorescein isothiocyanate (FITC, 1:200; Sigma-Aldrich, F9006), and goat anti-rabbit IgG FITC (1:200; Sigma-Aldrich, F1262). Cells were incubated with the secondary antibodies for 1 h at 37°C, and then washed twice with PBS that contained 0.1% Tween-20 for 5 min. We used 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) to stain the cell nuclei and cell analysis was conducted with a fluorescent microscope (Olympus). Immunofluorescence staining without primary antibodies was also used as a negative control. The SSEA1-positive cells (SSEA1-Alexa Flour-647 antibody, 1:200; Biolegend, 125607) were used as control PGCs for real-time reverse transcriptase (RT)–polymerase chain reaction (PCR) [23]. For this matter the posterior parts of 8.5 dpc embryos were dissociated to form a single-cell suspension by incubation with 0.05% trypsin and 0.02% EDTA for 10 min. After washing with PBS the cells were incubated in the presence of goat serum 10% for 30 min on ice. Afterward the cells were divided to two groups; one group was treated with the SSEA1-conjugated antibody and the other with the isotope antibody for 30 min on ice. Next, both groups were washed three times in PBS and after suspension in sorting buffer were sorted using a BD FACS cell sorter. For the double staining of PGCs with SSEA1 and Oct4 markers, the single cells of posterior fragment of the 8.5 dpc embryo were resuspended in PBS containing 5% goat serum and SSEA1 primary antibody for 1 h. After two washes with PBS the cell pellet was resuspended in BD Cytofix/Cytoperm solution (BD; 554722) and incubated for 30 min at 4°C. After washing twice with BD perm/wash buffer, the pellet was resuspended in 200 μL BD perm/wash buffer plus 10% goat serum and incubated for 30 min on ice. Then, the cells were incubated with Oct4 primary antibody overnight at 4°C. Thereafter, cells were washed twice with BD perm/wash buffer followed by 45-min incubation at 4°C for appropriate secondary antibodies (Alexaflour-488 anti-mouse IgM, Invitrogen A21042 and anti-mouse IgG-PE, Abcam ab97024). The isotype control group contained just the secondary antibodies. The cells were analyzed by BD FACS Calibur flow cytometer and the acquired data were assessed with CellQuest software.
Teratoma formation
To detect teratoma formation, we subcutaneously transplanted ∼2–3×106 EGCs into nude mice. At 15 days following injection, we observed teratoma formation; 2 weeks later, the teratomas were isolated, fixed in 4% Bouin for 48 h at room temperature, and then embedded in paraffin and sectioned into 6-μm-thick sections. The sections were stained with hematoxylin-eosin, after which we determined the presence of derivatives of three germ layers by using a brightfield microscope.
In vitro differentiation
For direct cardiomyocyte differentiation, embryonic bodies (EBs) from two different lines were formed by culturing 800 EGCs in 20 μL of ESC medium in hanging drops. ESC medium contained KO-DMEM, 15% FCS, 0.1% MEM nonessential amino acids, 2 mM L-glutamine, 0.1 mM β-mercaptoethanol, 100 U/mL penicillin, and 100 mg/mL streptomycin supplemented with 10−4 M ascorbic acid (vitamin C; Sigma-Aldrich, A4403) without LIF [24]. Thereafter, EBs were cultured in bacterial dishes in suspension mode for an additional 5 days. Subsequently, EBs were plated on 1% gelatin-coated tissue culture dishes for an additional 7 days to allow for attachment and expansion into beating cardiomyocytes. To assess the nature of EGC-derived cardiomyocytes, we performed immunofluorescence staining and RT-PCR for cardiomyocyte markers.
To induce direct differentiation of EGCs into a neural mode, we produced EBs by culturing 200 EGCs from two different lines in 20 μL of ESC medium in the absence of LIF, in hanging drops for 3 days. Afterward, EBs were cultured in the presence of retinoic acid (2 μM; Sigma-Aldrich, R2625) in suspension mode in an induction medium that contained DMEM/F12 medium supplemented with 1% KSR, 0.1% MEM nonessential amino acids, 4 mM L-glutamine, and 0.1 mM β-mercaptoethanol for an additional 4 to 6 days. At ∼day 8, EBs were plated onto poly-L-lysine-coated dishes in the previously mentioned induction medium where they were maintained for 2 more weeks to allow for additional differentiation of precursor cells into mature neurons [25].
Chimera production
Chimera generation was assessed with the intent to examine the in vivo differentiation capacity of EGCs. EGC colonies were trypsinized into single cells, after which ∼10–15 of the single cells were injected into 3.5 dpc blastocysts from superovulated female BALB/c mice. Approximately 43 injected blastocysts were transferred into the uterine horns of pseudopregnant BALB/c×C57 (F1) mice. Coat color appearance was the criteria for chimera generation. To test germ-line transmission, the chimeras were mated to the males or females from BALB/c strain.
Karyotype analysis
We assessed the chromosome integrity of EGCs by performing G banding according to standard methods. EGCs were synchronized by treatment with 0.66 μM thymidine (Sigma; T1895) at 37°C. After 16 h, the medium was replaced with EGC medium and incubated for 5 h at 37°C to enable the EGCs to enter the metaphase. Subsequently, the cells were arrested in M phase by the application of 0.15 μg/mL colcemid (Gibco; 15212-012) for 45 min at 37°C. After harvesting, cells were treated with a hypotonic solution, 0.075 M KCl (Merck; 1.04935) for 10 min at 37°C. We performed the fixation process three times with ice-cold methanol (Merck; 1.06008) and acetic acid (Merck; 1.00063) in a 3:1 ratio. Next, fixed cells were dropped on chilled slides and the G banding method was used for chromosomal analyses.
RNA isolation, reverse transcription, RT-PCR, and quantitative RT-PCR
Total RNA from the samples, with the exception of PGCs, were isolated by TRIzol reagent (T9424; Sigma) according to the manufacturer's protocol. Total RNA from the PGCs was isolated with the RNeasy Micro Kit (74004; Qiagen). We removed any contaminating genomic DNA by treating all RNA samples with DNase I (EN0521; Fermentas). The RevertAid™ H Minus First Strand cDNA Synthesis Kit (K1632; Fermentas) was used for cDNA synthesis, which was performed from 1 μg of total RNA. Gene expression was analyzed by the Rotor-Gene 6000 (Corbett Life Science) instrument. We used the relative standard curve method for data analyses [26]. Primer sequences, amplicon size, and gene bank accession numbers are listed in Supplementary Table S1(Supplementary Data are available online at
Statistical analysis
Real-time data were expressed as mean±standard deviation (SD) in which the data with symmetrical (normal) or nonsymmetrical distributions were analyzed with one-way ANOVA followed by the post hoc LSD or Dunnett's significant difference tests, respectively. P-values<0.05 were considered significant. The data for the efficiency significance of EGC derivation in different groups were analyzed by generalized linear model and the chi-square test.
Results
Induction of pluripotency in 8.5 dpc PGCs by SB+PD
We recovered PGCs from 8.5 dpc mouse embryos with the intent to evaluate the influence of SB+PD on PGC conversion to EGCs. The posterior embryo fragments that contained the caudal end of the primitive streak and allantois were isolated because PGCs are located in this section. As seen in Fig. 1A, following trypsinization of the embryo fragments, we transferred the single cells from each embryo to one well of a 24-well plate that contained day 0 medium (supplemented with bFGF and SCF) for 24 h. The plates were covered with MEFs or coated with gelatin to assess the effect of feeder cells on colony emergence. The following day we replaced the medium with defined medium (N2B27+LIF) that contained SB+PD combination. Medium refreshment was performed every other day. EGC colonies emerged after 7–10 days in both groups (MEFs or gelatin; Fig. 1B, C). These results showed that SB+PD provided an appropriate condition for EGC formation from PGCs in a chemically defined condition. Additionally, we showed that it was not necessary to use feeder layers such as MEFs for induction and derivation of EGC colonies. Thus, to further define this condition, we chose to continue this culture method on gelatin for the next steps.

The process of establishing mouse embryonic germ cells (EGCs).
SB+PD replaced the continuous need for SCF
SCF is the most important factor governing germ cell survival and motility from the time of their specification in the proximal epiblast until colonization into the genital ridge. This special factor is capable of preparing a suitable niche during migration [11]. Additionally, SCF affects PGC survival by inhibiting apoptosis during the first hours of primary culture [27]. Previous studies have used feeder cells as a source of SCF for in vitro culture of PGCs. The mentioned feeders include STO cells that express both the membrane and the soluble form of SCF or the Sl4-m220 cell line, a stromal cell line derived from bone marrow that constantly expresses only the membrane-bound form of SCF [22]. It has been shown that EGC derivation in the absence of bFGF and SCF in CHIR+PD condition was reduced significantly compared with parallel cultures initiated in the presence of SCF and bFGF [15]. In the next step we have sought to determine whether 1-day exposure to exogenous SCF was sufficient for PGCs to be reprogrammed efficiently. We observed the emergence of EGC colonies both in continuous presence of exogenous SCF and in 1-day SCF exposure (Fig. 1D) with no significant difference in efficiency (data not shown). Emerged EGC colonies tested positive for pluripotency marker proteins Oct4, Sox2, and Nanog and specific surface marker SSEA1 (Fig. 1D–F). SCF and bFGF were added for 1 day of the primary culture because the only growth factor present in the medium during the following days was LIF. By taking this data into consideration, for the next steps, we employed SCF for 1 day followed by N2B27 medium supplemented with SB+PD and LIF for the remainder of the culture period.
SB+PD showed the most efficiency for EGC derivation compared with other chemically supplemented conditions
It has been shown that the CHIR+PD condition (GSK3 and ERK inhibitors) supports EGC derivation [15]. In addition, A83+CHIR+PD (A83 as the TGF-β inhibitor) leads to a more efficient EGC emergence compared with the CHIR+PD mode [13]. However the previously mentioned disadvantages of using the GSK3 inhibitor in both protocols motivated us to assess the effect of SB+PD on the efficiency of EGC colony formation.
On the other hand, it demonstrated that the effect of A83+PD combination on EGC derivation was significantly less than the efficiency of CHIR+PD group [13]. Moreover, in our previous study, we observed that A83+PD combination has a lower efficiency in generation of mouse ESCs when compared with SB+PD [18]. Therefore, A83+PD combination was not included in the present study. Our findings documented the efficiency of EGC emergence from total embryos used in each group as 47% (SB+PD), 20% (A83+CHIR+PD, as positive control [13]), and 11% (CHIR+PD) (Fig. 2A and 2B). To confirm the effect of TGF-β inhibition on EGC formation, we used an alternative small molecule for SB, the ALK5 inhibitor (ALK5i). ALK5i is a selective and ATP-competitive inhibitor of the TGF-β family type I receptor activin receptor-like kinase (ALK5) [28]. Our data have demonstrated that ALK5i+PD had the capability to reprogram PGCs to EGCs with the same efficiency as the SB+PD group (42%, Fig. 2B). For additional evaluation of TGF-β inhibition, we eliminated PD from our culture. We observed that the presence of SB or ALK5i alone did not affect EGC induction (Fig. 2B). However, EGCs generated under SB+PD conditions could be propagated for over five passages in the medium that contained only SB and LIF (data not shown). We concluded that SB+PD was a more efficient compound for PGC reprogramming compared with CHIR+PD and A83+CHIR+PD. More importantly, in contrast to the previously mentioned studies, GSK3 inhibition was not necessary for this conversion.
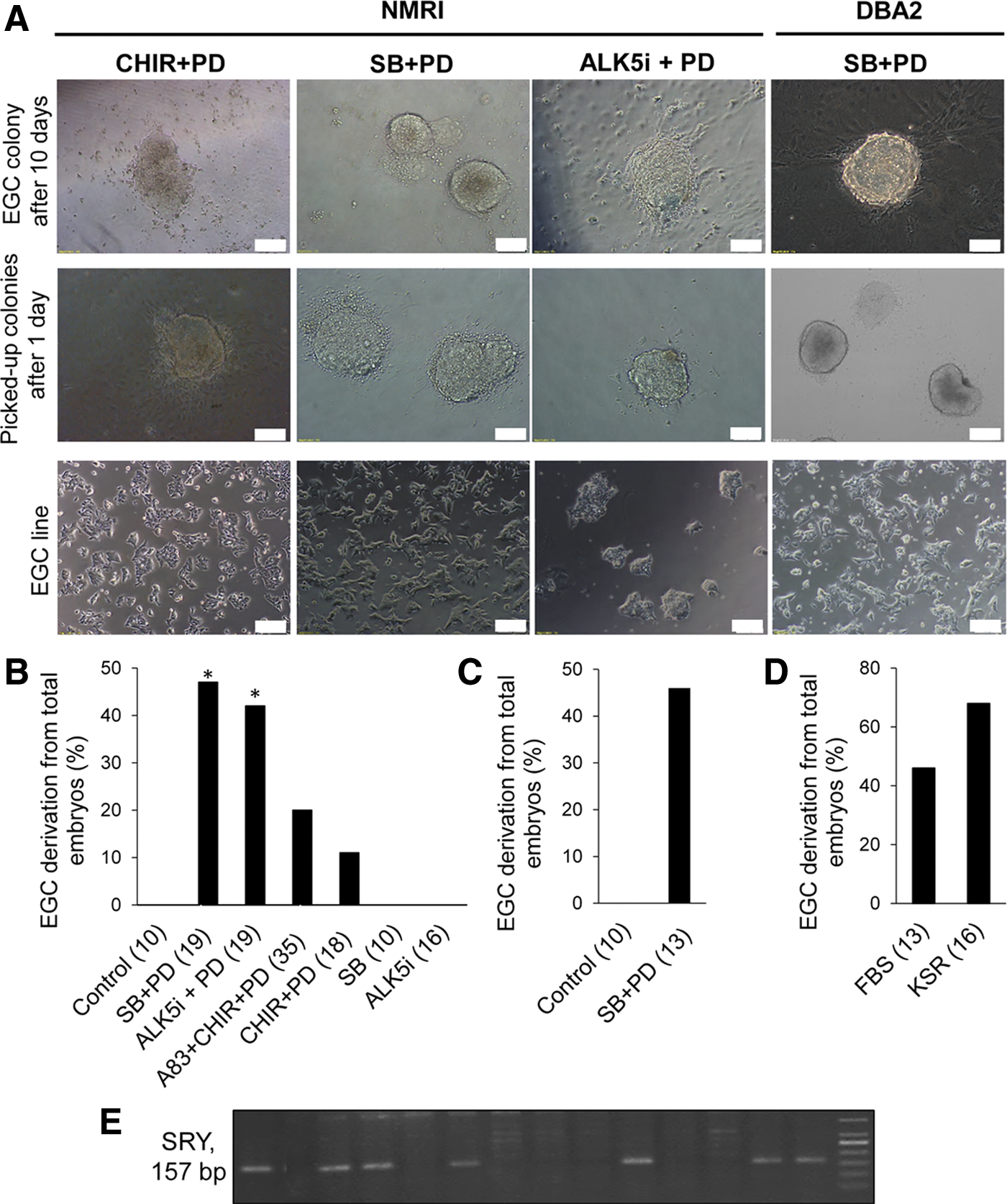
Induction of pluripotency in 8.5 dpc primordial germ cells by SB+PD.
The universality of SB+PD effect on EGC derivation
It has been proven that genetic variations in mice could affect the efficiency of ESC derivation; therefore, some mouse strains are more permissive to ESC derivation whereas others such as NMRI [18] and DBA2 [29] are considered refractory. To confirm the universality effect of SB+PD on the derivation of EGC lines, we performed the exact same protocol on the inbred DBA2 mouse strain. Our findings illustrated that SB+PD induced efficient reprogramming of PGCs to pluripotent EGCs in this strain also. The results were similar to those of the NMRI strain in which EGCs were derived at ∼46% efficiency in the presence of SB+PD (Fig. 2C). Therefore, it seems that simultaneous inhibition of the TGF-β and ERK1/2 pathways would promote EGC emergence in two different genetic backgrounds.
Exploitation of the fully chemically defined medium supplemented with SB+PD for EGC derivation
Since FBS is a biological material its quality varies from batch to batch. KSR, however, is a synthetic and defined combination that has less variability from lot to lot. This formulation is optimized to grow and support undifferentiated ESCs in vitro [30]. As previously suggested, KSR can promote reprogramming compared with FBS that leads to the improvement of ESC and iPS cell generation [1]. Therefore, to further define our medium, we used KSR instead of FBS in day 0 medium at the same concentration (2%). Surprisingly, the presence of KSR in day 0 medium increased the efficiency of EGC derivation up to 68% compared with FBS that had 46% efficiency (Fig. 2D). As a consequence with the use of KSR for just 1 day and N2B27 medium supplemented with SB+PD for the subsequent days, we documented a fully defined medium for a more efficient EGC derivation.
Evaluation of the sex bias on (SB+PD)-derived EGCs
It was previously mentioned that EGC derivation from migrating PGCs was more common from male embryos compared with female embryos [12]. We assessed the sex bias on EGC emergence in the SB+PD condition by evaluating SRY gene expression in the EGC colonies obtained from 8.5 dpc PGCs. Fourteen EGC lines derived from 14 embryos (independent biological samples) were assessed for SRY expression. The SRY expression was observed in 7 out of 14 EGC lines. Our results showed no significant difference between male and female embryos in terms of EGC colony derivation (Fig. 2E). The emergence of EGC colonies in the SB+PD condition did not appear to be affected by embryo's sex.
SB+PD supported EGC derivation from 12.5 dpc embryos
Many PGC characteristics, such as proliferative capacity, change during the developmental process. It has been shown that after entering the embryonic gonad on 12.5 dpc, PGCs meet with cell cycle arrest and at that time EGC derivation becomes more complicated [13]. Under the CHIR+PD condition, derivation of EGCs was observed only from 8.5 dpc PGCs, a time point that has a greater efficiency of EGC emergence. The capability of CHIR+PD for EGC formation from 12.5 dpc embryos was not investigated [15]. However, it was shown that A83+CHIR+PD in 15% KSR medium effectively derived EGCs from 11.5 dpc embryos [13]. Here we investigated the SB+PD potency in EGC derivation from 12.5 dpc embryos. It was demonstrated that the efficiency of EGC derivation was 31% in SB+PD condition and 14% in CHIR+PD condition (Fig. 3A, B). It is of note that the EGC derivation rate from total 12.5 dpc embryos was less than 8.5 dpc embryos (Fig. 3C). This result has shown that similar to 8.5 dpc embryos, EGCs can be derived from 12.5 dpc embryos in SB+PD condition with higher efficiency compared with CHIR+PD condition.
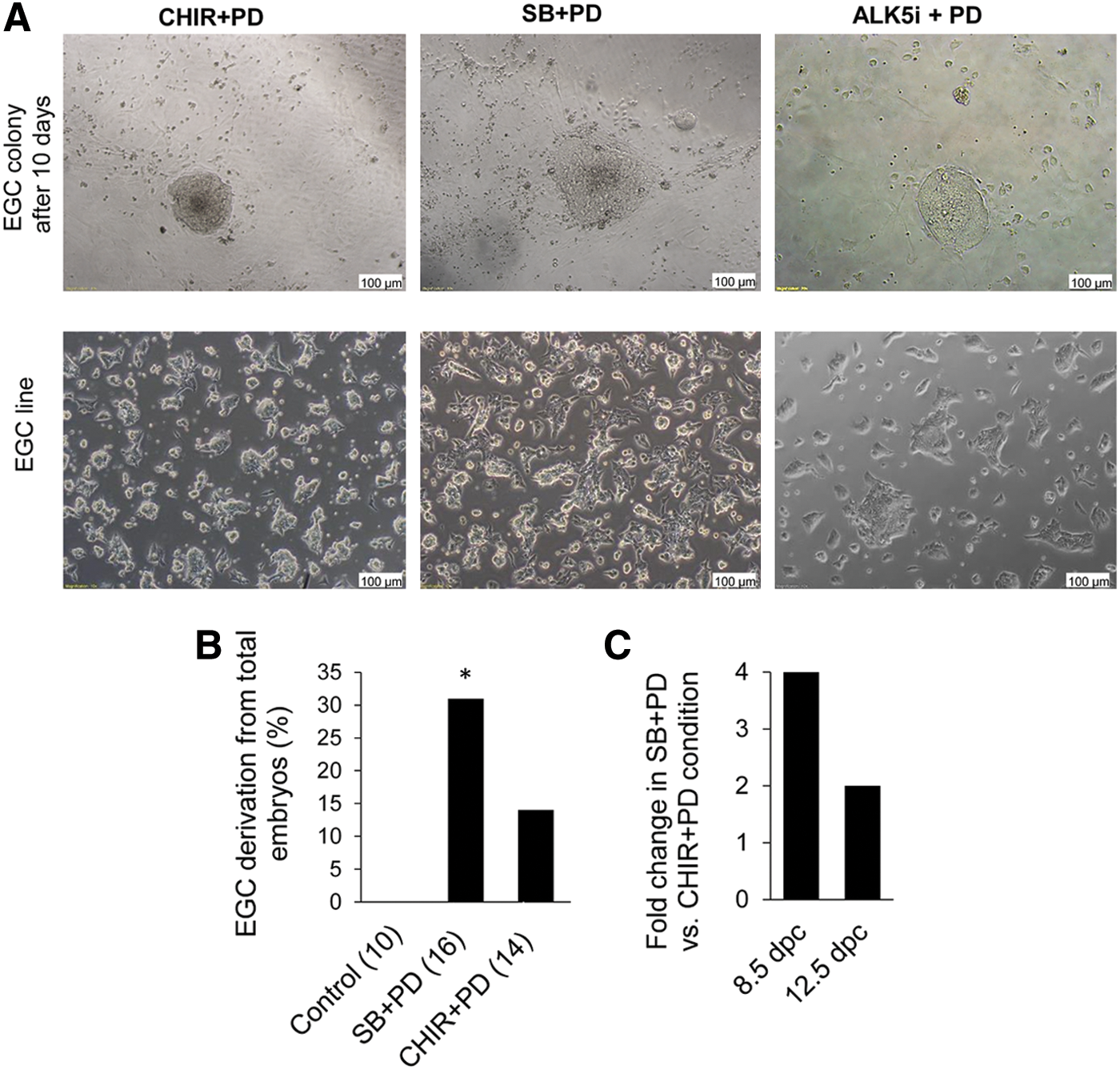
Phase-contrast picture of EGC colonies and EGC lines from 12.5 dpc NMRI mice.
Characteristics of (SB+PD)-derived EGC lines
Because EGC lines are considered to have most of the characteristics of pluripotent stem cells, we have investigated a number of these characteristics in our (SB+PD)-derived EGCs. The established EGC lines from NMRI and DBA2 strains were proven to have passageability criteria for at least 20 passages and high nuclear-cytoplasmic ratios. One line was chosen from each mouse strain for additional assessment of pluripotency characterization (EGRN line from the NMRI strain and EGRD line from the DBA2 strain). mRNA expression of these two EGC lines showed that the investigated pluripotency genes remained active in PGCs and EGCs, whereas levels of germ cell markers such as Blimp1 and Prdm14 reduced after reprogramming of PGCs to EGCs. In confirmation of the previous studies, Klf4 was not expressed in PGCs. However, after reprogramming to EGCs, its expression was augmented (Fig. 4). We evaluated our lines by immunostaining for the pluripotency markers Oct4, Nanog, Sox2, and SSEAl (Fig. 5A, B). Both lines showed the expression of the above proteins. Thirty metaphase spreads were counted for each line based on G-banding. Two independent replicates were prepared for karyotyping. The karyotype analysis showed a normal chromosomal content for the four (SB+PD)-derived EGC lines as well as nondetectable structural aberration (Fig. 5C).
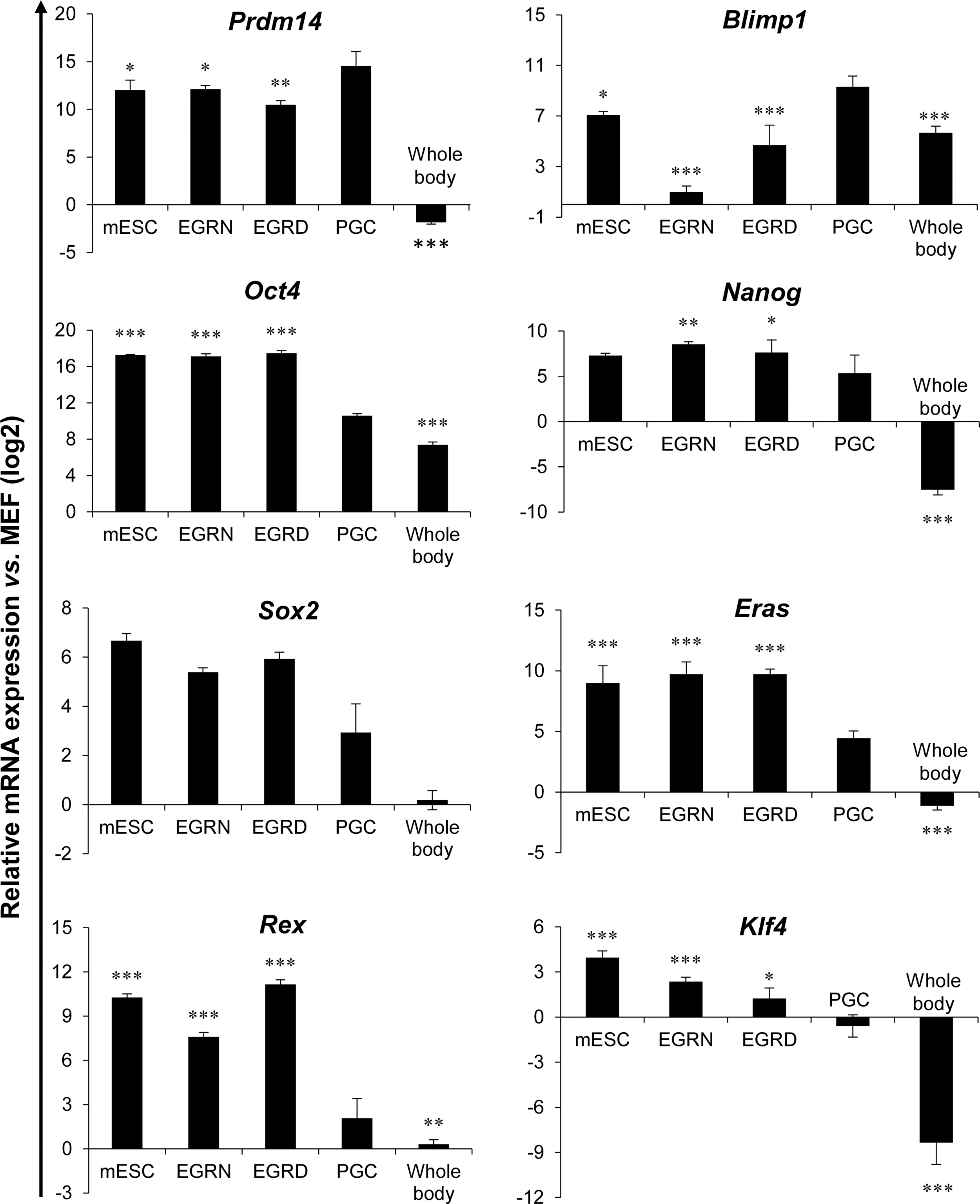
Quantitative reverse transcriptase (RT)–polymerase chain reaction of pluripotency and germ-cell-specific genes. Assessment of pluripotency and germ-cell-specific gene expression in EGRN and EGRD lines from 8.5 dpc embryos after 15 passages. PGCs from 8.5 dpc embryos were sorted by FACS based on the expression of SSEA1 and were used as control samples and the whole bodies of 8.5 dpc embryos that excluded posterior region were used as negative control. The data analysis and sorting of SSEA1-positive cells and their immunohistofluorescence staining in the embryo frontal section were presented in Supplementary Figs. S1 and S2, respectively. Relative expression levels normalized to the housekeeping gene b-Tub and MEFs at passage 1. Data show the mean±SD of three independent replicates. Asterisks indicate significance of each group toward PGCs. *P<0.05, **P<0.01, and ***P<0.001.
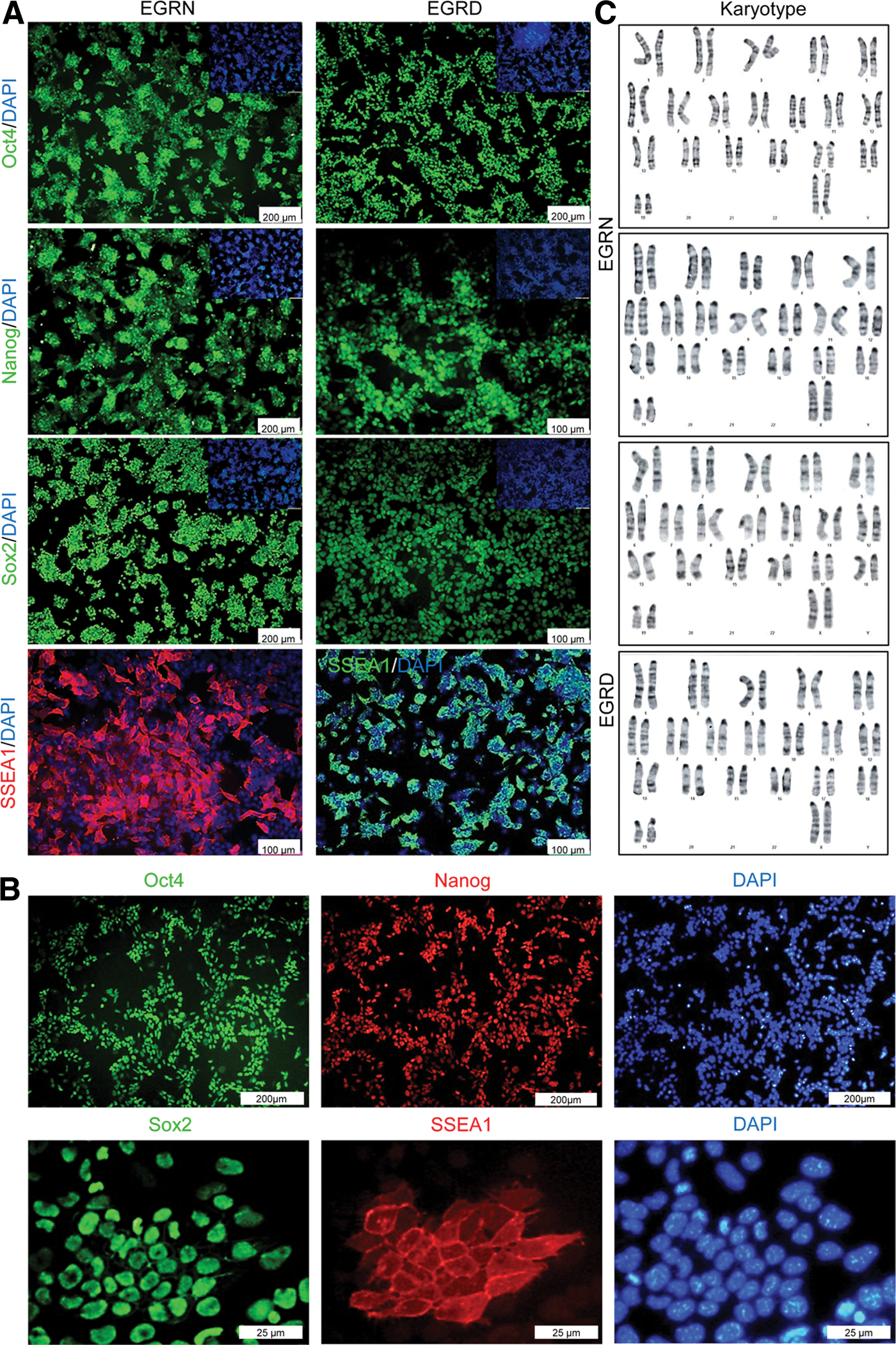
Characteristics of the established (SB+PD)-derived EGC lines from NMRI (EGRN) and DBA2 (EGRD) strains.
Next, we investigated directed differentiation of the generated EGC lines into cardiomyocytes and neuronal cells. Expression of neural (Gfap and Map2) and cardiac genes (Gata4 and Mlc2a) was evaluated (Fig. 6A). Cells that differentiated into cardiomyocytes demonstrated beating (Supplementary Movie S1). Immunofluorescent staining showed expression of cardiac markers α-Mhc, Mef2c, and Connexin 43 in the differentiated cells. In addition, directed neural differentiation of EGCs led to the generation of neurons that were positive for Tuj1 and Map2 in addition to the generation of astrocytes that expressed the Gfap marker (Fig. 6B).
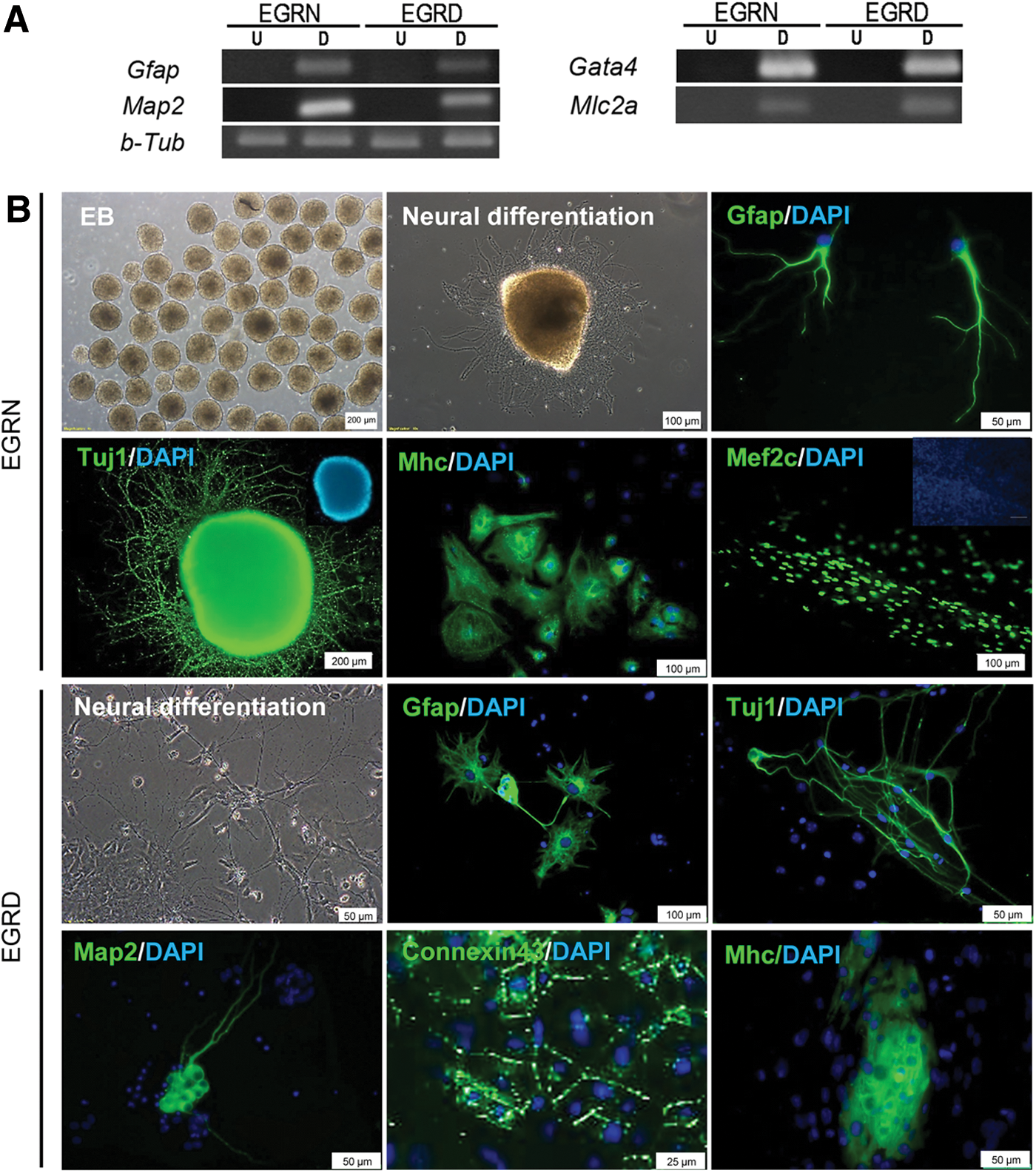
Evaluation of in vitro differentiation capacity in (SB+PD)-derived EGC lines.
To explore further the differentiation capacity of these cells, we subcutaneously transplanted 2–3×106 EGCs from NMRI and DBA2 strains into nude mice. After three weeks post-injection, EGCs gave rise to teratomas that contained all three germ layers (Fig. 7A). Histological analysis showed the presence of neural structures and epidermis (as ectodermal derivatives), bone structure and adipose tissue (as mesodermal structures), and gut structure (as endodermal derivatives) in the teratoma sections. Later, we evaluated the capacity of the DBA2-derived EGC line to contribute in chimeric mice. We injected the mentioned EGC line into 3.5 dpc blastocysts of BALB/c strain mice. Approximately 43 injected blastocysts were transferred to four pseudo-pregnant female mice. There were 17 live pups obtained from pregnant foster mothers of which 9 were chimeric (Fig. 7B) and showed germ-line transmission (Fig. 7C). Overall, gene and protein expression analyses confirmed the pluripotent identity of (SB+PD)-derived EGCs. Considering the above in vitro and in vivo differentiation assays, we confirmed that the EGC lines generated by SB+PD showed stem cell characteristics.
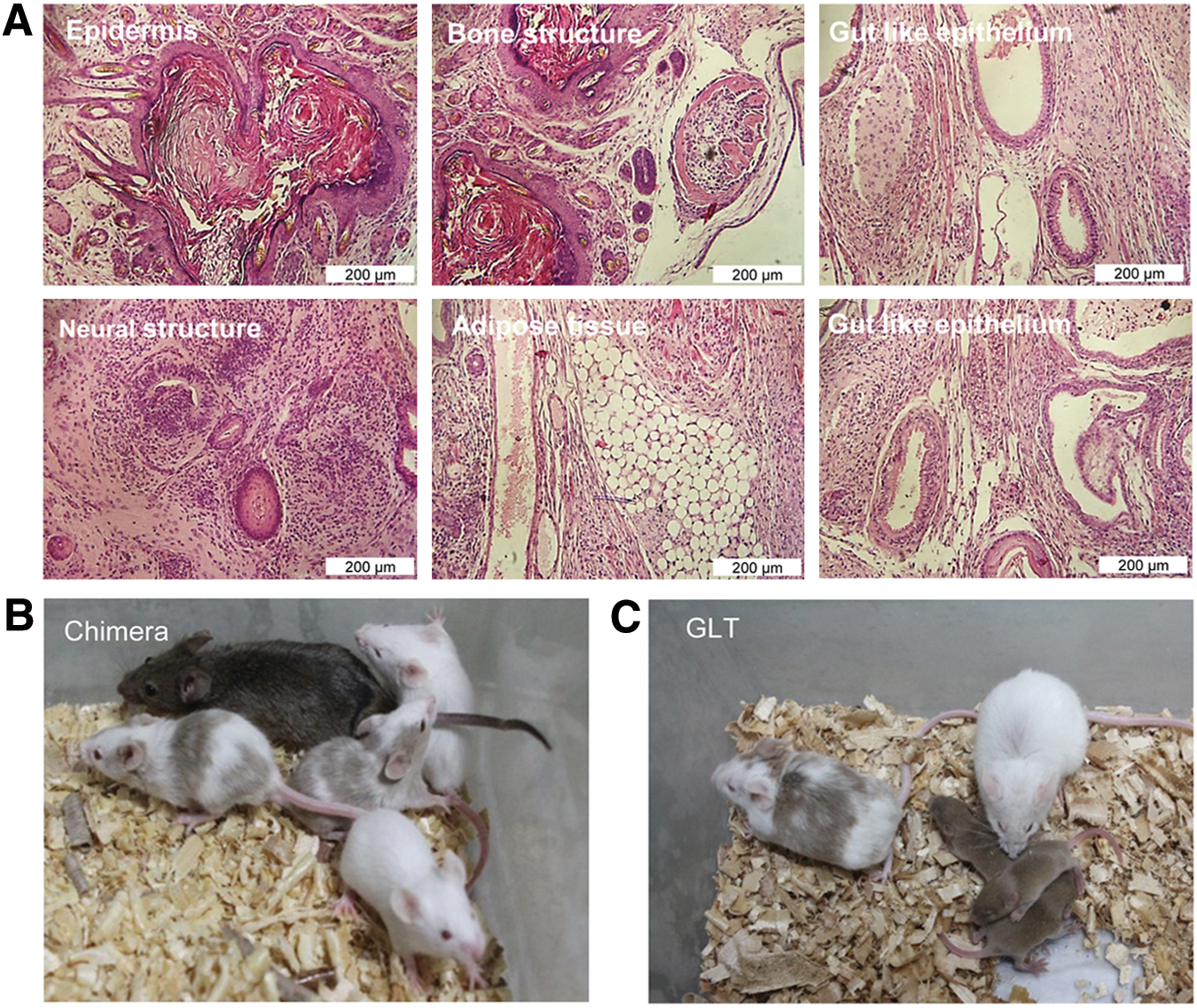
In vivo differentiation capacity of (SB+PD)-derived EGCs.
Discussion
In this study we successfully generated EGCs by using inhibitors of the TGF-β and ERK pathways (by combination of SB431542 plus PD0325901 named as SB+PD). Our findings revealed that EGC derivation and expansion could be performed under the SB+PD condition with greater efficiency compared with other chemically EGC-inductive media, such as CHIR+PD and A83+CHIR+PD.
It has been shown that specific culture conditions in vitro have the capability to force reprogramming on PGCs, leading their transformation into pluripotent stem cells [31]. Approximately 2%–20% of 8.5 dpc PGCs have the capability for transformation toward EGCs. This conversion has been performed under a conventional culture method by culturing PGCs on feeder cells and their exposure to serum and three cytokines, including LIF, bFGF, and SCF [5,9]. Previous reports have shown that the PGC developmental program begins to change after 1 day in culture for which the presence of bFGF in the culture for 24 h is necessary for efficient EGC derivation [32], although it was reported that even in the absence of this factor in CHIR+PD condition EGC colonies can emerge but its efficiency compared with bFGF-containing medium was very low [15]. Therefore, we have used bFGF for 24 h in our culture. It has been previously mentioned that PGCs need supporting cells that produce SCF; their conversion into EGCs occurs with constant exposure to this cytokine [5]. However, in our method, only a 1 day presence of this cytokine was adequate for PGC reprogramming, while the efficiency of this transformation was considerable. The MEF-included condition makes the medium more sophisticated; therefore, its elimination in our protocol could be of benefit with regards to a defined reprogramming condition. However we could not exclude any potential contribution of paracrine factors in the culture from contaminating somatic cells.
To obtain a more defined condition, we used medium containing 2% KSR supplemented with bFGF and SCF for 1 day that followed by substitution of these factors with SB+PD in N2B27 medium for the remaining 9 days. With this method we were able to efficiently induce pluripotency in PGCs compared with the CHIR+PD and A83+CHIR+PD conditions. Of note, this method for EGC derivation appears to be universal and can be generalized to different mouse strains. There are a number of developmental changes that occur during PGC development that result in a more complicated reprogramming with increased age [13]. According to our findings, in our medium, EGC derivation was achievable from not only 8.5 dpc embryos but also from 12.5 dpc fetuses, a more difficult age for EGC derivation. A previously successful attempt at EGC derivation from 11.5 dpc PGCs in A83+CHIR+PD condition has been shown with the use of 15% KSR for the entire culture period [13]. In the current study we have successfully derived EGCs from 12.5 dpc embryos by the use of 2% KSR for 1 day followed by a more defined medium, N2B27+SB+PD, for the remaining days. By using this culture method PGC age was no longer a barrier for transformation in that we have reprogrammed PGCs of 8.5 and 12.5 dpc ages.
The importance of inhibiting the TGF-β signaling pathway in self-renewal and maintaining stem cell pluripotency has been demonstrated [33]. Several reports have shown the positive effects of suppressing this pathway on the generation of iPS cells from mouse fibroblasts [21]. Additionally, we have previously shown that SB+PD application could be beneficial in effective derivation of mouse ESCs from nonpermissive strains [18,19]. In support, it has been documented that inhibition of the TGF-β pathway by A83-01 (A83) accompanied by CHIR+PD increases in EGC derivation compared with the CHIR+PD condition [13]. TGF-β signaling showed an inhibitory role on PGC proliferation [34]. Due to this inhibitory effect, we have predicted that suppression of this pathway would promote PGC proliferation and subsequently enhance the chances of its transformation toward EGC in the SB+PD condition. Our findings demonstrated that SB+PD presented another way to support PGC transformation toward EGCs and particularly clarify the reprogramming process in these cells.
Gene expression of EGCs derived in the SB+PD condition also confirmed these data. We examined expression patterns of eight genes specific for germ cells or pluripotent cells in PGCs, EGCs, ESCs, and MEFs, as the negative control. Our (SB+PD)-derived EGCs showed gene expression profiles similar to those of ESCs, yet they were distinct from the original PGCs. Germ cell markers such as Blimp1 and Prdm14 were highly expressed in PGCs; however, after their reprogramming a significant reduction of these markers was observed in EGCs. Although some of the pluripotency genes, such as Oct4, Nanog, Sox2, and Eras, have high expression levels in PGCs, their expression increases during conversion to EGCs. As another pluripotency marker, Klf4 did not show any expression in PGCs; however, its expression was significantly augmented immediately following the dedifferentiation process, which validated previous reports that the expression of key pluripotency genes Oct4, Nanog, and Sox2 in PGCs was insufficient to push them toward a pluripotent state. It has been shown that the expression of Klf4 along with c-Myc is a prerequisite for manifesting a pluripotency mode. It is believed that the activation of Klf4 and c-Myc expression is actually triggered by downregulation of Blimp1 [31].
Preserving and maintaining genome integrity would be one of the most prominent criteria for an ideal culture condition. In our experiment we have observed a stable cell karyotype under the SB+PD condition. Moreover, SB+PD preserve mouse ESCs with higher genomic integrity following long-term cultivation compared with CHIR+PD [19]. It has been postulated that GSK3 inhibitors have the potential to induce chromosome instability which may in turn promote tumor formation [35]. Eliminating the inhibition of GSK3 in the culture condition in this study was of considerable importance and would obviate the possibility of any chromosomal abnormality. From another perspective more than 40 direct substrates have been reported for GSK3. GSK3 contributes to the control of different signaling pathways, such as Wnt, Notch, mTOR, and insulin [17,36], and partial inhibition of GSK3 in CHIR+PD compound (3 μM) [37] makes the explanation of pluripotency even more complicated. This fact has shown that inhibition of this pathway in the process of EGC derivation will complicate clarification of the reprogramming mechanism toward pluripotency. Recently, we demonstrated that TGF-β inhibition resulted in the efficient production of homogenous and naive ESCs through augmenting BMP4 signaling pathway, as determined by the global transcriptome analysis and the detrimental effect of BMP4 suppression by its agonist noggin on pluripotency [19].
To summarize, we have shown that EGCs can be established and efficiently propagated under the chemically defined SB+PD condition. Elucidating the PGC transformation process and discovering its mechanism of action has some merits that would encourage future research. Somatic cell reprogramming of fibroblasts into iPS cells entails genomic integration [4]. However, of interest, PGCs per se are capable of expressing some pluripotency factors [31]; reprogramming these cells occurs without any need of inserting exogenous factors over a shorter period of time [9]. The determination of this conversion mechanism would shed light on understanding the reason for some tumors that have germ cell origins, the process involved in somatic cell reprogramming, and different aspects regarding stem cells and pluripotency. Future studies will determine whether generation of EGCs by inhibiting the earlier mentioned pathways could be applied to other mammals, particularly rats and humans.
Footnotes
Acknowledgment
This study was funded by grants provided from Royan Institute, the Iranian Council of Stem Cell Research and Technology and the Iran National Science Foundation (INSF).
Author Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.