
Abstract
Skeletal development and cartilage formation require stringent regulation of gene expression for mesenchymal stem cells (MSCs) to progress through stages of differentiation. Since microRNAs (miRNAs) regulate biological processes, the objective of the present study was to identify novel miRNAs involved in the modulation of chondrogenesis. We performed miRNA profiling and identify miR-29a as being one of the most down-regulated miRNAs during the chondrogenesis. Using chromatin immunoprecipitation, we showed that SOX9 down-regulates its transcription. Moreover, the over-expression of miR-29a strongly inhibited the expression of chondrocyte-specific markers during in vitro chondrogenic differentiation of MSCs. We identified FOXO3A as a direct target of miR-29a and showed a down- and up-regulation of FOXO3a protein levels after transfection of, respectively, premiR- and antagomiR-29a oligonucleotides. Finally, we showed that using the siRNA or premiR approach, chondrogenic differentiation was inhibited to a similar extent. Together, we demonstrate that the down-regulation of miR-29a, concomitantly with FOXO3A up-regulation, is essential for the differentiation of MSCs into chondrocytes and in vivo cartilage/bone formation. The delivery of miRNAs that modulate MSC chondrogenesis may be applicable for cartilage regeneration and deserves further investigation.
Introduction
D
Recently, microRNAs (miRNAs) have emerged as key regulators of diverse biological functions, including differentiation processes. miRNAs are small noncoding RNAs of about 22 nucleotides length, which act by annealing to mRNAs and trigger their degradation or repress translation [3]. Currently, ∼1,500 miRNAs are considered to be expressed in humans. Each miRNA putatively targets hundreds of mRNAs, suggesting a regulatory role in gene expression pathways rather than single genes. The first evidence of the impact of miRNAs on chondrogenesis emerged from experiments with knock-out mice, where the Dicer gene was conditionally disrupted in chondrocytes [4]. Dicer deficiency induced defects in skeletal growth, resulting from a reduced number of proliferating chondrocytes through two distinct mechanisms: decreased proliferation and accelerated differentiation into postmitotic hypertrophic chondrocytes. Since then, a few miRNAs have been identified [5]. Most of them have been studied in chondrocytes and shown to be involved in maintaining cartilage integrity and homeostasis [6]. Only a few miRNAs, such as miR-140, −145, −194, and −199a were described as being modulated during the chondrogenic differentiation of MSCs. The best characterized miRNA is miR-140, which was first shown to be up-regulated during the differentiation of human MSCs (hMSCs) and down-regulated in osteoarthritic (OA) cartilage [7]. Next, the same group generated miR-140−/− mice that were characterized by short stature and age-related OA-like changes; conversely, transgenic mice over-expressing miR-140 in cartilage were resistant to antigen-induced arthritis [8]. Interestingly, miR-145, −194, and −199a were shown to repress chondrogenesis [9 –11].
Several miRNAs have been reported as modulating MSC differentiation along other lineages. The miR-29 family is one of the best characterized families of positive regulators of osteogenesis. The expression of all three members (miR-29a, -b, and -c) increased during osteogenesis, and miR-29a and −29c were induced by canonical Wnt signaling [12,13]. MiR-29 members contribute to osteogenic differentiation by targeting fibrosis-associated markers (collagens I, III, and osteonectin) and inhibitors of osteoblast differentiation [Dikkopf-1 (DKK1), secreted frizzled-related protein 2 (sFRP2), histone deacetylase 4 (HDAC4), and transforming growth factor (TGF)-β3,…] [12 –14]. Up-regulation of miR-29b and −29c also plays important roles during myogenesis by inhibiting up-regulation of HDAC4, an inhibitor of muscle differentiation and SMAD3 expression [15 –17]. In myoblasts, miR-29 is negatively regulated by a complex containing Yin Yang1 (YY1) and on differentiation toward myotubes, the derepression and accumulation of miR-29 leads to YY1 inactivation through a regulatory feedback loop [17].
In the present study, a transcriptomic analysis revealed the down-regulation of several members of the miR-29 family during chondrogenesis, with miR-29a being the mostly highly modulated. We reported that the transcription of miR-29a was negatively regulated by SOX9, and subsequent analyses demonstrated that over-expression of miR-29a inhibited the differentiation of hMSCs toward chondrocytes in vitro. Furthermore, FOXO3A was identified as a new direct target of miR-29a. Our findings support a previously uncharacterized function of miR-29a to suppress chondrogenic differentiation of hMSCs likely by repressing FOXO3A and its downstream signaling.
Materials and Methods
Cell culture
Cartilage, primary chondrocytes, and bone marrow-derived MSCs were isolated from healthy or OA patients after informed consent and approval by the Local ethics committee (registration number: DC-2009-1052). Primary human and C3H10T1/2 murine MSCs were cultured as previously described [18]. Typically, primary human MSCs were characterized by the expression of classical markers: CD13, CD73, CD90, and CD105 and the absence of hematopoietic and endothelial markers: HLA-DR, CD11b, CD14, CD31, CD34, CD45, and CD106. They were also shown to differentiate into the three main skeletal lineages: adipocytes, osteoblasts, and chondrocytes (data not shown). Chondrogenesis of MSCs was induced by culture in pellets in the presence of 10 ng/mL TGF-β3 for 21 days, and the osteogenesis of MSCs was initiated by culture in inductive medium as already reported [19].
Chondrocytes were isolated from cartilage cut into small pieces and treated with 2.5 mg/mL pronase (Sigma, L'Isle d'Abeau, France) in Dulbecco's modified eagle medium (DMEM) at 37°C for 1 h followed by 250 U/mL collagenase type II (Sigma) at 37°C overnight (3 mL enzymatic solution/g cartilage). The cell suspension was filtered through a cell strainer (pore size: 70 μm; BD Biosciences, Le Pont-De-Claix, France), centrifuged at 300 g for 10 min, and plated at 2×106 cells/75 cm2. Primary chondrocytes and the cell line C-20A/4 [20] were cultured in DMEM that was supplemented with 10% fetal calf serum, 2 mM glutamine, and 100 U penicillin/streptomycin (Lonza, Amboise, France).
miRNA arrays and data processing
Total RNA was extracted from hMSCs at day 0 or from chondrogenesis-induced cell pellets at day 3 using miRNeasy kit (Qiagen S.A., Courtaboeuf, France). RNA samples were labeled with Cy3 or Cy5, and paired samples were hybridized to dual-channel microarray containing probes from Sanger miRBase Release version 11.0 using a service provider (LC Sciences, Houston, TX). Raw data were normalized by the service provider, and additional data analysis was performed as previously described [21]. Differential regulation of miRNAs in prechondrocytes at day 3 was expressed as a fold change (FC) according to the following formulae: Log2 prechondrocytes signal/MSC signal (d0); P<0.01. A list of differentially expressed miRNA transcripts was produced with a threshold of 1.4 (Supplementary Table S1; Supplementary Data are available online at
RNA isolation and real time-quantitative polymerase chain reaction
RNA was extracted using the miReasy kit according to the manufacturer's instructions (Qiagen). Total RNA (500 ng), including miRNAs, was polyadenylated using E. coli poly(A) polymerase (NEB, Evry, France). After acid phenol/chloroform purification, RNA was reverse transcribed with a poly(T) adapter using the MMLV reverse transcriptase (Life technologies, Saint Aubin, France) and used in SYBR Green PCR (Applied Biosystem, Meylan, France) using the miRNA-specific forward primer and the sequence complementary to the poly(T) adapter as the reverse primer [22]. Briefly, 50 ng cRNA were amplified using specific primers with the SYBR Green PCR kit. The following primers were used: miR-29a, F-TAGCACCATCTGAAATCGGTTA and R-GCGAGCACAGAATTAATACGACT; miR-29b, F-TAGCACCATTTGAAATCAGTGTT and R-GCGAGCACAGAATTAATACGACT; miR-29c, F-TAGCACCATTTGAAATCGGTTA and R-GCGAGCACAGAATTAATACGACT; RPS9, F-AAGGCCGCCCGGGAACTGCTGAC and R-ACCACCTGCTTGCGGACCCTGATA. For differentiation markers, the primers used were as follows: Collagen 2a1.2, F- CAGACGCTGGTGCTGCT and R-TCCTGGTTGCCGGACAT; Collagen 10a1, F-TGCTGCCACAAATACCCTTT and R-GTGGACCAGGAGTACCTTGC; Aggrecan, F-ATGCCCAAGACTACCAGTGG and R- TCCTGGAAGCTCTTCTCAGT; YY1, F-GGCAACAAGAAGTGGGAGCA and R-CCGTGGGTGTGCAGATGTTT; SOX9, F-AGTGCTCAAAGGCTACGAC and R-GTAATCCGGGTGGTCCTTCT; FOXO3A, F-AGTGGATGGTGCGCTGTGT and R-CTGTGCAGGGACAGGTTGT; p21, F-ACCGAGGCACTCAGAGGAG and R-CAGGTCCACATGGTCTTCCT; p27, F-GGACCAAATGCCTGACTCGT and R-GGACCAAATGCCTGACTCGT. Analysis of mRNA expression level was performed using the Roche LightCycler® 480 software1.5. The expression level of cDNA samples was normalized to the expression of reference RPS9 mRNA using the formulae 2−ΔCt or as FC using the formulae 2−ΔΔCt.
DNA constructs and cell transfection
MSCs were transfected with 50 nM of premiR, antagomiR, or siRNA oligonucleotides using Oligofectamine™ reagent following the manufacturer's instructions (Life Technologies). Transfection was done twice, on days −4 and −1 before inducing the differentiation of hMSCs on day 0, by culture in pellets with 10 ng/mL TGF-β3 for 21 days.
C-20/A4 chondrocytes (50,000 cells) were transfected with 1 μg expression plasmids or reporter plasmids using Lipofectamine™ reagent (Life Technologies). SOX9- and YY1-expressing vectors (pcDNA3-SOX9 and pcDNA3-YY1-HA) were previously described [23,24]. FOXO3A 3′UTR was amplified using the platinium taq polymerase kit (Life Technologies) and primers carrying restriction sites for XhoI (forward primer 5′-CCGCTCGAGGCAGTGAGAAATGTGCGAAG) and NotI (reverse primer 5′-ATAAGAATGCGGCCGCTCCCAACTATTCCGTCTACCA) and cloned into the psiCHECK-2™ reporter plasmid (Promega, Charbonnières, France). Mutated FOXO3A 3′UTR was obtained by mutating the miR-29a seed sequence (5′-GTTGTGTTTTCAAAAAGTTATAATATTGTATAGGTGCTTCTGTTTAACCTTGTGAAAGTGTGATTATATTCG), using the QuickChange® Site-Directed Mutagenesis Kit according to the manufacturer's recommendations (Stratagene, Massy, France). Luciferase activity was assessed using the Dual Luciferase® Reporter Assay System (Promega) and expressed as the mean ratio of Firefly luciferase to β-galactosidase activity. For premiR or antagomiR over-expression, chondrocytes were transfected with 50 nM oligonucleotides using Lipofectamine reagent (Life Technologies).
Chromatin immunoprecipitation
C-20/A4 cells were transfected with pcDNA3-SOX9 or pcDNA3-YY1-HA vectors for 48 h and fixed with 1% paraformaldehyde. Nuclei were lysed using RIPA buffer [10 mM Tris-HCl pH8, 1 mM ethylene diamine tetracetic acid (EDTA), 500 μM EGTA, 140 mM NaCl, 1% triton×100, 0.1% Na-deoxycholate, 0.1% SDS, protease inhibitor cocktail (Sigma)] followed by chromatin sonication. Chromatin was immunoprecipitated with 2 μg of antibodies that were specific for SOX9 or HA (Cell Signaling, Ozyme, Saint-Quentin en Yvelines, France) and 25 μL of protein A coupled agarose beads (Sigma), at 4°C, overnight. DNA was then digested with 20 μg/mL of proteinase K (Sigma) and purified by phenol/chloroform extraction (Sigma). Immunoprecipitated DNA (1/100) was used as a template for real time-quantitative polymerase chain reaction (RT-qPCR). The sets of primers were as follows: SOX9-I, F-GCATCAAATTAAAGCTGTGGGTACA and R-CAGCAAGGGGAGGGTTAGGA; SOX9-II, F- CAGGCCCCAAGGAAAGTGAC and R-TGCATGGCAAAAGCTGAACA; SOX9-III, F-CTCAAACCCCAGGGGAAGTG and R-CGCCAGGTACTCCTGTGTGG; SOX9-IV, F-GGAGCAGTGTATGTATGGGTGGTT and R-TTCCTTTTTATCATGTTTTGCTTT; YY1, F-CACTGGTTCAGATGGCTTCATCA and R-GAAATCGCGCCACTGCAC; negative Ctrl, F-TCCTAGGAGGTTGCAGGGACA and R- GGAAGCTGCTGGGAAGTCCT.
Immunoprecipitation and western blot analysis
Cells were lysed using immunoprecipitation (IP) buffer [20 mM Tris-HCl pH8, 137 mM NaCl, 1% Nonidet P-40, 2 mM EDTA, and protease inhibitor cocktail (Sigma)] and incubated at 4°C, overnight, with 2 μg of anti-SOX9 or anti-HA antibodies and 25 μL of protein A-coupled agarose beads (Sigma). After five rinses in IP buffer, 20 μg proteins were loaded on a 10% poly-acrylamide gel and allowed to migrate before transfer onto a PVDF membrane. Western blot analysis was performed using the following antibodies: anti-SOX9 (1:1,000; Cell Signaling), anti-HA (1:1,000; Cell Signaling), anti-FOXO3a (1:500; Abcam, Paris, France), and anti-phosphoFOXO3a (1:500; Millipore, Molsheim, France). Membranes were incubated at 4°C, overnight, with primary antibody in 5% milk (SOX9) or BSA (others), 0.1% tween 20, and, subsequently, with horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h.
Cell cycle analysis
Human MSCs were transfected with specific pre-miRs, antagomiRs, or siRNA as described earlier. Forty-eight hours later, cells were detached with 0.05% trypsin and 0.53 mM EDTA. Subsequently, the cells were fixed with 70% ethanol for 15 min on ice. DNA was stained with 50 μg/mL propidium iodide as previously described [25]. FACS analysis was performed using a Gallios cytometer (Beckman Coulter, Villepinte, France), and results were expressed as the percentage of cells in the G0/G1 or S+G2/M phases of the cell cycle.
Statistical analysis
Statistical analysis was performed with GraphPad Software (San Diego, CA). Values are given as mean±SEM of separate experiments. A comparison between several groups used one-way analysis of variance followed by Dunnett post-hoc test or a Student's t-test for two groups. Differences were considered significant when P<0.05.
Results
Identification of miR-29a as a down-regulated miRNA during early chondrocyte commitment
We investigated the miRNAs that might be involved in the early chondrocyte commitment, using MicroRNA Arrays. We compared miRNA expression profiles between undifferentiated hMSCs (day 0) and prechondrocytes (day 3) and found that among the 109 miRNAs which were significantly modulated, 33 were up-regulated and 76 were down-regulated at day 3 (Fig. 1A and Supplementary Table S1). The mostly highly down-regulated miRNA was miR-29b with an FC: −5.16; while the other two members of the family, miR-29c and miR-29a, were down-regulated with an FC of −3.14 and −1.42, respectively. However, miR-29a was the single member of the family, which was highly expressed in hMSCs (Supplementary Table S1), and this result was confirmed by RT-qPCR (Fig. 1B). We next compared its expression level in hMSCs and primary chondrocytes and found fourfold more miR-29a in hMSCs than in normal or OA cartilage, validating its down-regulation in chondrocytes (Fig. 1C). In agreement with published data, we confirmed that the expression of miR-29a increased by a 5- to 10-fold factor during the osteogenic differentiation of hMSCs and showed that globally, the level was maintained from day 3 till day 21 (Fig. 1D). Finally, we explored the expression kinetics of miR-29a during TGF-β3-induced chondrogenic differentiation of hMSCs. The time-dependent down-regulation of miR-29a was associated with up-regulation of aggrecan (ACAN) and collagen type II variant 2 (COL2A1Δ2), which is the transcript that is specifically expressed in chondrocytes (Fig. 1E–G). The up-regulation of the master regulator of chondrogenesis SOX9 required for the transcription of these genes [26] occurred at day 3 of chondrogenesis and declined thereafter (Fig. 1H). Indeed, the expression of miR-29a is specifically down-regulated during the chondrogenic differentiation of hMSCs.
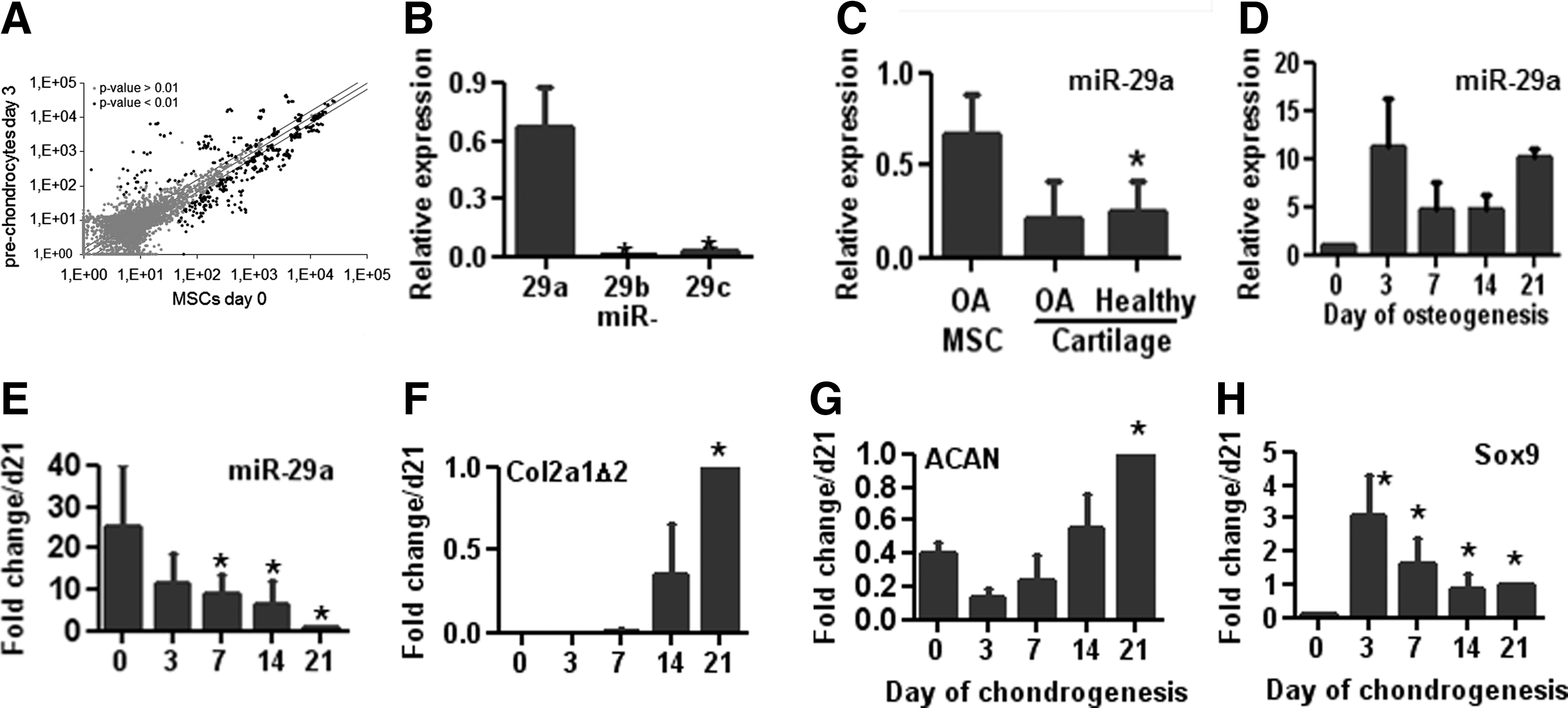
miR-29a is down-regulated during chondrogenesis.
Negative regulation of miR-29a by Sox9 and YY1
To identify potential TFs regulating the expression of miR-29a, we analyzed the regulatory regions of miR-29a promoter using the MIR@nt@n software [27]. Analysis revealed one putative binding sequence for YY1 and four for SOX9 (Fig. 2A); two TFs whose expression was shown to be up-regulated at day 3 of chondrogenesis in a previous analysis [19]. YY1 is a transcriptional repressor of chondromodulin I, a cartilage-specific gene [29]. We, therefore, postulated that the expression of these TFs might down-regulate miR-29a levels. To investigate the role of these TFs, we used the C-20A/4 chondrocyte line, in which the machinery of gene expression is tightly regulated. After the transfection of SOX9 alone or along with YY1, we observed a huge up-regulation of SOX9 (Fig. 2B). Similarly, the up-regulation of YY1 was detected when YY1 was transfected alone or with SOX9 (Fig. 2C). Interestingly, significant down-regulation of miR-29a expression was obtained after the transfection of SOX9 or YY1 but a dramatic effect was observed when both TFs were over-expressed (Fig. 2D). To confirm the regulation of miR-29a by the TFs and determine whether direct binding of the TFs to the miR-29a promoter exists, we performed chromatin immunoprecipitation experiments using anti-SOX9 or anti-HA-tagged YY1 antibodies. As a control, we used COL2 A1 as a known target gene of SOX9. While YY1 did not physically bind to its putative binding sequence, SOX9 bound to three out of four binding sites in the miR-29a promoter (Fig. 2E, F). However, using co-immunoprecipitation technique, we revealed that YY1 co-immunoprecipitated with SOX9, suggesting that these two proteins physically interact (Fig. 2G). Altogether, these results demonstrated that SOX9 binds to the promoter region of miR-29a and negatively regulates its transcription via an additive action of YY1.
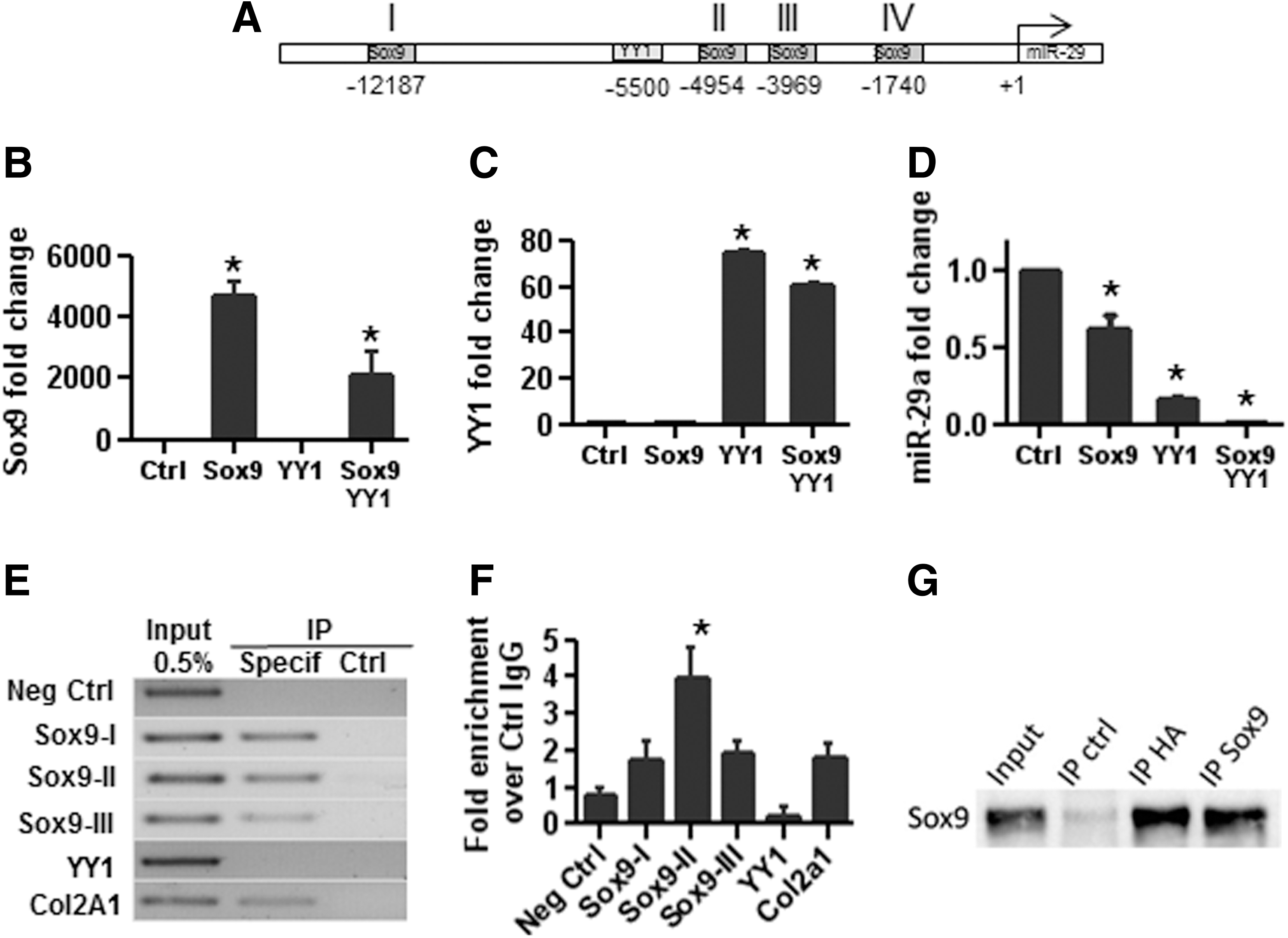
The transcription of miR-29a is repressed by SOX9 and YY1.
Inhibitory role of miR-29a in chondrogenesis
To evaluate the functional role of miR-29a, we transfected hMSCs with specific premiR and antagomiR oligonucleotides twice on days −4 and −1 before initiating the chondrogenic differentiation on day 0. Transfection of premiR-29a induced huge up-regulation of miR-29a at day 7 and 14 in the three individual samples, though variability between samples was observed (Fig. 3A). This was related to a significant inhibition of the chondrocyte markers ACAN and COL2 A1 Δ2 by day 14 and a trend to reduced expression of these markers and of that of COL1 0A1 at day 7 (Fig. 3A). A twofold decrease in the expression of SOX9 was also detected as soon as day 0 after premiR-29a transfection as compared with premiR-Ctrl (Fig. 3B). Moreover, after initiating the differentiation, a huge down-regulation of SOX9 expression was observed at day 1 (100-fold factor) and day 3 (200-fold factor). Using the antagomiR approach, we observed no significant induction of chondrocyte markers, although the average down-regulation of miR-29a by more than 105-fold factor was detected from day 0 to 14 (data not shown). This is likely due to the high down-regulation of miR-29a observed when hMSCs are cultured in chondrogenic conditions.
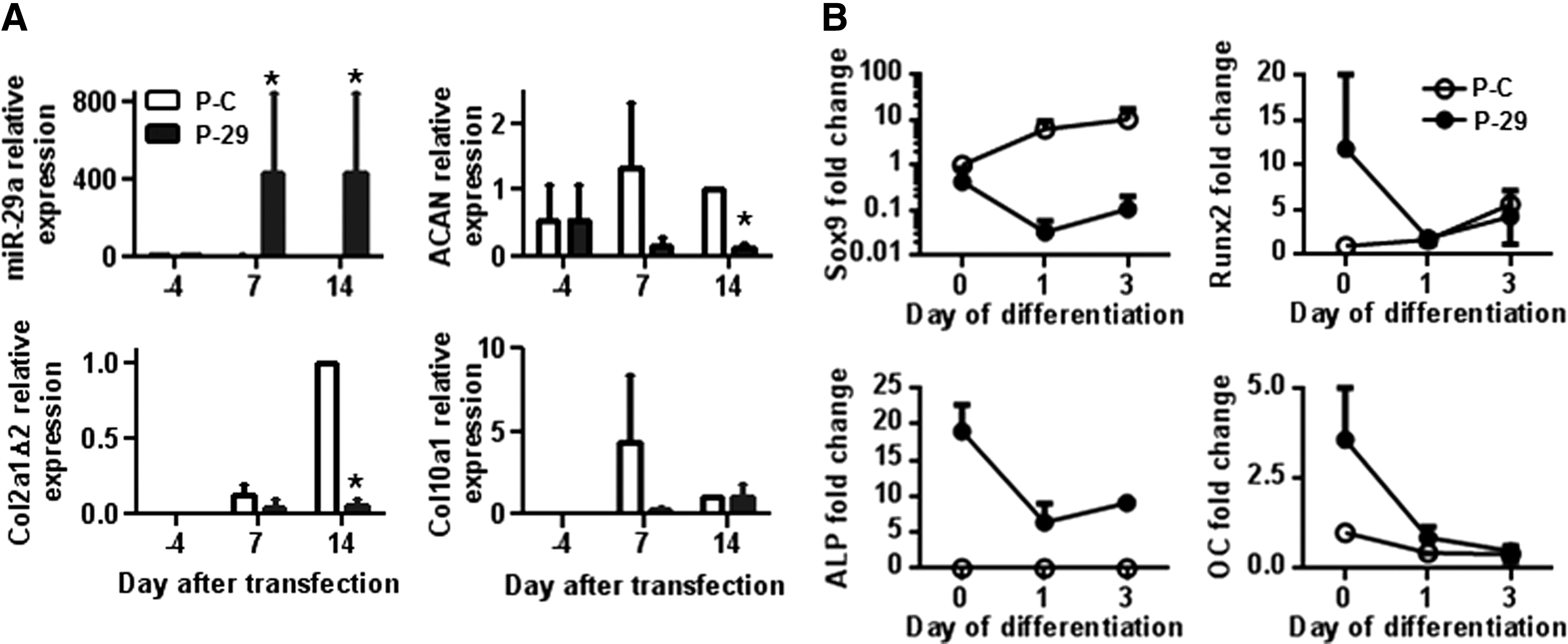
miR-29a inhibits the differentiation of MSCs toward chondrocytes.
Since premiR-29a has been reported to be pro-osteogenic [12,13], we investigated the expression of osteoblastic genes during the early stages of chondrogenic differentiation. As expected, we noticed a high up-regulation of RUNX2, ALPL, and OC after the transfection of premiR-29a (day 0) (Fig. 3B). However, as soon as chondrogenesis was initiated, all these genes were rapidly down-regulated with expression levels similar or close to those observed with premiR-Ctrl. Indeed, the pro-osteogenic effect of premiR-29a was not sufficient to counterbalance the down-regulation of the osteogenic program by chondrogenesis-inducing culture conditions. These data suggest that during chondrogenic differentiation, the inhibitory effect of miR-29a is not associated with a switch of differentiation toward the osteoblast lineage but rather to a inhibition of differentiation.
miR-29a targets Foxo3a and downstream signaling
To understand the molecular signaling underlying the regulation of MSC differentiation by miR-29a, we evaluated the expression of several target genes that have been already validated. We confirmed that over-expressing miR-29a led to the down-regulation of COL 1A1, COL 4A2, and SPARC expression; while the expression of VEGFA was not modulated (Fig. 4). These results confirmed the down-regulation of the osteogenic program (decrease of COL 1A1 and SPARC), while other target genes, such as DKK1 or SMAD3, were not regulated by miR-29a over-expression (data not shown). We also searched for potential new targets and identified FOXO3A using the TargetScan prediction algorithm. To determine whether FOXO3A is a direct target of miR-29a, we cloned the wild-type or mutated 3′UTR seed sequence for FOXO3A-binding site into the 3′UTR of a luciferase reporter plasmid. The reporter plasmids were cotranfected with premiR-29a oligonucleotides into C-20A/4 chondrocytes, and luciferase activity was quantified. We found that miR-29a ectopic expression highly reduced luciferase activity of the wild-type 3′UTR reporter plasmid as compared with that of the control or mutated reporter plasmids, implying that FOXO3A is a direct target of miR-29a (Fig. 5A). To further confirm that miR-29a negatively regulated FOXO3A expression, we used a reporter plasmid where the luciferase expression was dependent on an FOXO3A-responsive promoter [29]. We, therefore, co-transfected this reporter plasmid along with premiR-29a, antagomiR-29a, or siFOXO3A oligonucleotides into C-20A/4 chondrocytes. In these experiments, we revealed the down-regulation of luciferase activity with premiR-29a and siFOXO3A compared with that observed with respective controls (Fig. 5B). The transfection of antagomiR-29a resulted in opposite results. At the protein levels, the transfection of premiR-29a oligonucleotides into hMSCs led to a significant down-regulation of endogenous FOXO3a; while phosphoFOXO3a proteins were increased as compared with premiR-Ctrl. The transfection of antagomiR-29a significantly up-regulated expression of both forms (Fig. 5D–E).
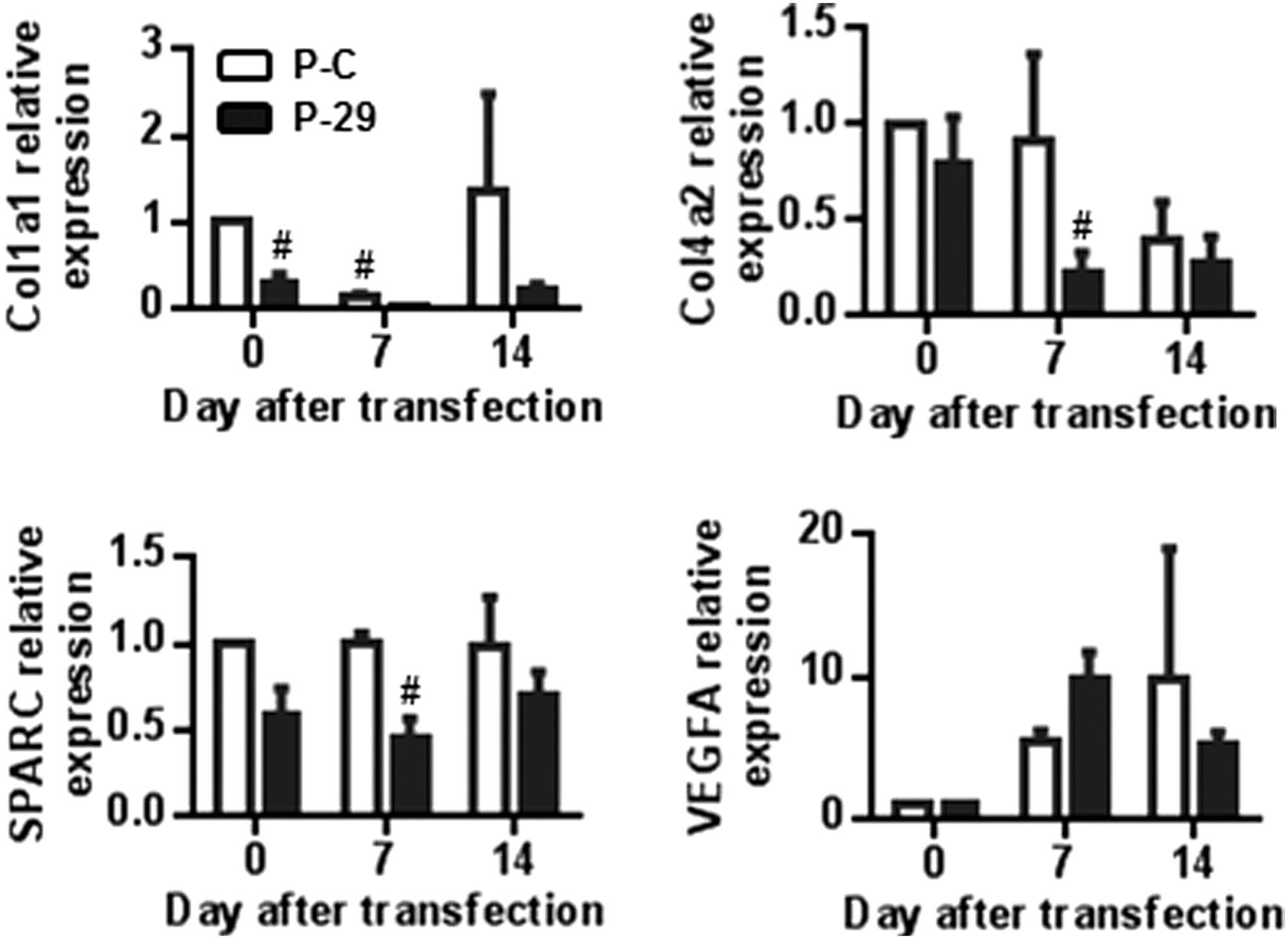
Differential expression of target genes of miR-29a during chondrogenesis. Expression of already validated target genes for miR-29a [collagens type I and type IV (COL1A1 and COL4A2); osteonectin (SPARC); and vascular epithelial growth factor alpha (VEGFA)] at different time points during the chondrogenic differentiation of three individual hMSC samples previously transfected with premiR-control (P-C) or premiR-29a (P-29). # P<0.01.

FOXO3A is a target of miR-29a.
In parallel, we evaluated the kinetics of FOXO3A expression during chondrogenesis. We previously described that the expression of FOXO3A progressively increases during chondrogenesis of murine MSCs [30]. Here, we confirmed the up-regulation of FOXO3A, which was negatively correlated with miR-29a expression, during the chondrogenic differentiation of hMSCs (Fig. 5C). Finally, we showed a negatively correlated expression of miR-29a and FOXO3A in hMSCs transfected with premiR-29 as compared with premiR-Ctrl and cultured in chondrogenic conditions (Fig. 5G, H). In these experiments, even though the down-regulation of FOXO3A was not significant due to variability in transfection efficiencies of primary MSCs from different individuals, the expression of FOXO3A tended to decrease by a 5.7-fold factor. Together, these results suggest that FOXO3A is a target of miR-29a.
Down-regulation of Foxo3a inhibits chondrogenic differentiation
Finally, we aimed at evaluating how miR-29a-mediated down-regulation of FOXO3A may result in chondrogenesis inhibition. First, we wanted to confirm the effect of FOXO3A down-regulation on chondrogenesis by investigating the effect of siRNAs against FOXO3A on the differentiation of hMSCs. We found that siFOXO3A transfection resulted in impaired formation of cartilaginous pellets from hMSCs. Pellets were flat and yellowish instead of round and glossy as observed with control siRNA (Fig. 6A). Accordingly, FOXO3A down-regulation inhibited chondrogenesis with lower expression of the chondrocyte specific markers, COL2A1 Δ 2 and ACAN (Fig. 6B). In subsequent experiments, we observed that FOXO3A over-expression in C-20A/4 chondrocytes up-regulated SOX9 expression, while miR-29a was repressed (Fig. 6C). We previously showed that SOX9 inhibited miR-29a, which, in turn, inhibited FOXO3A; the overall effect of SOX9 is, therefore, FOXO3A induction. These results suggested that FOXO3A expression is important for SOX9 regulation and exerts a positive feedback loop to enhance SOX9 levels and, as a result, FOXO3A during chondrogenesis. Interestingly, FOXO3A up-regulation and related enhanced SOX9 expression was associated with increased expression of FOXO3A-targeted genes, p21 and p27, which exhibit anti-proliferative function. Due to this and data reporting that deletion of FOXO3A leads in some stem cell populations to increased cell cycle progression (for review, see Ref. [31]), we evaluated the effect of premiR-29a or siFOXO3A on the cell cycle of hMSCs. Using both approaches, the down-regulation of FOXO3A resulted in enhanced cell proliferation as shown by lower percentage of cells in the G0/G1 phases (Fig. 6D) and higher percentage of cells in the S+G2/M phases (Fig. 6E). These results enabled us to conclude that FOXO3A is required for MSC commitment toward chondrocytes by repressing cell cycle progression.
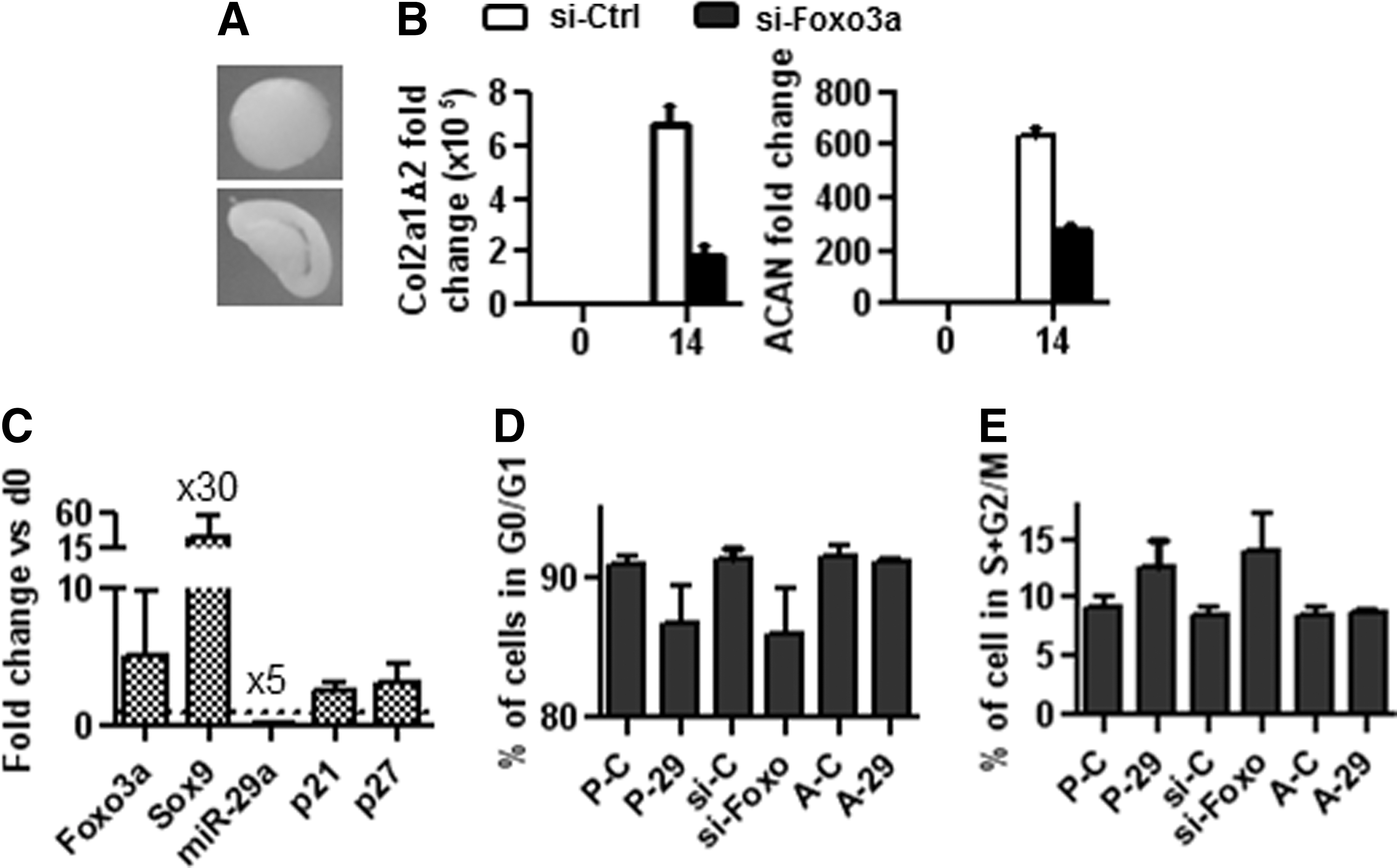
FOXO3A is required for chondrogenic differentiation.
Discussion
Here, we report a previously uncharacterized role of miR-29a in the inhibition of chondrogenic differentiation of MSCs. The key finding of our study is that miR-29a negatively regulates chondrogenesis likely by targeting FOXO3A, which mediates the survival and proliferation of MSCs. In other words, miR-29a expression and FOXO3A inhibition are required for the maintenance of the undifferentiated, proliferative state of MSCs.
There are four miR-29 members: miR-29a and −29b1, on the one hand, and miR-29c and miR-29b2, on the other hand, which are transcribed within two different pri-miRNAs. miR-29b1 and miR-29b2 have identical mature sequences, which are together called miR-29b. All mature miR-29s share identical seed sequences, and, therefore, predicted target genes for the family members largely overlap [32]. There is now strong evidence that the members of the miR-29 family play important roles in regulating fibrosis by targeting several genes related to extracellular matrix, immune response, apoptosis/cell proliferation through the inhibition of oncogenes or anti-apoptotic genes, and differentiation. For example, miR-29s were previously attributed a pro-osteogenic function in MSCs or osteoblast progenitors as well as a pro-myogenic effect in primary muscle cells [13 –15], while they inhibit the differentiation of naïve T lymphocytes toward Th1 pro-inflammatory lymphocytes [33]. Indeed, the expression of miR-29 increases during osteoblastic differentiation, and miR-29 over-expression up-regulates osteogenesis [12,13]. The activation of osteogenic differentiation functions primarily by directly targeting inhibitors of signaling pathways that are required for osteogenesis (TGF-β3, HDAC4, DKK-1, KREMEN2, and SFRP2) and attenuating collagen expression (COL 1A1, COL 4A2, and COL 5A3) in mature osteoblasts to maintain the differentiated phenotype [13]. During myogenesis, the expression of miR-29 is also up-regulated [15]. The increased expression of miR-29 reduces HDAC4 protein, which acts as a co-repressor of muscle regulatory factors through the direct inhibition of MEF2. Furthermore, treatment with TGF-β1, a well-characterized inhibitor of myogenesis, induces increased expression of HDAC4 by down-regulating miR-29 expression. Interestingly, in that study, the authors found that miR-29 can dominantly suppress TGF-β signaling through the inhibition of Smad3 expression, explaining the fact that miR-29 can reduce HDAC4 expression required for induction of the myogenic program despite potentially high levels of TGF-β. Its role during the differentiation of MSCs toward chondrocytes has not been addressed. In the present study, we, therefore, investigated the role of miR-29 during the chondrogenic differentiation of MSCs. We found that the expression of miR-29a was high in MSCs and progressively decreased during chondrogenesis, while markers of chondrocytes appeared. We further demonstrated that over-expression of miR-29a resulted in the inhibition of chondrogenesis in vitro and of endochondral ossification in vivo (data not shown). We, therefore, provided evidence that miR-29a is a negative regulator of the chondrogenic differentiation of MSCs.
One important finding of our study relates to the transcriptional regulation of miR-29a in chondrocytes. Previous studies have reported the negative regulation of miR-29 expression by various transcriptional regulators and signaling pathways such as C-MYC, HEDGEHOG, WNT, or NF- KКB signaling [32]. On the other hand, CCAAT/enhancer binding protein alpha (CEBPA) was reported to activate miR-29 transcription [34]. Here, we demonstrated that SOX9 can physically bind to at least three out of its four putative binding sites within the proximal miR-29a/b1 promoter and negatively regulate miR-29 transcription. This finding strongly suggests that SOX9 is the direct regulator of miR-29a transcription. During the chondrogenesis of MSCs, SOX9 likely acts as the missing link between TGF-β-induced Smad3 signaling, as shown in muscle cells [15], and negative regulation of miR-29a. Moreover, we found that SOX9 acts in synergy with the TF YY1 to suppress miR-29a expression. Although we observed the regulation of miR-29 transcription by YY1, we were unable to detect direct binding of YY1 within the promoter of miR-29. However, during chondrogenesis, YY1 may recruit SOX9, as suggested by the co-immunoprecipitation experiment, and act as a transcriptional corepressor of miR-29. This last hypothesis is further supported by one study reporting that in MSCs, YY1 binds to the core-promoter region of chondromodulin (ChM)-I, which is a specific gene for cartilage tissue, and represses ChM-I transcription; whereas in chondrocytes, the transcriptional repression of YY1 is relieved by p300 [28,35]. We, thus, may hypothesize that, during chondrogenic differentiation, the binding of YY1 to the miR-29 promoter is displaced and YY1 preferentially binds to SOX9 and acts as a transcriptional co-repressor of miR-29a, resulting in chondrocyte differentiation.
The other new finding of our study is the demonstration that FOXO3A is a direct target of miR-29a. Several targets of miR-29s have already been described in other cell models that may explain their effect on chondrogenesis. Indeed, the regulation of extracellular matrix genes, in particular several collagen isoforms and integrin β1, which have important functions for the generation and stability of the chondrocyte phenotype as well as a role in fibrosis occurrence, has been reported [36]. In addition, miR-29s target known inhibitors of osteoblast differentiation, among which are TGF-β3, activin A receptor IIA, DUSP2, and antagonists of the pro-osteoblastic canonical WNT pathway [12,13]. The importance of this signaling pathway was reported several years ago when genetic inactivation of β-catenin was reported to cause ectopic formation of chondrocytes at the expense of osteoblast differentiation [37]. Therefore, after the inhibition of miR-29a by SOX9, WNT-induced signaling pathways will be shut off by enhanced expression of antagonists, resulting in osteogenesis inhibition. Interestingly, miR-29a directly targets HDAC4, which inhibits the TFs RUNX2 and MEF2 C, which are involved, respectively, in osteogenic and myogenic differentiation of mesenchymal progenitors as well as terminal differentiation of chondrocytes toward hypertrophy [38,39]. Moreover, in the presence of TGF-β1, HDAC4 was reported to speed up and maintain high levels of chondrogenesis [40]. In the present study, we confirmed the down-regulation of some of the miR-29a targets during chondrogenesis that might participate in controlling chondrogenesis inhibition. However, in addition to the mechanisms earlier, we propose that miR-29a may inhibit chondrogenesis by controlling the expression of FOXO3A. Although FOXO3A is the only member of the FOX family having a putative target sequence for miR-29a, we also checked that at least two other family members, FOXA3 and FOXO1A, are not regulated by miR-29a (data not shown). As a result of miR-29a inhibition by SOX9, FOXO3A expression increased during chondrogenesis, and we have previously reported that it was correlated with increased apoptosis [30]. In this last study, the down-regulation of FOXO3A resulted in increased differentiation. We confirmed here, using human MSCs, the up-regulation of FOXO3A during chondrogenesis, which was negatively correlated with miR-29a expression. Moreover, the forced expression of miR-29a reduced FOXO3A mRNA expression at early steps of MSC differentiation, and this resulted in chondrogenesis inhibition. While this result may appear contradictory with our previous report, it is likely that transient down-regulation of FOXO3A by RNA interference did not lead to a similar effect of stable expression of an siRNA in cell clones. Moreover, we demonstrate that the down-regulation or over-expression of FOXO3A, respectively, resulted in the up- or down-regulation of SOX9, ACAN, and COL 2A1. These three genes were identified as putative targets, as they contain FOXO3A-binding sequences on their genomic sequences. Although we cannot exclude that FOXO3A and miR-29a may use different mechanisms of action, we demonstrate, for the first time, that FOXO3A is required for the chondrogenesis of adult MSCs.
FOXO3 expression in osteoblasts has been shown to be indispensable for skeletal homeostasis in mice [41]. The deletion of FoxO3 resulted in decreased number of osteoblasts and bone formation. Conversely, the over-expression of a FoxO3 transgene in mature osteoblasts increased bone formation. However, the function of FoxO3a during chondrogenesis is not fully clarified, even though some studies have been performed on mesenchymal cells isolated from mouse embryos at stages E12.5 or E15.5 [42]. In that study, the authors nicely showed that the Akt-FoxO pathway enhanced chondrocyte proliferation but inhibited chondrocyte maturation and cartilage matrix production. More recently, another study reported that continued expression of Sox9 in differentiated chondrocytes was essential to sustain survival mechanisms by binding to the Pik3ca promoter, inducing AKT phosphorylation [43]. During the chondrogenic differentiation of adult MSCs, in parallel to the activation of the PI3K-Akt pathways, Sox9 might up-regulate FoxO3a expression via the inhibition of miR-29a. This dual activation could result in an enhanced phosphorylated form of FOXO3a, inducing survival mechanisms and differentiation. The fact that over-expressing FOXO3A results in SOX9 up-regulation strongly suggests that it may control chondrogenesis, but FOXO3A may also act via a regulatory feedback loop mechanism and maintain SOX9 expression (Fig. 7). Indeed, although not formally proved in the present study, we could hypothesize that SOX9 induction of cell survival mechanisms through FOXO3A might be a regulator for chondrogenic differentiation.
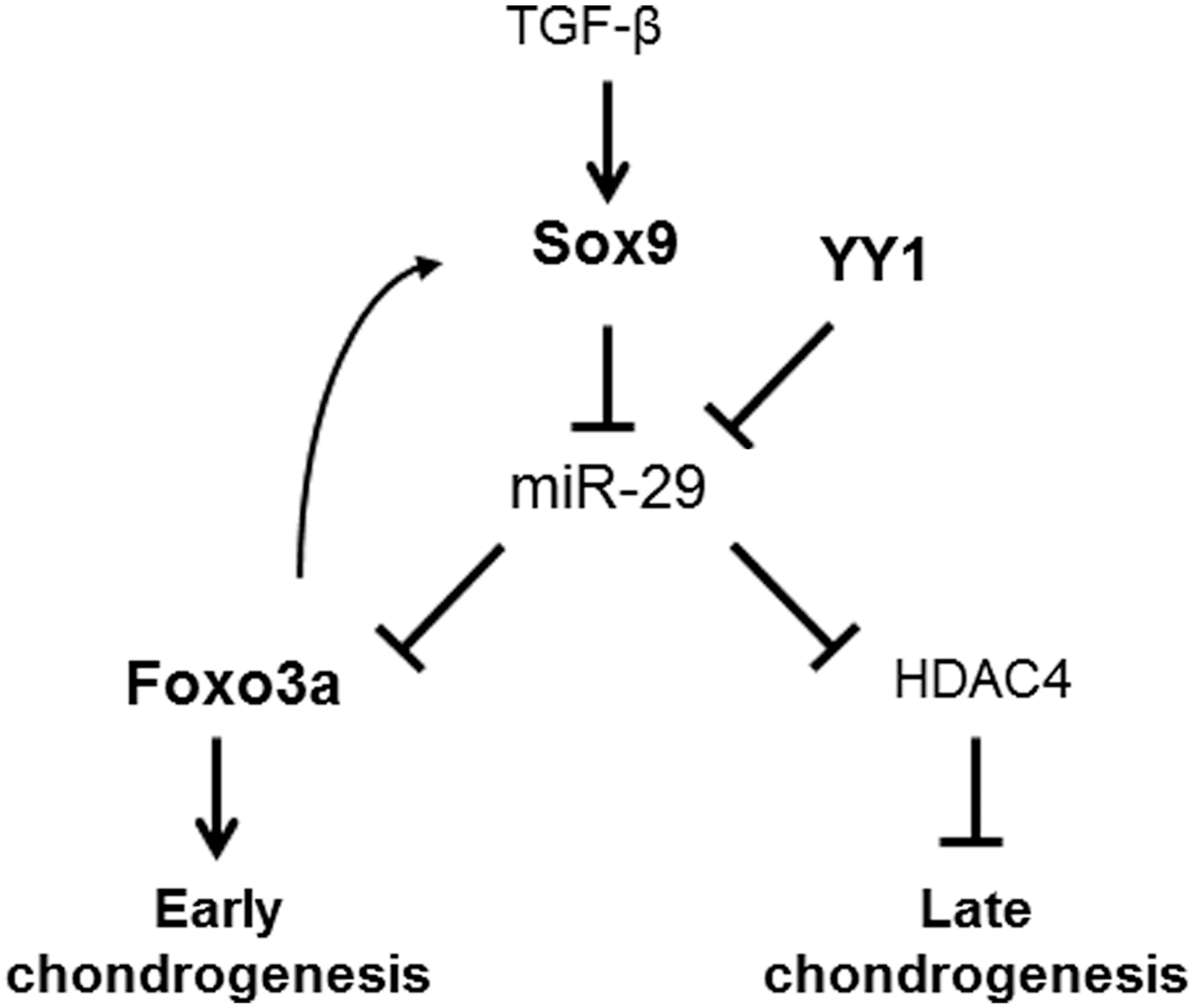
A proposed model of the mechanisms by which miR-29a expression controls chondrogenesis commitment. The inhibitory effect of TGF-β on miR-29 expression through Smad3-dependent pathway and up-regulation of HDAC4 expression, as previously reported 15 (normal case), is directly mediated via SOX9 during chondrogenesis (bold case). YY1 cooperates with SOX9 and inhibits miR-29 expression, resulting in FOXO3A induction and chondrogenesis (bold case). HDAC4, histone deacetylase 4; TGF, transforming growth factor.
In conclusion, we report here that the down-regulation of miR-29a by SOX9 is required for MSCs to differentiate into chondrocytes, and this might be mediated through FOXO3A up-regulation. Both over-expression of miR-29a and inhibition of FOXO3A, using a specific siRNA, in MSCs causes a decrease of chondrocyte markers in vitro. These findings imply that FOXO3A inhibition is likely to be involved in the maintenance of the multipotent state of MSCs. As a consequence, the increase of FOXO3A expression is required for chondrogenesis. This may be of relevance for the use of MSCs in tissue engineering approaches.
Footnotes
Acknowledgments
The authors are very grateful to Charles Le Cellier (CNRS UMR5535, Montpellier, France) for helpful discussions and A. Brunet (Stanford University, California), Ph. Blache (UMR 5237, Montpellier, France), and D.C. Guttridge (Ohio State University, Ohio) for kindly providing, respectively, the FKHRL1, Sox9, and YY1-HA plasmids. Work in the laboratory Inserm U844 was supported by the Inserm Institute, the University of Montpellier I and funding was provided by the Fondation pour la Recherche Médicale (FRM). D. Philipot was granted by “Région Languedoc-Roussillon and University of Montpellier I.” The authors thank the “Réseau des Animaleries de Montpellier” animal facility, the “Réseau d'Histologie Expérimentale de Montpellier” histology facility for processing their animal tissues, and the “Montpellier RIO Imaging” platform.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.