
Abstract
The gene Lrrc34 (leucine rich repeat containing 34) is highly expressed in pluripotent stem cells and its expression is strongly downregulated upon differentiation. These results let us to suggest a role for Lrrc34 in the regulation and maintenance of pluripotency. Expression analyses revealed that Lrrc34 is predominantly expressed in pluripotent stem cells and has an impact on the expression of known pluripotency genes, such as Oct4. Methylation studies of the Lrrc34 promoter showed a hypomethylation in undifferentiated stem cells and chromatin immunoprecipitation–quantitative polymerase chain reaction analyses of histone modifications revealed an enrichment of activating histone modifications on the Lrrc34 promoter region. Further, we could verify the nucleolus—the place of ribosome biogenesis—as the major subcellular localization of the LRRC34 protein. We have verified the interaction of LRRC34 with two major nucleolar proteins, Nucleophosmin and Nucleolin, by two independent methods, suggesting a role for Lrrc34 in ribosome biogenesis of pluripotent stem cells. In conclusion, LRRC34 is a novel nucleolar protein that is predominantly expressed in pluripotent stem cells. Its altered expression has an impact on pluripotency-regulating genes and it interacts with proteins known to be involved in ribosome biogenesis. Therefore we suggest a role for Lrrc34 in ribosome biogenesis of pluripotent stem cells.
Introduction
T
Most pluripotency regulators like Oct4 [2], Nanog [3], and Sox2 [4] are known to act as transcription factors thereby controlling the network of pluripotency-regulating genes. Therefore we suggest putative pluripotency-regulating genes to be endowed with protein domains known to be responsible for DNA recognition. Today, transcription factor proteins are divided into eight groups (for review see Luscombe et al. [5]): (1) helix-turn-helix proteins, (2) zinc-coordinating proteins, (3) zipper-type proteins, (4) other α-helix proteins, (5) β-sheet proteins, (6) β-hairpin/ribbon proteins, (7) other proteins, and (8) enzymes.
The in silico analysis of mouse and human Lrrc34 genes and deduced protein structures reveal two leucin rich repeat (LRR) domains each harboring several leucin repeats (Fig. 1A). LRR domains with typically 20–29 leucin repeats are known to play a role in diverse biological functions (for review see Kobe and Kajava [6]). Proteins of the LRR family were shown to be frequently involved in protein–protein interactions. However, also a possible function as transcription factor is described. It was shown that the LRR motifs are highly conserved in evolution and appear in a variety of transcription factors from fungi, plants, and animals. If acting as transcription factor, then one distinct subdomain of the motif acts as a leucin zipper mediating dimerization of the protein while the other basic region interferes with the DNA (for review see Pabo and Sauer [7]). There are many subfamilies of LRR proteins known and four are described in animals: the RI-like subfamily, the SDS22-like subfamily, the cysteine-containing subfamily, and the typical subfamily. They vary in the length of the LRR and the cellular localization [6].
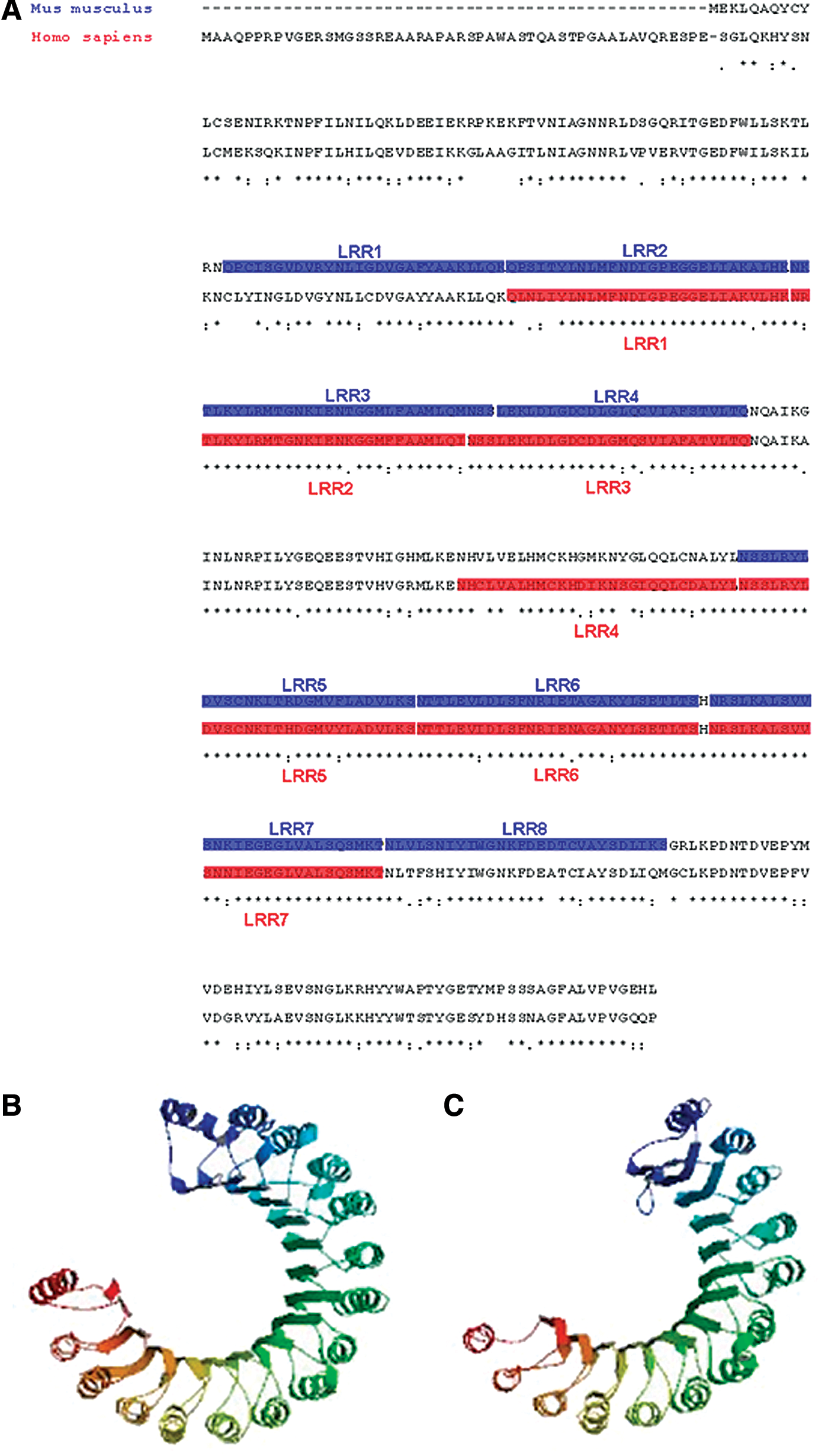
Comparison of the amino acid sequences of mouse and human LRRC34 protein including domain structures.
Lrrc34 is a predicted member of the RI-like subfamily. The crystal structure of the RI-LRR protein shows that it consists of β-strands with α-helices (Fig. 1B) [8]. A prediction of the mouse Lrrc34 protein structure using the ModBase Database of Comparative Protein Structure Models (
Materials and Methods
Cell culture
ESC and maGSC lines were cultured as previously described [11]. For differentiation, cells were plated on 0.1% gelatin-coated dishes without leukemia inhibitory factor and in the presence of 1 μM retinoic acid (RA) for 20 days (Fig. 2B) or indicated time points (Fig. 3B). All other cell lines were cultured in standard conditions in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum.
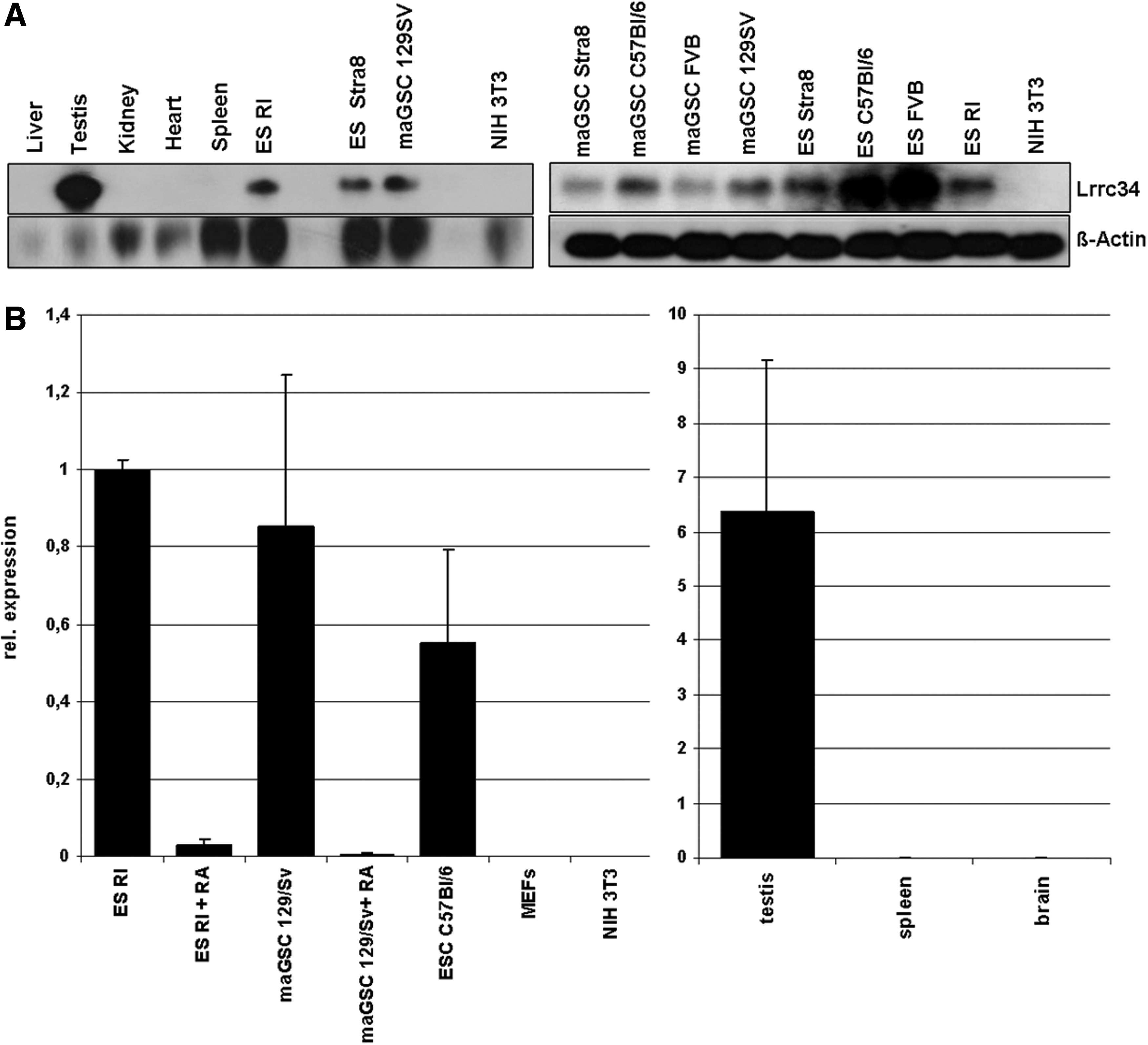
Expression analysis of mouse Lrrc34.
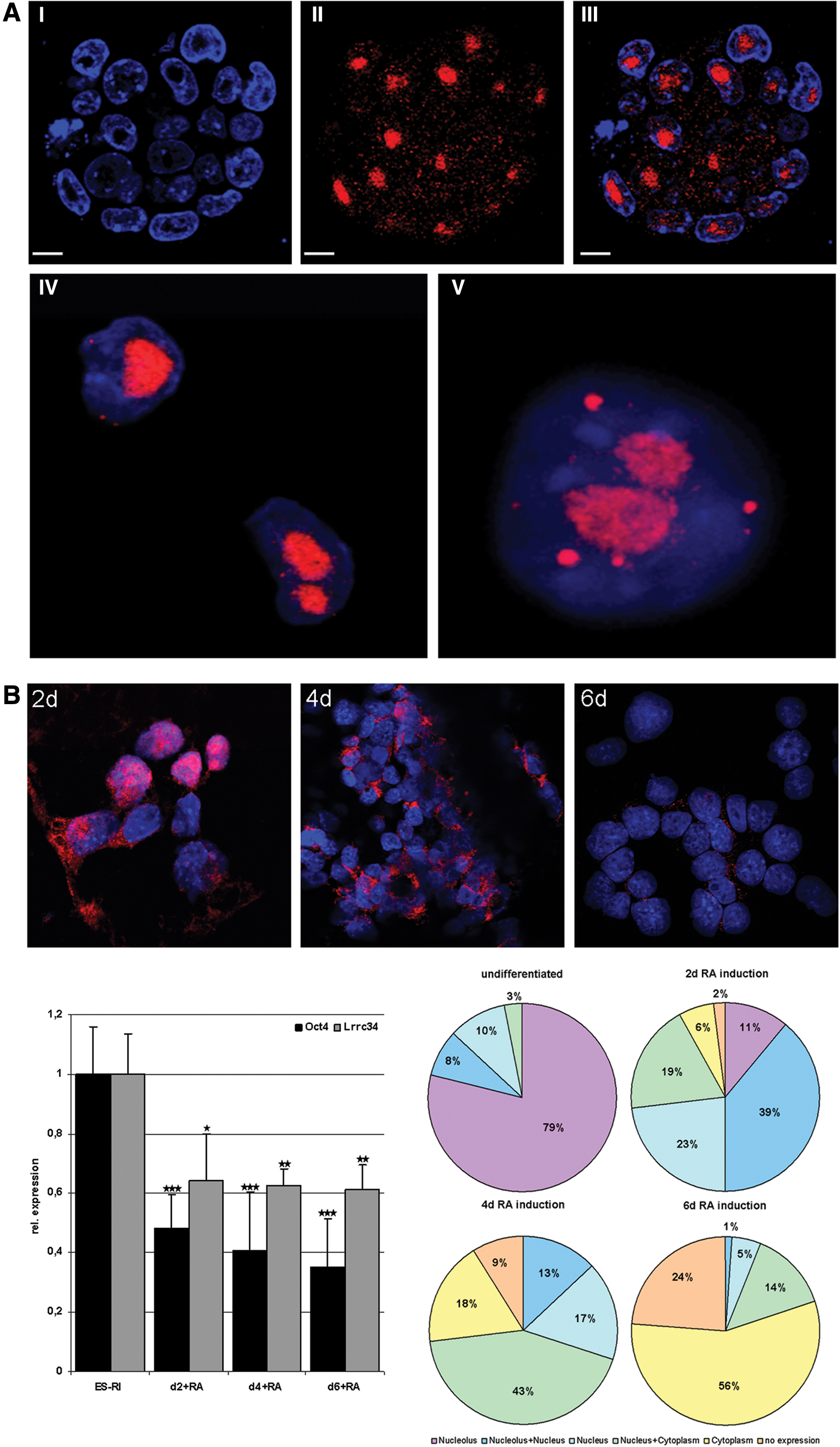
Localization of the LRRC34 protein in undifferentiated and differentiated ESCs.
Proliferation assay
To investigate the proliferation capacity of the knockdown (KD) cell lines, 4×104 cells of each cell line were plated on a six-well plate with mouse embryonic fibroblasts (MEFs). Cells were harvested and counted after 3 and 5 days, respectively. The experiment was performed in two biological with each two technical replicates to reach statistical significance.
Colony forming assay
To investigate the capacity of colony forming of the KD cell lines, 4×104 cells of each cell line were plated on a six-well plate with MEFs. After 4 days of culture, an alkaline phosphatase (AP) staining (Sigma REF: 86C-1 KT) was performed. AP-positive colonies were counted in three different fields of vision and the colony size was measured at the obvious longest side of each particular colony. From each cell line 100 colonies were measured.
Transfection of stem cells with siRNA or shRNA plasmids
Transfection of pluripotent cell lines with siRNA oligonucleotides at a final concentration of 60 nM was performed using Lipofectamine™2000 (Invitrogen) according to manufacturer's instructions. In brief, 500,000 cells per well were plated on six-well tissue culture plates 1 day prior to transfection. On the next day 10 μL of siRNA (Invitrogen) (sequences can be found in Supplementary Table S1; Supplementary Data are available online at
Construction of plasmids and derivation of stable cell lines
To obtain a vector for the overexpression of Lrrc34, the human elongation factor 1 alpha (hEF1α) promoter was cut out from the pBOS vector (BD Bioscience) using HindIII/EcoRI and cloned into pEGFP-1 (Clontech). The resulting phEF1α-EGFP vector served as a control in all experiments. From this vector, the ORF (open reading frame) of EGFP was replaced by the PCR-amplified ORF of Lrrc34 tagged with E2 using BamHI/NotI sites resulting in phEF1α-Lrrc34-E2. Stable cell lines were generated after electroporation and G418 selection by picking individual clones and culture them further.
Gene expression studies
Total RNA was extracted from cells or preimplantation embryos by using Trifast Reagent (Peqlab) according to manufacturer's protocol. Prior to reverse transcription using Superscript II cDNA synthesis kit and oligo(dT) primer (Invitrogen), RNA was treated with DNase I (Sigma) to avoid any genomic DNA contamination. qRT-PCR was performed on ABI Prism 7900 HT Fast Detection System (Applied Biosystems, Inc.) as previously described [12]. Each 10-μL reaction was performed in 384-well format using SYBRgreen PCR Master Mix (Qiagen) and 3 μM of each PCR primer. All reactions were performed from two biological replicates and analyzed in one run in triplicate and runs were repeated three times (n=18). Levels of mRNA expression were normalized to those of the mouse housekeeping genes Sdha (succinate dehydrogenase) and HPRT (hypoxanthine-guanine phosphoribosyltransferase). Oligonucleotide primers for qRT-PCR were obtained from Eurofins MWG Operon. Primer sequences used for gene amplification can be found in supplementary material (Supplementary Table S2). Conventional RT-PCR amplification was performed using specific primers for HPRT and Lrrc34. RT-PCR was achieved after 33 cycles at 94°C, 30 s; 50°C–62°C, 30 s; and 72°C, 45 s, depending on the primer sets. Primer sequences are given in Supplementary Table S2. For northern blot analysis, 20 μg of total RNA was separated on a 1.0% agarose-formaldehyde gel and transferred to a nitrocellulose membrane (Hybond C; Amersham). A 0.4-kb, mouse Lrrc34-mRNA-specific probe was radiolabeled using the Amersham Megaprime Labelling System (RPN1606) according to the manufacturer's protocol. The membrane was hybridized to the labeled probe in 8 mL of hybridization solution (KPL) and 150 μL of denatured salmon sperm DNA (10 mg/mL) at 65°C overnight.
Immunostaining of cells and blastocysts
Cells were grown on culture slides till 80% confluence and then fixed with 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature. Preimplantation embryos were collected from superovulated wild-type mice by flushing the uterus. Embryos were washed in PBS and then also fixed in 4% PFA in PBS for 10 min at room temperature. Fixed cells as well as fixed embryos were washed twice with PBS and then incubated for 1 h in PBS containing 0.1% bovine serum albumin (BSA) and 0.1% Tween20. Then, cells were incubated with primary antibodies (Lrrc34: Abcam, ab107820; Npm1: Abcam, ab10530; and Ncl: Santa Cruz, sc-8031) diluted in blocking buffer in a humidity chamber at 4°C overnight. Cells were rinsed three times and incubated in the appropriate Cy3- or Alexa488-conjugated secondary antibody (Sigma and Invitrogen, respectively). For negative control, cells were incubated with IgG as first antibody. All antibody incubation steps were performed in PBS (pH 7.4) with 5% BSA and 0.1% Triton X-100. For nuclear staining, fixed cells were imbedded with 40,60-diamidino-2-phenylindole (DAPI; Sigma). Microscopy was performed with an Olympus confocal laser scanning microscope. Pictures were taken as individual confocal stacks.
Isolation of proteins and western blot
Cells were resuspended in lysis buffer [10 mM Tris/HCl (pH 8.1), 1 mM EDTA, and 2.5% sodium dodecyl sulfate (SDS)] supplemented with 1 mM phenylmethanesulphonylfluoride (PMSF) and protease inhibitors and were thereafter sonicated. Protein extracts (20 μg) were denatured at 70°C in NuPage SDS sample buffer (Invitrogen), separated on a NuPage 10% Bis-Tris Gel (Invitrogen), and transferred to a Hybond-C Extra membrane (GE Healthcare Europe). Blots were blocked for unspecific binding with 5% nonfat dry milk in blocking buffer (25 mM Tris, 0.15 M NaCl, and 0.1% Tween20) and were incubated overnight at 4°C with primary and—after washing in blocking buffer for 1 h at 4°C—with secondary horseradish-peroxidase-conjugated antibody. Protein bands were visualized using enhanced chemiluminescence as described by the manufacturer (Santa Cruz Biotechnology).
Gene-specific methylation analysis
Genomic DNA was isolated from maGSC and male ESC lines by isopropanol precipitation and the promoter of Lrrc34 was analyzed by bisulfite pyrosequencing as described previously [13].
Chromatin immunoprecipitation RT-PCR assays
Assays were performed as published previously [14]. In brief, to crosslink the cells, they were cultured for 10 min at room temperature in 1% formaldehyde in culture medium. Crosslinking was stopped by glycine at a final concentration of 125 mM. Cells were lysed by incubation in buffer containing 10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 3 mM MgCl2, 0.5% IGEPAL, and 1 mM PMSF followed by centrifugation and incubation in lysis buffer supplemented with 1 mM CaCl2 and 4% IGEPAL. The chromatin was then sonicated to obtain an average DNA fragment length of ∼200–500 bp. The desolved chromatin was incubated with and without antibody (3–5 μg) directed against H3K4me3 (39159; Active Motif), H3K9me3 (07-442; Millipore), H3K9ac (07-352; Millipore), and H3K27me3 (07-449; Millipore), respectively, for 3 h at 4°C and complexed with protein-A sepharose beads overnight at 4°C. Next, the beads were washed in different wash buffers (buffer I: 20 mM Tris-HCl (pH 8.0), 2 mM EDTA, 1.0% Triton X-100, 150 mM NaCl, and 1 mM PMSF; buffer II: wash buffer I supplemented with 0.1% SDS and 500 mM NaCl; and buffer III: 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 250 mM LiCl, 0.5% IGEPAL, and 0.5% deoxycholate). Finally, elution was performed using the elution buffer (25 mM Tris-HCl, 10 mM EDTA, and 0.5% SDS) and incubation for 1 h at 65°C. Reverse crosslinking was conducted by proteinase K followed by incubation for 1 h at 65°C. The DNA was then extracted, purified, and eluted in 40 μL of TE buffer using the Wizard SV Gel and PCR clean-up System (Promega). Quantification was done by qRT-PCR analysis. Primer sequences are given in Supplementary Table S2. The qRT-PCR data of two biological and two independent technical replicates were calculated and expressed as percentage of input DNA.
Proximity ligation assay
ES-RI cells cultured on chamber slides were washed in PBS and then fixed with 4% PFA for 20 min. After fixation, the slides were washed two times for 5 min with PBS and incubated for another 10 min with 0.1% Triton X-100 diluted in PBS for permeabilization of the cell membrane. Subsequently, the slides were incubated for 20–30 min in blocking buffer (3% BSA in PBS). After blocking, the primary antibodies, diluted in blocking buffer in the appropriate concentration, were added to the cells and the slides were incubated overnight at 4°C in a humidity chamber.
The following day the proximity ligation assay (PLA) probes (Duolink In Situ PLA Probe Anti-Rabbit PLUS; Duolink In Situ PLA Probe Anti-Mouse MINUS) were diluted 1:5 in the blocking buffer (provided by manufacturer) and rested for 20 min at room temperature. The cells were washed twice for 5 min with PBS before PLA sample solution was added. The slides were incubated in a preheated humidity chamber for 1 h at 37°C. Subsequently, the PLA sample solution was removed and the cells were washed twice with PBS. The ligation mixture was prepared according to manufacturer's advices and added to the slides. The slides were incubated for 30 min at 37°C in a preheated humidity chamber. Afterward, the cells were washed twice for 2 min with PBS. The amplification mixture was prepared according to manufacturer's advices and added to the cells. The slides were incubated for 100 min in the dark. Following the amplification reaction, the mixture was removed from the slides and the cells were washed twice for 10 min with 1× washing buffer B and for 1 min with 0.01× washing buffer B. The slides were briefly dried in a dark chamber prior to mounting using DAPI. The analysis of the results was performed using a confocal laser scanning microscope.
Coimmunoprecipitation assay
Cultured ES-Lrrc34-E2 cells were washed twice in ice-cold PBS and resuspended in lysis buffer followed by 40-min incubation on ice. The cell lysate was centrifuged for 30 min at 4°C by 13,000 rpm. After centrifugation 25 μL from the supernatant was transferred into a fresh tube and stored at −20°C to be used as input (positive control).
The rest of the supernatant was divided into two fresh tubes in order to perform a negative control at the same time. After washing of the agarose beads, they were resuspended in the antibody cell suspension and incubated for 1 h at 4°C. Subsequently, the samples were centrifuged for 5 min at 4°C by 2,000 rpm and the supernatant was transferred into a fresh tube and incubated for 2–3 h with either 5 μg of E2-antibody or IgG. The samples were centrifuged and the supernatant (SN fraction) containing any proteins nonbound to the beads was stored. The beads were washed and resuspended into 30 μL of 2× loading dye (CoIP). All samples (input, supernatant, and CoIP) were subjected to analysis using SDS–polyacrylamide gel electrophoresis.
Results
Lrrc34 was one of the genes showing the strongest downregulation after RA-induced differentiation of mouse ESCs and maGSCs [1]. Lrrc34 RNA expression could be found by RT-PCR analysis in all pluripotent stem cells tested and in testis (Supplementary Fig. S1A). These results could be confirmed by northern blot analysis (Fig. 2A). To further elucidate the cell type in the adult mouse testis expressing Lrrc34, we performed RT-PCR on testis RNA isolated from different testis developmental mutants (Supplementary Fig. S1B). W/WV mice are known to lack germ cells (review in de Rooij and de Boer [15]). Tfm/y mutant mice have a block in spermatogenesis after the stage of primary spermatocytes [16], Leyl−/− (Insl3) mutant mice after secondary spermatocytes [17], olt/olt mutant mice after the stage of round spermatids [18], and qk/qk mutant mice at the stage of elongated spermatids [19]. Since no expression could be detected in the testis of W/Wv mutants, we suggested Lrrc34 to be germ cell specific. Further, the expression pattern of Lrrc34 RNA in mouse organs and cell lines could be confirmed in the human as well where Lrrc34 was only detectable in the testis (Supplementary Fig. S1C). By qRT-PCR, the results of the array [1], of RT-PCR experiments, and of northern blot analysis could be confirmed. Lrrc34 is strongly expressed in undifferentiated pluripotent stem cells and becomes downregulated upon differentiation whereas no expression could be detected in the negative control cells, MEFs, and NIH3T3. Further, we could detect a high expression in mouse testis but no expression in spleen and brain (Fig. 2B). Since all the results till now fit with our hypothesis that Lrrc34 might play a role in establishment and maintenance of pluripotency, we analyzed Lrrc34 expression during the time course of reprogramming by qRT-PCR (samples used for this study are described in [20] Supplementary Fig. S2). Lrrc34 RNA expression could be detected on day 10 of classical Yamanaka reprogramming—exactly at the stage where also endogenous Oct4 expression becomes detectable. However, although Lrrc34 increases in the same manner as Oct4, at day 12 there was no further increase visible till the stage of fully established iPSCs. These results let us to suggest that Lrrc34 might play a role in the early steps of reprogramming and later in the fully established pluripotent stem cells.
We next aimed to clarify the subcellular localization of the LRRC34 protein. Since most pluripotency-regulating genes are known to be transcription factors and therefore are located in the nucleus, we expected LRRC34 to be located in this compartment as well. Specificity of the antibody used for all immunocytological studies was confirmed by transfecting NIH 3T3 cells with an Lrrc34 construct that produces a fusion protein with an E2 tag. Costaining with an E2 antibody and the Lrrc34 antibody showed a perfect match (Supplementary Fig. S3) and therefore we conclude that the antibody used for our studies is specific for the LRRC34 protein. However, we found LRRC34 to be dominantly located in the nucleolus and only in a minor fraction in the nucleus where it shows a dot-wise staining pattern (Fig. 3A; for nucleolar verification see also Fig. 6A). Immunocytological staining of partly differentiated ESCs by RA treatment for 2, 4, and 6 days suggests the LRRC34 protein to translocate from the nucleolus to the nucleus and later to the cytoplasm upon stem cell differentiation. Differentiation status of the cells was confirmed qRT-PCR that showed a decrease of Oct4 and Lrrc34 expression (Fig. 3B, bar diagram). Quantification was performed by counting 100 DAPI-positive cells per differentiation time point (Fig. 3B, circle diagrams).
Since it is well known that pluripotency-regulating genes are already expressed in preimplantation embryos, we performed immunostaining on different stages of preimplantation development. LRRC34 protein is detectable already in unfertilized oocytes (Supplementary Fig. S4 IA) where it shows a cytoplasmic localization. In two-cell embryos, LRRC34 shows cytoplasmic as well as nuclear localization (Supplementary Fig. S4 IC). No signal was detectable in the negative controls using IgG as first antibody (Supplementary Fig. S4 IB, D). Analysis of four- and eight-cell-stage embryos confirmed the results from two-cell stages (Supplementary Fig. S4 II). In morula-stage embryos, LRRC34 seems to be predominantly localized in the nucleolus (Supplementary Fig. S4 IIIA) whereas the staining pattern in the different cell types of a blastocyst-stage embryo could not be specified as specific to nucleolus or nucleus since a signal was detectable in both compartments (Supplementary Fig. S4 IIIC). To further elucidate the cellular compartment, in which LRRC34 is localized in the adult mouse testis, we performed immunocytological analysis and could show that LRRC34—as expected from our RT-PCR results on mouse testis development mutants—is expressed in all germ cell development stages despite spermatozoa (Supplementary Fig. S5A). However, LRRC34 seems to be strongly expressed in the nucleus and nucleolus of some cells located close to the outer layer of the seminiferous tubuli where the most undifferentiated spermatogonia are located. With the further differentiation of the germ cells, LRRC34 expression in the nucleus becomes weaker before translocating to the cytoplasm. These results fit to our previous finding that Lrrc34 translocates from the nucleolus in pluripotent stem cells to the nucleus and later to the cytoplasm upon differentiation. To further support this hypothesis, we used two different permanent cell lines resembling different stages of germ cells. The cell line GC1 resembles the stage of spermatogonia [21] whereas GC4 are known to be spermatocyte-stage cells [22]. Immunostaining revealed LRRC34 localization in the nucleolus in spermatogonial (GC1) cells whereas in spermatocyte (GC4) cells the protein is located in the nucleus (Supplementary Fig. S5B). This indicates that also in these germ cell lines the protein translocates with the status of differentiation. These results confirm our assumption that LRRC34 translocates from the nucleolus to the nucleus and further to the cytoplasm upon differentiation of both, germ cells and pluripotent stem cells.
To get an idea about the function of Lrrc34 in the pluripotency network, we performed downregulation and overexpression experiments in mouse ESCs. For stable downregulation, shRNA plasmids targeting Lrrc34 mRNA were transfected in the mouse ESC line ES-RI. After positive selection with G418, several clonal cell lines were established and analyzed for Lrrc34 expression (data not shown). Two cell lines (No. 4 and No. 6) showing the highest downregulation were selected and cultured further. After three and five passages, RNA was isolated in order to obtain two biological replicates for qRT-PCR analyses (Fig. 4A, left). Both cell lines showed a downregulation of Lrrc34 mRNA up to 70%. This leads to a significant downregulation of the pluripotency genes Oct4 and Klf4 whereas Nanog expression was not affected. To check, whether the downregulation of Oct4 is also detectable on the protein level, we performed western blot analysis and indeed could show that the Oct4 protein is downregulated in the two respective Lrrc34-shRNA clones compared with the shRNA control cell line. α-Tubulin served as loading control. To further investigate the effect of Lrrc34 KD, we checked the expression of differentiation marker genes (Fig. 4B) by qRT-PCR and observed no regulation of the mesodermal marker Vimentin whereas the ectodermal marker Nestin and the endodermal marker GATA4 were significantly downregulated. In addition, we could not see any differentiation-specific morphological changes upon Lrrc34 KD (Fig. 4C). We conclude that the downregulation of Oct4 resulting from the downregulation of Lrrc34 does not induce differentiation of cells. However, during the culture of the KD cell lines, we observed a reduced proliferation capacity and quantification resulted in a significant downregulation of the proliferation rate in KD cell lines (Fig. 4D). Further, colony forming assay using AP staining (Fig. 4E–G) showed a decrease in the colony size and amount of colonies in the KD cells. Taken together, we have shown that the downregulation of Lrrc34 expression has an effect on pluripotent stem cells. The functional reason for this needs to be elucidated in future experiments.
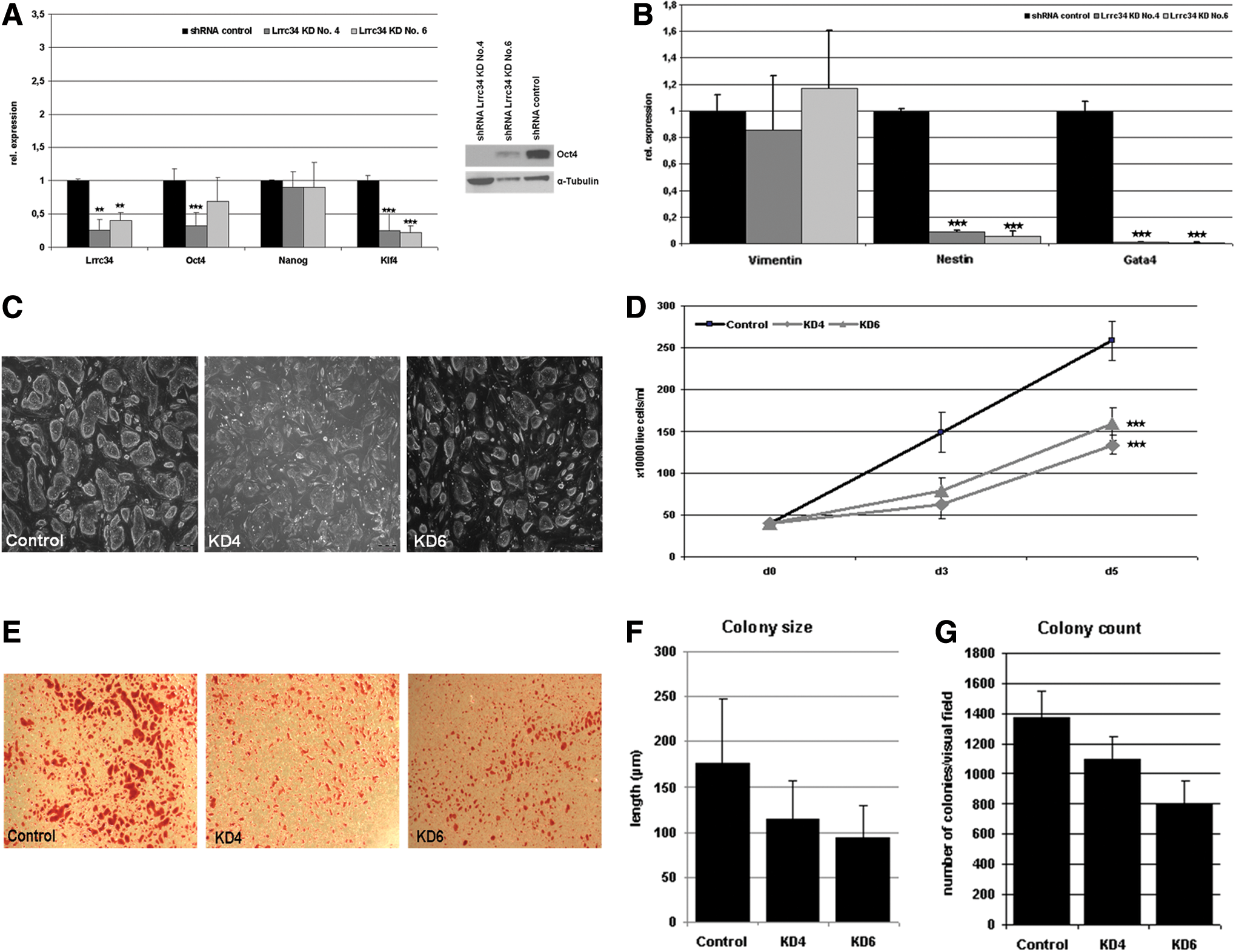
Consequences of Lrrc34 KD on ESCs.
To test whether this is a short-term or long-term effect, we performed transient downregulation of Lrrc34 using the siRNA-mediated KD approach (Supplementary Fig. S6A). Oct4 as well as Klf4 and Zfp206 already showed a significant downregulation after 48 h of siRNA transfection against Lrrc34. In contrast to the stable downregulation of Lrrc34, Nanog was also significantly downregulated after 80% KD of Lrrc34. The Lrrc34-siRNA-mediated KD of Oct4 could be confirmed by western blot analyses using an Oct4 antibody. However, the KD on the protein level became first visible after 72 h posttransfection (Supplementary Fig. S6B). To elucidate whether the KD of Oct4 has an effect on Lrrc34 expression, we performed siRNA-mediated KD of Oct4 and analyzed the expression of Lrrc34 in these cells (Supplementary Fig. S6C). A downregulation of Oct4 indeed resulted in a clear downregulation of Lrrc34. Taken together, these results suggest a rapid regulation of Oct4 expression after the KD of Lrrc34 and vice versa.
To see the effect of Lrrc34 overexpression on the pluripotency network, we generated an overexpression construct using the hEF1α promoter to drive Lrrc34 expression. We additionally tagged the protein with an E2-tag. We performed transfection into ES-RI cells and established several clonal cell lines after positive selection with G418. Multiple clones were analyzed for Lrrc34 expression (data not shown) and two cell lines (No. 15 and No. 18) showing the highest overexpression were selected and cultured further. After three and five passages, RNA was isolated in order to obtain two biological replicates for qRT-PCR analyses (Supplementary Fig. S7A). An up to threefold overexpression of Lrrc34 resulted in the significant overexpression of Klf4; however, Oct4 expression was moderate to significantly downregulated. The same result could be obtained for Nanog expression. Although these results were not expected, there is still a regulation of the pluripotency network visible upon overexpression of Lrrc34. To confirm the regulation of Oct4 expression on the protein level, we performed western blot analyses and indeed could show a downregulation of Oct4 even on the protein level (Supplementary Fig. S7B).
Since it is well known that pluripotency-regulating genes showed typical epigenetic marks, we analyzed the methylation of the putative Lrrc34 promoter in undifferentiated and differentiated ESCs and maGSCs from the same genetic background by bisulfite sequencing (Fig. 5A). As expected and known for other pluripotency-regulating genes, the Lrrc34 promoter shows a hypomethylation in undifferentiated stem cells and becomes hypermethylated upon differentiation. Further, we analyzed the Lrrc34 promoter for the enrichment of activating and repressive histone marks by chromatin immunoprecipitation (ChIP) qRT-PCR analysis and could show that the Lrrc34 promoter is enriched for the activation histone marks 3meH3K4 and acH3K9 and lacks the repressing chromatin marks 3meH3K9 and 3meH3K27 (Fig. 5B). This pattern is also known for other pluripotency-regulating genes like Nanog and Oct4 whereas the promoters of developmental genes like Hoxa11 are known to have a bivalent chromatin mark. Taken together, the analysis on the epigenetic level also revealed Lrrc34 to be a pluripotency gene.
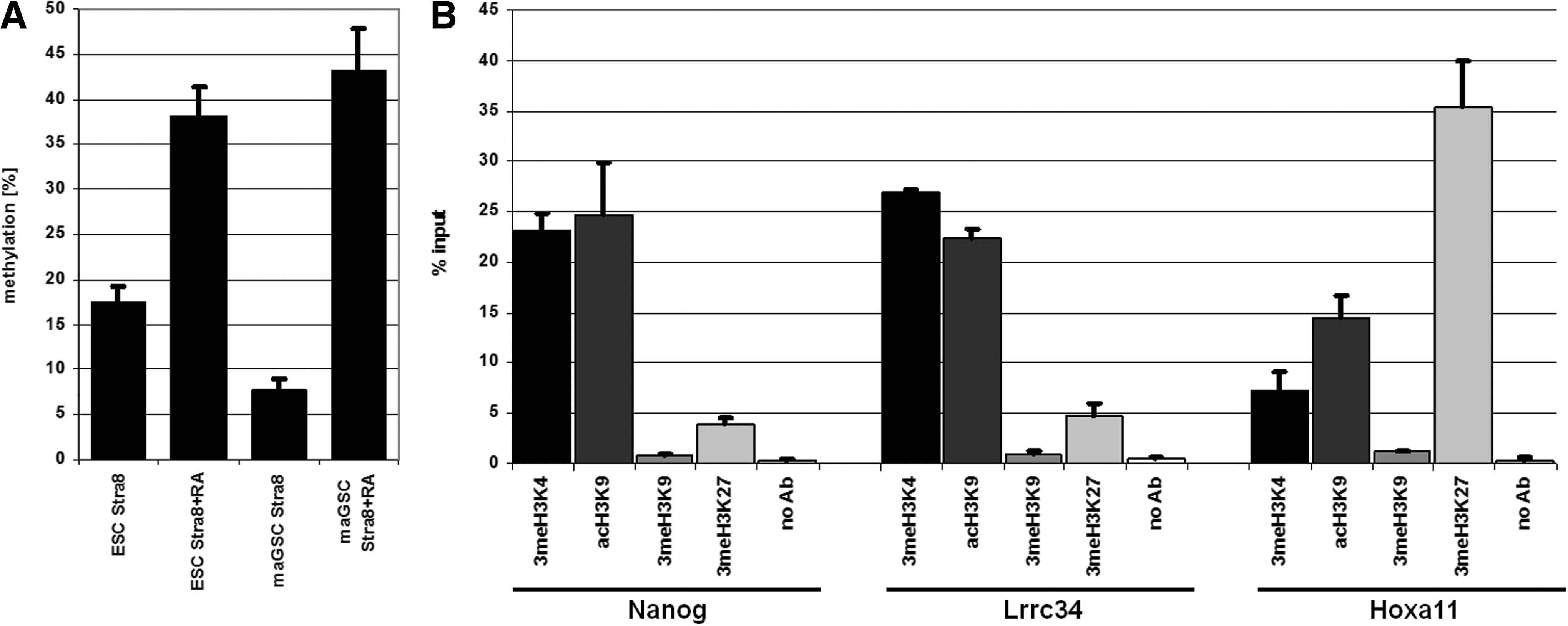
Epigenetic analysis of the putative promoter of Lrrc34.
Nucleolin (Ncl) and Nucleophosmin (Npm1) are known to be predominantly localized in the nucleolus and are therefore applicable to confirm the nucleolar localization of LRRC34. Costaining of ESCs using LRRC34 and Npm1- or Ncl-specific antibodies (Fig. 6A) confirmed the nucleolus as the prominent subcellular localization of LRRC34 as well as the colocalization with both putative interaction partners. For Npm1 we additionally performed colocalization studies on blastocysts and indeed could show the nucleolar colocalization of both proteins (Supplementary Fig. S8). Since both Npm1 and Ncl are known to be involved in pluripotency, we assume them to directly interact with the LRRC34 protein. Coimmunoprecipitation experiments using the stable Lrrc34-E2 ESC line could indeed confirm the interaction of LRRC34 with both NPM1 and NCL (Fig. 6B, C, upper part). To confirm the interaction further, we performed PLA using LRRC34 and NPM1 or NCL antibodies, respectively. PLA signal could be detected in both cases (Fig. 6B, C, lower part) but not in the appropriate controls (data not shown). However, the PLA signal was predominantly detectable in the nucleus instead of the nucleolus. This is suggested to be due to technical limitations of the method since all antibodies used for this study showed a clear nucleolar localization of the respective proteins before.

Analysis of the interaction of LRRC34 with the nucleolar proteins NPM1 and NCL.
Discussion
The novel LRR protein LRRC34 is predominantly expressed in mouse pluripotent stem cells as well as in mouse and human testes. In the adult mouse testis, the expression is restricted to premeiotic germ cells. Other pluripotency-regulating genes, like Oct4 [2], Zfp206 [23], and Sall4 [24], showed the same expression pattern making it a criterion for new candidate genes for the regulation of pluripotency. The connection of germ cells and stem cells is indeed a well-discussed topic. Very recently, Leitch and Smith postulated that germ cells represent a continuous cycle of pluripotency distinguishable just by a naive state in the epiblast and a latent in the germline but not through the expression of pluripotency-regulating genes [25]. Indeed, we could also show that germ cell marker genes are expressed in pluripotent stem cells and arise early in reprogramming [20]. Since Lrrc34 expression arises also very early in the reprogramming time course, it can be a hint for both pluripotency regulator and germ cell gene. Further we showed that there is a difference between germ cell marker genes and premeiotic marker genes on the epigenetic level. The promoters of germ cell marker genes are endowed with activating chromatin marks in ESCs and iPSCs whereas promoters of premeiotic marker genes are endowed with bivalent chromatin marks. The promoter region of Lrrc34 is endowed with the activating chromatin modifications 3meH3K4 and acH3K9 and since this is also known for the key regulator genes Oct4, Sox2, and Nanog [26 –28] it is a strong hint for Lrrc34 to be a pluripotency as well as germ cell marker gene.
Altered expression of pluripotency regulators resulted in the altered expression of the whole network and/or differentiation of the cells. For Zfp206, for example, it could be shown that the KD of the gene could not induce differentiation of the cells but that the cells respond earlier to differentiation-inducible conditions [23]. For other genes like, for example, Oct4 and Sox2, it is known that the KD itself is enough to induce directed differentiation of ESCs into trophoectoderm and other lineages, respectively [29,30]. The altered expression of Lrrc34, however, resulted in the significant downregulation of some but not all core pluripotency regulators. We could not observe any differentiation in the cells but a reduced proliferation and a reduction of colony size and number. Therefore we suggest Lrrc34 not to be a key regulator of pluripotency but it indeed has an impact on pluripotent stem cells. To address the question through which mechanisms Lrrc34 has this influence, further work is ongoing.
In the present study we could show that the LRRC34 protein is predominantly localized in the nucleolus of pluripotent cells. The nucleolus is the most prominent substructure in the nucleus of eukaryotic cells. It is the place of ribosomal RNA (rRNA) transcription and processing, as well as coordinating ribosome assembly. Nucleoli are built by the fusion of nucleolar-organizing regions from different chromosomes that can partially disassemble in metaphase and rebuilt in late telophase [31]. Despite the main function in ribosome biogenesis, there are many results that suggest nucleoli to have additional functions, such as regulation of the cell cycle, cellular aging, signal recognition, particle biosynthesis, small RNA processing, mRNA transport, and even p53 regulation (reviewed in Pederson and Boisvert et al. [32,33]).
Nucleophosmin (Npm1) was first reported as a nonribosomal nucleolar phosphoprotein that is localized within the nucleolus [34]. It is known to be involved in multiple biological processes, such as RNA processing [35], working as a chaperone [36], antiapoptotic activity [37], and ribosome biogenesis and transport [38]. Recently, it was found that Npm1 can form complexes with the core transcription factors Oct4, Sox2, and Nanog, suggesting a role for Npm1 in ESC maintenance [39]. An overexpression of Npm1 in ESCs resulted in higher proliferation rates [40] whereas a downregulation resulted in reduced proliferation rates [41]. The other candidate that was chosen, Nucleolin (Ncl), is also known to be predominantly expressed in the nucleolus [42] and involved in ribosome biogenesis [43] as well as in rRNA synthesis [44]. Further, Ncl plays a role in the proliferation of cells supported by the fact that it is highly expressed in actively dividing cells [45]. Recently, it was shown that Ncl is highly expressed in undifferentiated ESCs and rapidly downregulated upon their differentiation. Further, Ncl was shown to be important for the self-renewal of ESCs as a regulator of the p53-dependent pathway. A KD of Ncl resulted in an upregulated p53 expression and subsequently in the induction of cell cycle arrest, apoptosis, and differentiation [46]. There are further several interaction partners for Ncl known, including Npm1 [47] as well as the pluripotency regulator Oct4 [40]. During the present study both nucleolar proteins could be identified as interaction partners of LRRC34, suggesting that there might exist a protein interaction network of Oct4, Npm1, Ncl, and Lrrc34. Whether these proteins interact at the same time or whether there are different situations where diverse interactions occur needs to be elucidated in the future.
Conclusion and Outlook
Here we characterize the novel pluripotency marker gene Lrrc34 and show that it has an impact on the expression of key pluripotency-regulating genes and the growth behavior of ESCs. Further we identified the nucleolus as the major subcellular compartment where the LRRC34 protein is localized. Using the two independent methods we could identify two nucleolar proteins that are known to be involved in the control of ribosome biogenesis, as interaction partners of LRRC34. These results let us to hypothesize a putative role of LRRC34 in the regulation of ribosome biogenesis in pluripotent stem cells what would imply a special regulatory mechanism in stem cells compared with others where Lrrc34 is not expressed. However, whether Lrrc34 indeed has an impact and whether this impact is on the expression of pre-rRNA or the processing needs to be elucidated in future experiments.
Footnotes
Acknowledgments
The authors would like to thank Dr. Sandra Meyer (Institute of Reconstructive Neurobiology, University of Bonn) for interpretation of array data suggesting Lrrc34 as a candidate, Diana Riethmüller and Janine Ulrich for excellent technical assistance, and Nadine Mellies for proofreading of the article. This work was supported by the German Research Foundation (DFG) in the Priority Programme SPP1356; grant to J.N. (no. 941/1-2) and ZE442/4-2.
Author Disclosure Statement
The authors declare that they have no competing interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.