
Abstract
Hypoxia inducible factor-1α (HIF-1α) plays an important role in angiogenesis-osteogenesis coupling during bone regeneration, which can enhance the bone healing capacity of mesenchymal stem cells (MSCs) by improving their osteogenic and angiogenic activities. Previous studies transduced the HIF-1α gene into MSCs with lentivirus vectors to improve their bone healing capacity. However, the risks due to lentivirus vectors, such as tumorigenesis, should be considered before clinical application. Dimethyloxaloylglycine (DMOG) is a cell-permeable prolyl-4-hydroxylase inhibitor, which can activate the expression of HIF-1α in cells at normal oxygen tension. Therefore, DMOG is expected to be an alternative strategy for enhancing HIF-1α expression in cells. In this study, we explored the osteogenic and angiogenic activities of adipose-derived stem cells (ASCs) treated with different concentrations of DMOG in vitro, and the bone healing capacity of DMOG-treated ASCs combined with hydrogels for treating critical-sized calvarial defects in rats. The results showed that DMOG had no obvious cytotoxic effects on ASCs and could inhibit the death of ASCs induced by serum deprivation. DMOG markedly increased vascular endothelial growth factor production in ASCs in a dose-dependent manner and improved the osteogenic differentiation potential of ASCs by activating the expression of HIF-1α. Rats with critical-sized calvarial defects treated with hydrogels containing DMOG-treated ASCs had more bone regeneration and new vessel formation than the other groups. Therefore, we believe that DMOG enhanced the angiogenic and osteogenic activity of ASCs by activating the expression of HIF-1α, thereby improving the bone healing capacity of ASCs in rat critical-sized calvarial defects.
Introduction
T
In previous studies, mesenchymal stem cells (MSCs) were widely used with bone substitutes to promote new bone repair and regeneration [4,5]. These cells have osteogenic differentiation potential and can differentiate into osteoblasts to promote osteogenesis [6,7]. They also secrete many angiogenic growth factors, which promote local revascularization. Angiogenesis is a fundamental step in new bone formation, and neovascularization promotes bone regeneration [8,9]. However, large numbers of seed cells are needed for the successful repair of a large defect. Due to this requirement, treated cells that have enhanced osteogenic and angiogenic capacity are beneficial in the application of cells for bone regeneration in tissue engineering studies.
Hypoxia inducible factor-1α (HIF-1α) is a crucial mediator of the adaptive cell response to hypoxia, and plays an important role in angiogenesis-osteogenesis coupling during bone regeneration [10]. It controls the activity of a broad array of genes and modulates cell proliferation, differentiation, and pluripotency [11]. HIF-1α can promote the secretion of vascular endothelial growth factor (VEGF) in MSCs, is a potent mitogen for endothelial cells (ECs), and plays a central role in neovascularization and angiogenesis [12]. Angiogenesis helps support MSCs and osteoblasts that are necessary for bone repair, and is coupled with osteogenesis in bone repair and regeneration [13 –15]. VEGF also has direct effects on osteoblast lifespan and function, and regulates the communication between osteoblasts and ECs [16 –18]. It was also reported that HIF-1α improves the osteogenic differentiation capacity of MSCs and enhances their osteogenic activity [19]. Therefore, overexpression of HIF-1α in MSCs enhances their osteogenic and angiogenic activities and promotes the repair of large bone defects.
Previous studies transduced the HIF-1α gene into MSCs with lentivirus vectors, and found that HIF-1α transgenic MSCs had better osteogenic and angiogenic capacity in vitro and in vivo, leading to better bone healing potential in large bone defects [20 –22]. However, once transduced into cells, the HIF-1α gene will, theoretically, be overexpressed throughout the cell's life. In addition, the risks of lentivirus vectors, such as tumorigenesis, should be seriously considered before clinical application. Dimethyloxaloylglycine (DMOG) is a small molecular drug, and is a cell-permeable prolyl-4-hydroxylase inhibitor. At normal oxygen tension, hypoxia-inducible factor prolyl hydroxylase (HIF-PH) hydroxylates a specific proline residue of HIF-1α, which promotes HIF-1α binding to the Von Hippel-Lindau tumor suppressor, and causes the degradation of HIF-1α. DMOG can inhibit the effect of HIF-PH, and thus stabilize the expression of HIF-1α in cells [23]. Therefore, DMOG is expected to be an alternative strategy for enhancing HIF-1α expression in cells, which may be safely used in the clinic.
In previous studies, by activating the expression of HIF-1α, DMOG has been successfully used to attenuate post-ischemic myocardial injury [24] and renal injury in remnant kidney [25], induce angiogenesis in ischemic skeletal muscles [26], and provide neuroprotection in a middle cerebral artery occlusion model [27]. However, to our knowledge, there are no reports on the effects of DMOG on the bone healing capacity of MSCs. Adipose-derived stem cells (ASCs) are an important type of MSCs, which are easy to harvest with liposuction and are abundant, leading to minimal donor site morbidity and a reduced period of in vitro expansion. In this study, we explored the influence of DMOG on the osteogenic and angiogenic potential of ASCs, and its bone healing capacity in the treatment of critical-sized calvarial defects in rats.
Materials and Methods
Cell harvest and culture
All procedures adhered to the recommendations of the U.S. Department of Health for the care and use of laboratory animals and were approved by the Ethics Committee of Shanghai Jiao Tong University. Primary ASCs were harvested from the adipose tissue of F344 rats (Shanghai Animal Experimental Center, Shanghai, China) as previously reported [28]. Briefly, adipose tissues were harvested from the animals under anesthesia (pentobarbital sodium; Sigma, St. Louis, MO) and then digested with 0.1% collagenase I (Sigma) for 60 min at 37°C. The tissue was filtered through a 100-μm nylon mesh (Shanghai Bolting Cloth Manufacturing Co., Ltd, Shanghai, China), and the filtrate was centrifuged at 1,500 rpm (363g). The cells were then plated on culture dishes (Corning, Tewksbury, MA) and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) at 37°C in a humidified 5% CO2 incubator. The culture medium was replaced every 3 days and cells were passaged when they reached ∼80% confluence with 0.25% trypsin (Invitrogen). Flow cytometry (BD Biosciences, San Jose, CA) was used to characterize ASCs with CD29, CD31, CD34, CD44, CD45, and CD90 (BD Biosciences). To avoid contamination by hematopoietic cells in earlier passages and differentiated cells in later passages, cells of four to six passages were used for the following experiments.
Cell proliferation and survival evaluation
To determine the influence of DMOG on ASCs proliferation, cells were seeded on plates and then treated with different concentrations of DMOG (Sigma; 0, 200, 500, and 1,000 μM) for 24, 48, and 72 h. Cell numbers were measured using the Cell Counting Kit-8 (CCK-8; Beyotime Institute of Biotechnology, Shanghai, China) and a Microplate Reader (450 nm; Bio-Rad, Hercules, CA). Trypan blue staining was performed to assess the cell death ratio, a possible indication of the toxicity of DMOG. Briefly, trypan blue (Invitrogen) was added to the medium at a final concentration of 0.05%. After 10 min of incubation, images of six different randomly chosen fields per well were taken under the microscope. Cell death was calculated by the ratio of trypan-blue-positive cells to total cells. The influence of DMOG on cell survival in serum deprivation conditions was also evaluated using trypan blue staining. ASCs were plated in six-well plates (Corning) and cultured in regular medium for 12 h. The cells were then washed with phosphate-buffered saline (PBS) and cultured in serum-free medium with different concentrations of DMOG. After 24, 48, and 72 h, trypan blue staining was performed.
HIF-1α shRNA construction and transfection
To inhibit the expression of HIF-1α in ASCs, the HIF-1α shRNA (forward oligo: 5′ T CCAGTTGAATCTTCAGATA TTCAAGAGA TATCTGAAGATTCAACTGG TTTTTT C 3′, reverse oligo: 5′ TCGAGAAAAAA CCAGTTGAATCTTCAGATA TCTCTTGAA TATCTGAAGATTCAACTGG A 3′; Invitrogen) was constructed by the pll3.7(Lentilox 3.7)-Zsgreen vector (Addgene, Cambridge, MA). For infection, ASCs were plated on a 12-well plate (Corning) at 5×104 cells and infected with shRNA lentivirus at an MOI of 100 in the presence of 8 μg/mL polybrene (Sigma). To determine the infection efficiency of the lentivirus, shHIF-1α ASCs were reflected by the GFP-positive proportion of cells detected by flow cytometry, and the results showed that more than 85% were GFP positive 48 h after infection.
Western blot analysis
To evaluate the influence of DMOG on the expression of HIF-1α and VEGF in ASCs, the cells were cultured in regular medium with DMOG (0, 200, 500, and 1,000 μM) under normal oxygen conditions. After 24 h, total protein was harvested from the cultured cells according to standard protocols. The protein concentration of the cells was measured with a BCA protein assay kit (Thermo, Rockford, IL). The cell lysates were then separated on SDS-PAGE (Bio-Rad) using 12% gels (Bio-Rad) and transferred to nitrocellulose membranes (Whatman). The membranes were incubated with primary antibodies of HIF-1α (Abcam, Cambridge, MA) and VEGF (Abcam) at a 1:800 dilution overnight, followed by incubation with infrared-conjugated secondary antibodies (Odyssey) at 1:10,000 for 1 h at room temperature. The membranes were scanned in an Odyssey Scanner (Li-COR Biosciences, Lincoln, NE), and bands were quantified using Odyssey software V3.0. The protein levels were normalized against β-actin. shHIF-1α ASCs cultured in regular medium with 1,000 μM DMOG were used as controls.
Enzyme-linked immunosorbent assay for VEGF
To analyze VEGF production by ASCs treated with DMOG, ASCs were cultured in regular medium with different concentrations of DMOG (0, 200, 500, and 1,000 μM). shHIF-1α ASCs exposed to 1,000 μM DMOG were also included in the study. After 1, 3, 7, 14, and 21 days, the cells were seeded into six-well plates at 300,000 cells per well. After incubation in regular medium with different concentrations of DMOG for 24 h, the medium was harvested and stored at −80°C until analysis of the VEGF protein using an enzyme-linked immunosorbent assay (ELISA) kit (Antibodies-Online, Aachen, Germany) according to the manufacturer's instructions. The total protein content of these cells was then determined for standardization of VEGF production with a BCA protein assay kit (Pierce Biotechnology, Rockford, IL).
Quantitative real-time polymerase chain reaction analysis
To verify whether DMOG improved the osteogenic differentiation potential of ASCs, cells (1×105) were plated on six-well plates and cultured in osteogenic differentiation medium (No. A10072-01; Gibco BRL) with different concentrations of DMOG (0, 200, 500, and 1,000 μM). shHIF-1α ASCs exposed to 1,000 μM DMOG were also included in this study. Total cellular RNA was harvested with TRIzol (Invitrogen) at days 1, 3, 7, 14, and 21 after osteogenic induction, according to the manufacturer's protocol. One microgram of purified RNA was then reverse transcribed using the PrimeScript RT reagent kit (Takara, Shiga, Japan) according to the manufacturer's protocol. The reverse transcription reaction was mixed with iQ SYBR Green super mix and amplified with the iQ5 real-time system. The product was quantified using a standard curve, and all values were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers were designed with the assistance of PrimerBank and are listed in Table 1.
Alkaline phosphatase activity and alizarin red-S staining
The in vitro osteogenic capacity of ASCs exposed to DMOG was then confirmed by determining the deposition of mineralized matrix with alizarin red-S (ARS) staining 21 days after osteogenic induction. Cell cultures were fixed in neutral-buffered formalin (Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) for 30 min and stained with ARS (Sigma) for 30 min. The cultures were then evaluated by light microscopy and calcium deposition was confirmed by the formation of red nodules. Quantification was performed using a colorimetric assay. The cultures were incubated with 20% methanol/10% acetic acid solution (Sinopharm Chemical Reagent Co., Ltd) for 15 min followed by optical density determination at 450 nm. The alkaline phosphatase (ALP) activity was also determined in cell lysates using an ALP kit (Catalog No. GTX85593; Gene Tex, Irvine, CA) 14 days after osteogenic induction, according to the manufacturer's instructions. Values were measured at 420 nm optical density and normalized against the protein concentration measured with a BCA protein assay kit (Pierce Biotechnology).
Preparation of transplanted composite
BeaverNano™ hydrogel (Cyagen, Guangzhou, China), a self-assembling peptide gel, was used as the scaffold to load the cells. By mixing with fluids containing polyvalent metal ions, the hydrogels can form three-dimensional (3D) nanofiber networks, which can support cell migration, proliferation, and differentiation within the network. The hydrogels were mixed with cells according to the manufacturer's protocol. Briefly, cells were suspended in 10% sucrose solution (Sinopharm Chemical Reagent Co., Ltd) at 4×106 cells/mL and evenly mixed with the hydrogels in an equal volume. PBS (Sinopharm Chemical Reagent Co., Ltd) was then added to the mixed hydrogel composites at 1:1 and incubated for 30 min to allow cross-linking. The cell/hydrogel composites were then applied to the critical-sized calvarial bone defects in rats. For groups implanted with normal or shHIF-1α ASCs treated with DMOG, cells were cultured in medium with 1,000 μM DMOG for 24 h before mixing with the hydrogel. To ensure the cells were exposed to DMOG in vivo, DMOG was added to PBS during the hydrogel mixing procedure and the mixed hydrogel composites in the two groups contained 1,000 μM DMOG.
Animal surgery and protocol
To evaluate the in vivo bone healing capacity of DMOG-treated ASCs, critical-sized calvarial defects in the parietal bones of rats were performed. The rats were randomly divided into four groups. Group I (n=12) underwent implantation of hydrogels alone. Group II (n=12) underwent implantation of hydrogels mixed with normal ASCs. Group III (n=12) underwent implantation of hydrogels mixed with shHIF-1α ASCs treated with 1,000 μM DMOG. Group IV (n=12) underwent implantation of hydrogels mixed with normal ASCs treated with 1,000 μM DMOG. Following anesthesia with intraperitoneal pentobarbital (3 mg/100 g; Sigma), two 5-mm defects in the frontal parietal bone were carefully made using a trephine drill (Nouvag AG, Goldach, Switzerland), avoiding damage to the dura mater. The hydrogels with or without cells were then implanted into the defects in the parietal bones. Eight weeks after surgery, rats were euthanized and perfused with Microfil (Microfil MV-122; Flow Tech, Carver, MA). In brief, the animals were anesthetized and the rib cage was opened. The descending aorta was clamped and the inferior vena cava was incised. Then, heparinized saline (Shanghai No.1 Biochemical & Pharmaceutical Co., Ltd, Shanghai, China) and Microfil were successively perfused into the left ventricle with an angiocatheter. Subsequently, the rats were stored at 4°C for 1 h to ensure polymerization of the contrast agent.
Evaluation of osteogenesis in vivo
The evaluation of osteogenesis in vivo was performed at 8 weeks after surgery. After harvesting the parietal bone, micro-CT scanning was performed to evaluate new bone formation in the calvarial defect area. The scan was performed using a high-resolution micro-CT II scanner (SkyScan 1076, Kontich, Belgium) at the following settings: X-ray voltage of 80 KVp, anode current of 450 μA, and an exposure time of 400 ms. The images were segmented with a low-pass filter to remove noise, and a threshold of 800 was defined as bone tissue. The parameters of new bone volume (BV)/total volume (TV) and bone mineral density (BMD) of the bone defect area were then calculated.
After micro-CT scanning, six samples from each group were dehydrated in alcohol from 70% to 100%, and embedded in polymethylmethacrylate (PMMA; Sinopharm Chemical Reagent Co., Ltd). The sagittal sections, which represented the central area of the defect, were cut with a microtome (Leica, Germany) and polished to a final thickness of ∼40 mm, as previously reported [29]. The sections were then stained with van Gieson's picrofuchsin according to standard procedures. To evaluate new bone formation, the surface areas of the newly formed bone were measured based on the entire defect area using a Leica digital imaging system and NIH image analysis software. Averages for each sample and each group were then calculated.
Evaluation of angiogenesis in vivo
After micro-CT scanning of bone regeneration, six samples from each group were fixed in 4% paraformaldehyde (Sinopharm Chemical Reagent Co., Ltd) and decalcified with 10% ethylene diamine tetra acetic acid (EDTA; Sinopharm Chemical Reagent Co., Ltd) for 3 days. Then, micro-CT was performed again to evaluate new vessel formation at a resolution of 18 μm per voxel. For segmentation of blood vessels from background, noise was removed by a low-pass Gaussian filter. The blood vessels were then included with semiautomatically drawn contours at each two-dimensional section by the built-in “Contouring Program” (SkyScan CTAn software) for automatic reconstruction of 3D images of vasculature. The number of vessels in the bone defect area was then counted, and the total vessel volume represented by all voxels in the specified Microfil range was calculated.
Immunohistochemistry
After micro-CT, the decalcified samples were embedded in paraffin and sectioned at 5 μm at the central area of the defect. Immunohistochemistry for HIF-1α (ab8366, 1:100; Abcam), osteocalcin (OCN, ab13420, 1:100; Abcam), and CD31 (ab24590, 1:200; Abcam) was then performed in the sections from each group. Briefly, the sections were rehydrated, treated with antigen retrieval, and incubated with the primary antibody at 4°C overnight. Subsequently, the biotinylated secondary antibody and ABC complex were applied and DAB substrate was used to stain the sections. The sections were then treated with hematoxylin and mounted. The results were analyzed with a light microscope.
Statistical analysis
All of the in vitro experiments were performed in triplicate and data were presented as mean±SD. Statistical analysis of the data was performed using ANOVA with an SNK post hoc analysis. P values<0.05 were considered statistically significant.
Results
DMOG suppresses ASC proliferation and enhances ASC survival
The ASCs showed the potential of osteogenic (Fig. 1A), adipogenic (Fig. 1B), and chondrogenic differentiation (Fig. 1C). It also had high expression of MSC markers CD29, CD44, and CD90, whereas low expression of the hematopoietic lineage markers CD31, CD34, and CD45 (Fig. 1D). To determine the influence of DMOG on ASCs proliferation, the numbers of ASCs treated with different concentrations of DMOG were measured with the CCK-8. ASCs proliferation was significantly suppressed after 48 and 72 h of incubation with DMOG, and this inhibition was dose dependent (Fig. 2A). ASCs death was detected using trypan blue staining after exposure to different concentrations of DMOG. There were no significant differences between the death ratio of cells with/without exposure to DMOG (Fig. 2B), which indicated that DMOG at these concentrations had no obvious toxicity in ASCs. The effects of DMOG on serum-deprivation-induced cell death were also determined. DMOG showed a dose-dependent reduction in cell death in serum deprivation conditions (Fig. 2C), which indicated that DMOG enhanced cell survival during cell stress.

The adipose-derived stem cell (ASC) potential of osteogenic, adipogenic, and chondrogenic differentiation was separately confirmed by alizarin red-S (ARS) staining
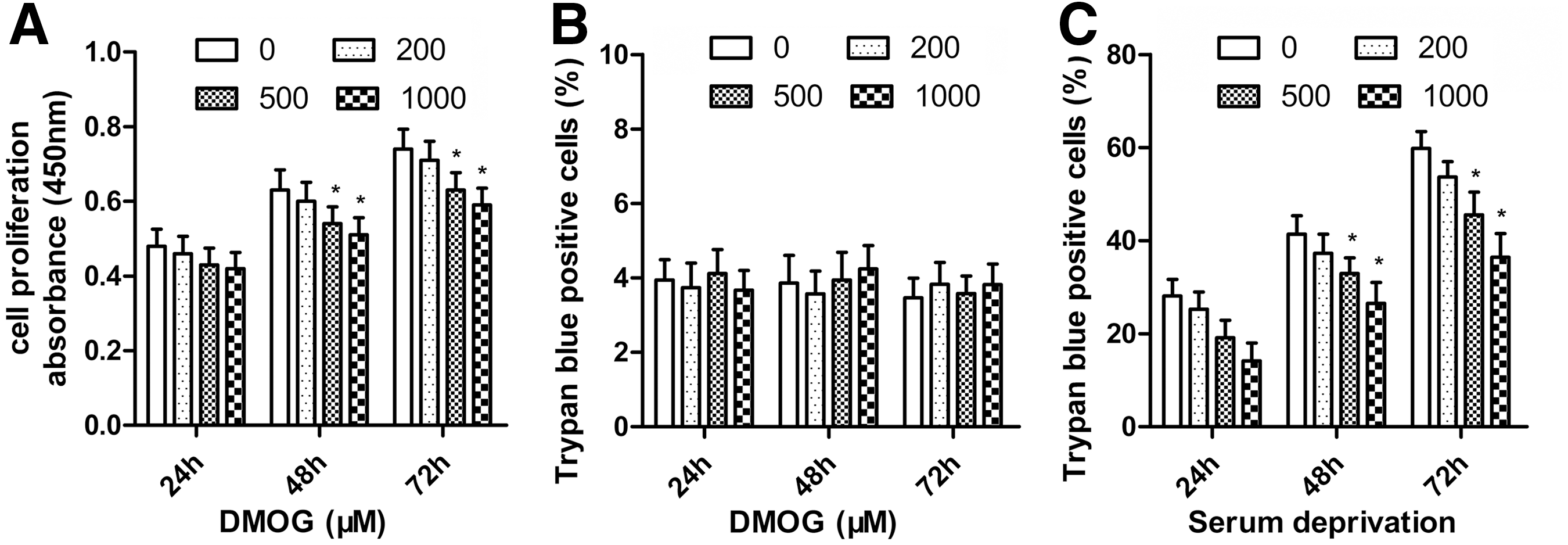
Effect of dimethyloxalylglycine (DMOG) on the proliferation and survival of ASCs.
DMOG promotes expression of VEGF
After treatment with different concentrations of DMOG, the expression of HIF-1α and VEGF in ASCs was detected by western blotting (Fig. 3A). The level of HIF-1α protein was enhanced in response to DMOG treatment in a dose-dependent manner. After treatment with DMOG for 24 h, the expression of HIF-1α in ASCs increased to stable levels, which were ∼2-, 4-, and 6-fold for 200, 500, and 1,000 μM DMOG, respectively. However, there was no notable increase in shHIF-1α ASCs treated with 1,000 μM DMOG (Fig. 3B). The level of VEGF protein was also enhanced after DMOG treatment in a dose-dependent manner. After treatment with DMOG for 72 h, the expression of VEGF in normal ASCs increased by ∼4-, 7-, and 9-fold for 200, 500, and 1,000 μM DMOG, respectively. There was also no significant increase in VEGF expression in shHIF-1α ASCs, which indicated that the increase in VEGF was due to the overexpression of HIF-1α stabilized by DMOG (Fig. 3C).
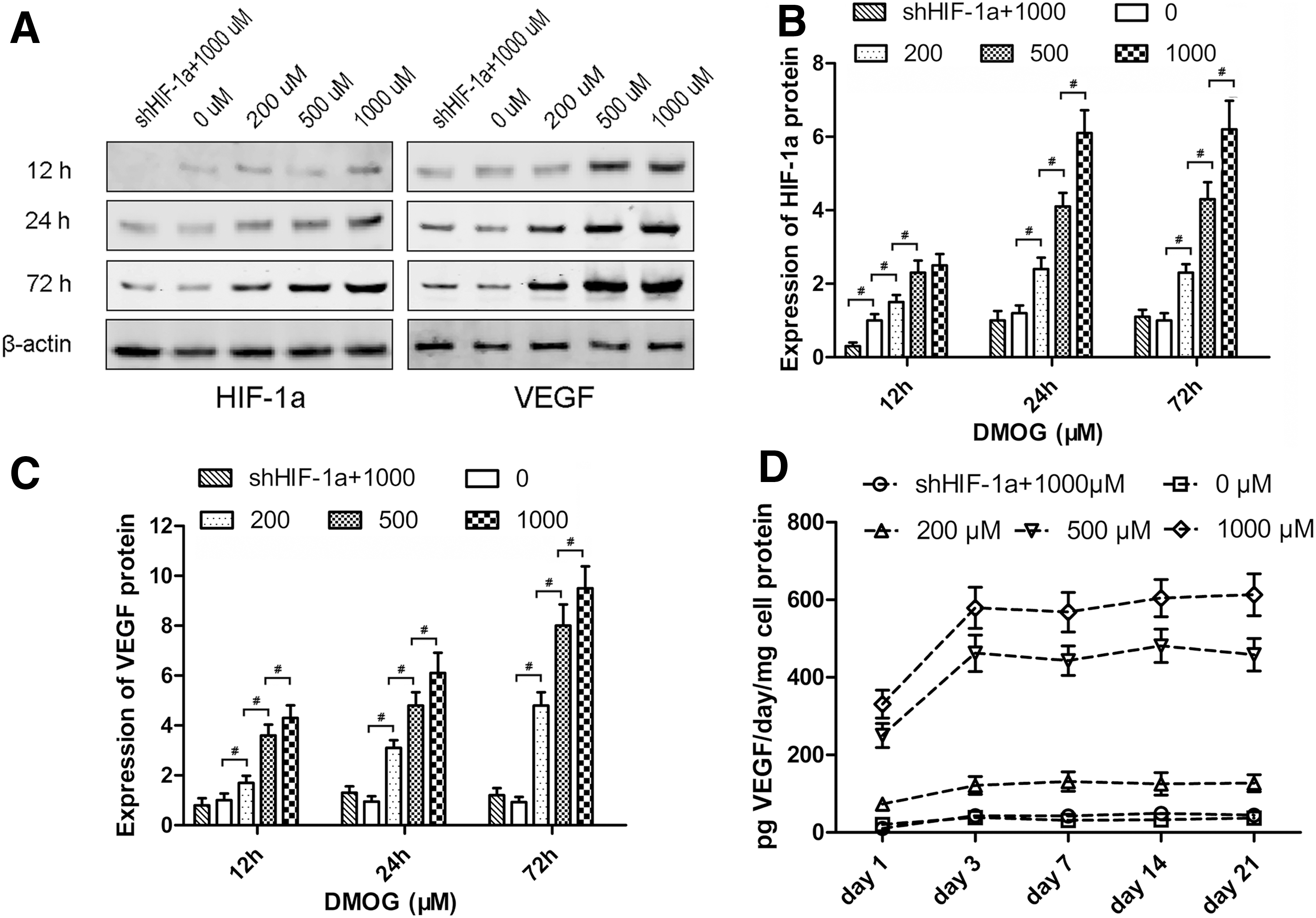
Effect of DMOG on the expression of hypoxia inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) in ASCs.
The levels of VEGF secreted from cells into the culture medium were measured by ELISA (Fig. 3D). VEGF production in normal ASCs was very little and did not change significantly. VEGF production in ASCs treated with DMOG markedly increased from day 1 to 3 and was then maintained at a higher level until day 21. The increase in VEGF production was also dose dependent, and VEGF production in shHIF-1α ASCs was maintained at a lower level.
DMOG improves the osteogenic differentiation potential of ASCs
To verify whether DMOG improves the osteogenic differentiation potential of ASCs, the expression of related osteogenic genes in ASCs, treated with different concentrations of DMOG, was determined by quantitative real-time polymerase chain reaction (qRT-PCR) (Fig. 4A). The transcript level for Runx-2, an early stage osteogenic marker, was upregulated from day 3 to 14 in normal ASCs, and the increase was significantly higher in ASCs treated with DMOG than in normal ASCs (P<0.05). The expression of OCN and ALP, mature-stage bone markers, showed continued upregulation from day 7 to 21, and the increase in ASCs treated with DMOG was also higher than that in normal ASCs during this period (P<0.05). However, the expression of these three osteogenic genes was not significantly enhanced in shHIF-1α ASCs treated with 1,000 μM DMOG, compared with that in normal ASCs.
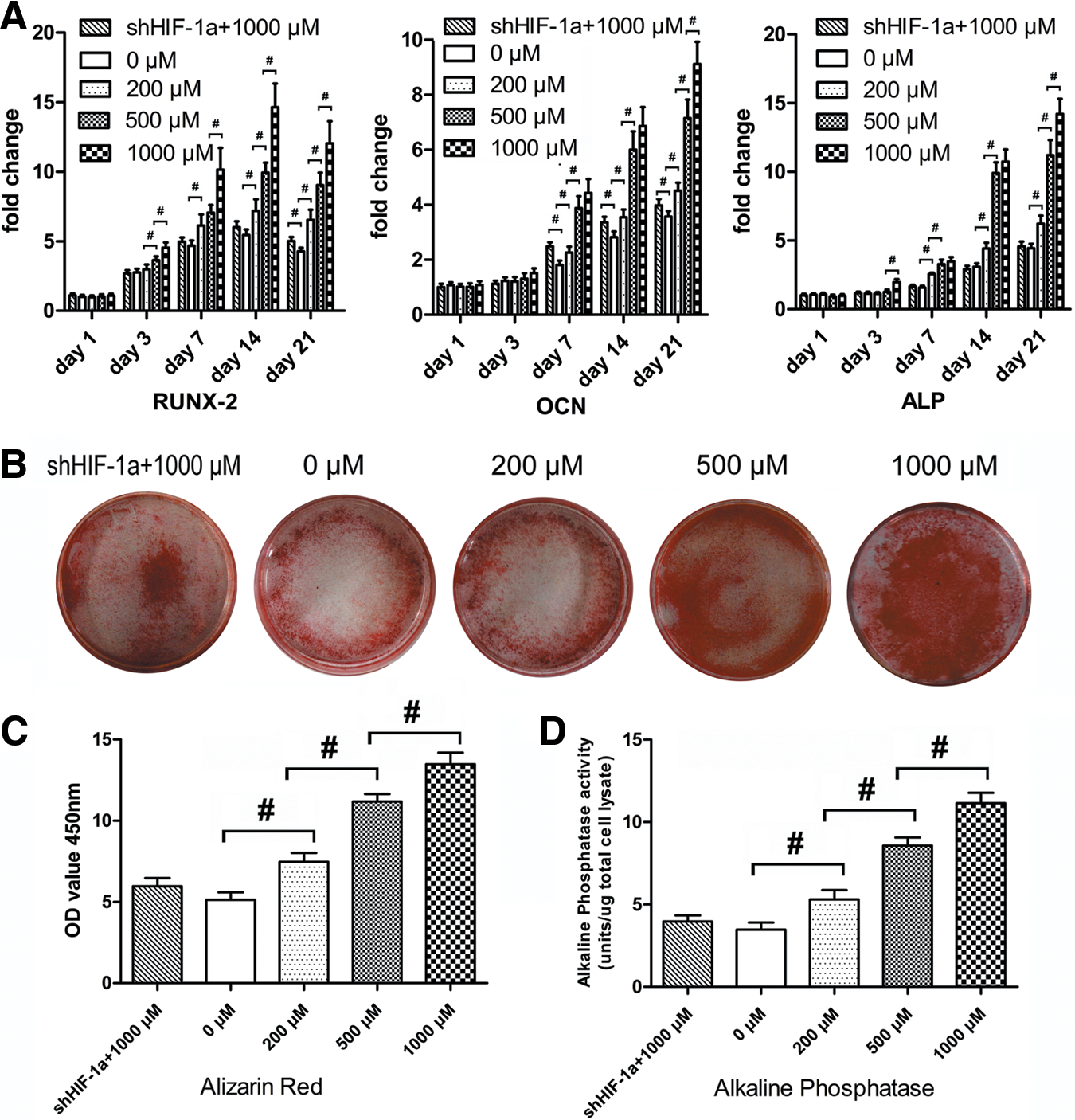
Effect of DMOG on the osteogenic differentiation potential of ASCs.
ARS staining showed more calcium deposits in ASCs exposed to DMOG than in normal ASCs (Fig. 4B). Spectromorphometric quantification showed that the amount of calcium deposits increased by ∼0.5-, 1.1-, and 1.4-fold after exposure to 200, 500, and 1,000 μM DMOG, respectively (P<0.05; Fig. 4C). The results of the ALP activity assay (Fig. 4D) showed that ALP activity in ASCs was significantly increased after exposure to DMOG (P<0.05). ALP activity in ASCs treated with 1,000 μM DMOG was ∼2.5-fold higher than that in normal ASCs. There was no significant difference between ALP activity in normal ASCs and shHIF-1α ASCs treated with 1,000 μM DMOG (P>0.05).
DMOG improves the bone healing capacity of ASCs in vivo
The morphology of the newly formed bone in the defect area of the parietal bone was determined with micro-CT scanning 8 weeks after surgery. In the group implanted with ASCs exposed to 1,000 μM DMOG, plate-like bone structure was formed and occupied a large part of the bone defect area observed on the coronal and sagittal surfaces (Fig. 5A). Quantitative analysis of the newly formed bone was performed using morphometric methods. There was almost no healing of the defect area in the group treated with hydrogel alone. About 23.3% of the defect area was regenerated in the group treated with normal ASCs, and the healing area was about 51.3% of the entire defect area in the group treated with ASCs exposed to 1,000 μM DMOG, which was significantly higher than that in the previously mentioned two groups. The healing area of the group treated with shHIF-1α ASCs exposed to 1,000 μM DMOG was only about 32.3% (Fig. 5B), which indicated that DMOG may improve the bone healing capacity of ASCs by enhancing the expression of HIF-1α. The BMD of the new bone in the four groups was also analyzed. The results demonstrated the same trends, in that BMD was highest in the group treated with ASCs exposed to 1,000 μM DMOG than in the other groups, and decreased significantly when HIF-1α expression in ASCs was inhibited by shRNA (Fig. 5C). Histological findings also showed that the undecalcified specimens in the group treated with ASCs exposed to 1,000 μM DMOG had a larger area of new bone formation in the defect than the other groups, which further supported the micro-CT data (Fig. 6).

Micro-CT evaluation of the repaired bone defect at 8 weeks after implantation.
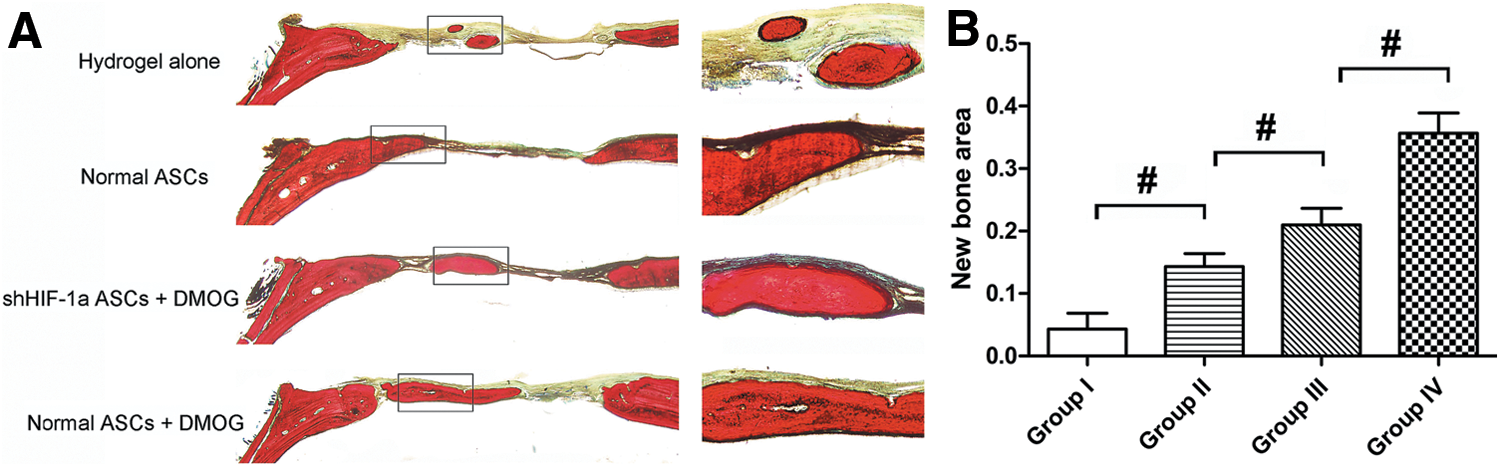
Histological analysis of newly formed bone in the defect area.
DMOG promotes revascularization in vivo
The effects of the implanted cells on vascularization of the defect area 8 weeks after implantation were determined by perfusing the vessels with Microfil and imaging with micro-CT. The reconstructed 3D micro-CT images directly showed newly formed blood vessels in the defect area. The vessels in the defect area of the group treated with ASCs exposed to DMOG were significantly denser than those in the other three groups (Fig. 7A). Quantification of the newly formed blood vessels was performed by morphometric analysis. The number and total volume of blood vessels in the group treated with ASCs exposed to DMOG were more than those in the groups treated with normal ASCs. However, the number and total volume of blood vessels in the group treated with shHIF-1α ASCs were less than those in the group treated with ASCs exposed to DMOG, but more than those in the group treated with normal ASCs (Fig. 7B, C). This may be due to DMOG contained in the hydrogels in Group III that affected autologous bone-marrow-derived mesenchymal stem cell (BMSC) and EC migration to the defect areas, which promoted local revascularization.

Micro-CT evaluation of revascularization in the defect area at 8 weeks after implantation.
Immunohistochemistry analysis
To detect HIF-1α expression in DMOG-treated ASCs implanted in the bone defect area, immunohistochemistry for HIF-1α was performed. In Groups I, II, and III, there were no obvious positive staining for HIF-1α 8 weeks post-surgery (Fig. 8A–C). However, positive brown staining was apparent in the bone defect area of Group IV (Fig. 8D). The expression of OCN, a late osteogenic marker, in the defect area was also analyzed using immunohistochemistry. The results showed that nearly no positive staining for OCN could be found in Group I, a few of positive staining observed in Groups II and III, and more positive staining in Group IV (Fig. 8E–H). Immunohistochemistry for CD31 was performed to evaluate new blood vessel formation in the defect area. Blood vessels were defined with the positive CD31 stain and their typical round or oval structure. The results showed that there were more new vessel formation in Group IV than in other three groups (Fig. 8I–L), which agreed with the micro-CT data.
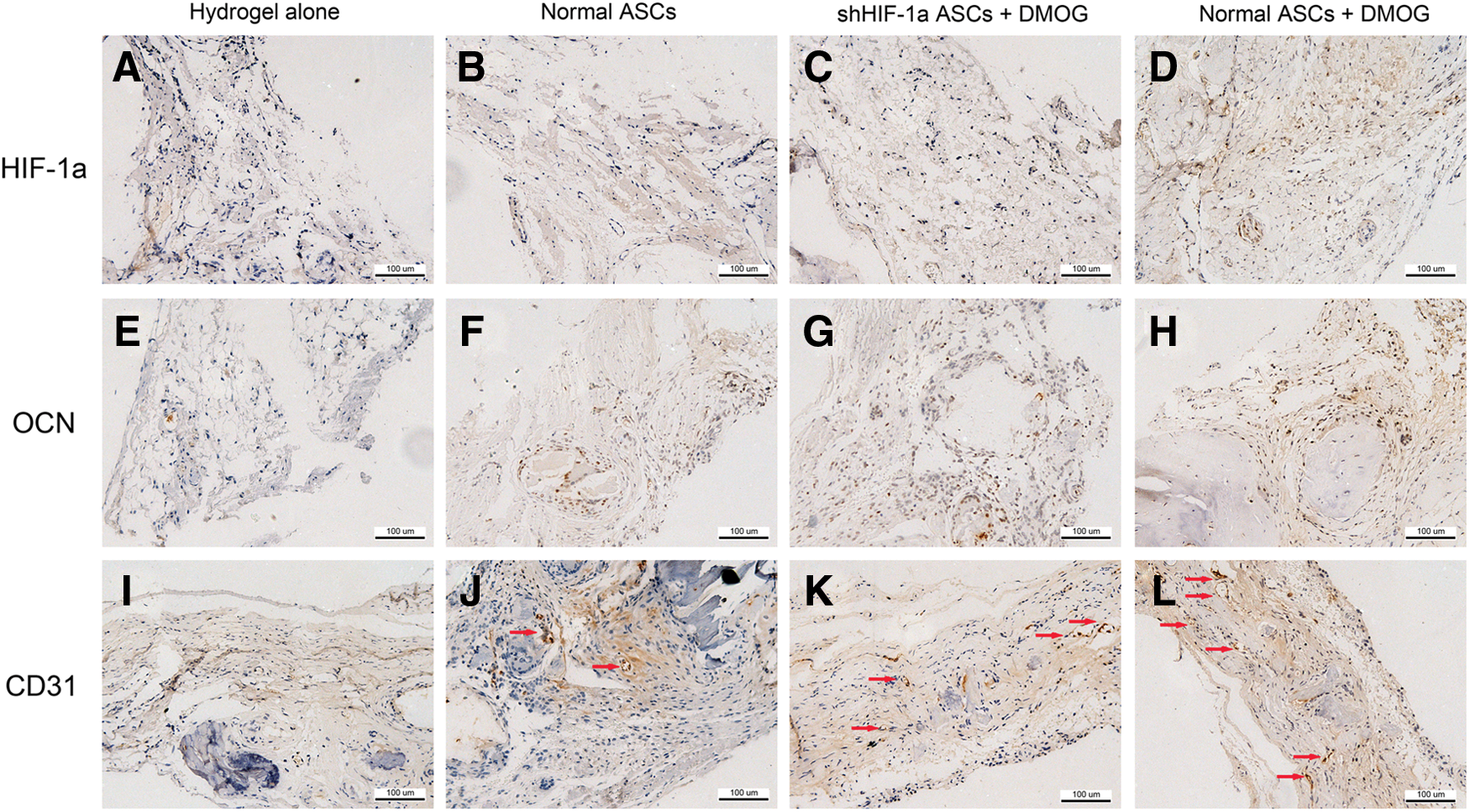
Immunohistochemical analysis of HIF-1α, OCN, and CD31 in the defect area of each group at 8 weeks after implantation. There were no obvious positive staining for HIF-1α in Groups I
Discussion
DMOG is a cell-permeable prolyl-4-hydroxylase inhibitor, which can upregulate the protein level of HIF-1α post-transcriptionally under normoxic conditions. HIF-1α belongs to the Per/Anrt/Sim subfamily of basic helix-loop-helix transcription factors, and plays an important role in bone development and regeneration [30,31]. It also regulates the expression of numerous genes and modulates stem cell proliferation, differentiation, and pluripotency [11]. In this study, we determined whether DMOG improved the osteogenic healing capability of ASCs by activating the expression of HIF-1α, and whether DMOG-treated ASCs would be more suitable as seed cells for bone repair and regeneration in tissue engineering.
Using CCK-8, we found that DMOG slightly inhibited ASCs proliferation. However, trypan blue staining showed that DMOG did not cause the death of ASCs at the tested concentrations (up to 1,000 μM), which indicated that DMOG had no obvious cytotoxic effects on ASCs. Previous studies have shown that HIF-1α upregulation in cancer cells and neurons participates in cell survival and regulation of cell apoptosis under hypoxic conditions or trophic factor deprivation [32 –34]. In this study, we demonstrated that DMOG also inhibited ASCs death induced by serum deprivation in a dose-dependent manner. It was reported that about 90% of transplanted MSCs died on the first day, and no more than 1% survived 4 days after transplantation [35,36]. Therefore, DMOG treatment may enhance ASCs viability in the bone defect area after transplantation, and thereby improve the bone healing capacity of ASCs.
Angiogenic activity of MSCs is regulated by the activation of various signaling pathways, one of which is the HIF-1 pathway. Under hypoxic conditions, HIF-1α is stabilized and accumulated, and subsequently binds to HIF-1β. The HIF-1 complex then activates the expression of VEGF. In the present study, western blotting demonstrated that the expression levels of VEGF in ASCs were also upregulated when cells were exposed to DMOG, and the increase was dose dependent. However, this increase was significantly inhibited in shHIF-1α ASCs, which indicated that DMOG enhanced the expression of VEGF by activating HIF-1α.
Using ELISA to detect VEGF, we found that the sustained supplement of DMOG led to an obvious increase in the secretion of VEGF in ASCs for at least 28 days in vitro. The slow and continued release of VEGF is thought to be crucial for angiogenesis in new bone [37,38]. The micro-CT data and immunohistochemistry for CD31 showed that there were more new vessel formation in the group treated with ASCs exposed to DMOG, which supports this point of view. It has been reported that the expression of other angiogenic factors, such as bFGF, Ang-1, SDF-1, PLGF, and SCF, could also be enhanced by HIF-1α [39 –41]. These factors might also be upregulated by DMOG, and aid VEGF in the promotion of new vessel formation in the defect areas. The effect of DMOG on these factors will be explored in a future study.
Previous studies found that HIF-1α overexpression could directly enhance the osteogenic differentiation potential of MSCs [20,21]. In this study, the results of qRT-PCR, ALP activity, and ARS staining demonstrated that DMOG could improve the osteogenic differentiation potential of ASCs in vitro by activating the expression of HIF-1α. DMOG-treated ASCs were implanted into critical-sized calvarial defects in rats to explore their capability of promoting bone healing in vivo. Micro-CT and histological examination showed that DMOG could obviously improve the osteogenic healing capability of ASCs in vivo. Immunohistochemistry showed that there were few of positive staining for HIF-1α in the bone defect area of group treated with ASCs exposed to DMOG, while nearly no positive staining in other three groups. This demonstrated that DMOG could also activate the expression of HIF-1α in vivo. More positive staining for OCN was observed in group treated with ASCs exposed to DMOG than other groups, which may be due to that DMOG promotes the osteogenic differentiation of implanted ASCs in vivo.
In this study, bone regeneration in group treated with DMOG-treated shHIF-1α ASCs was more than that in group treated with normal ASCs. This may be because the DMOG contained in hydrogels in Group III affected autologous BMSCs and ECs, which had not been interfered by HIF-1α shRNA and could migrate to the defect area to participate in the repair of bone defects. A previous study demonstrated that the delivery of DMOG from scaffold biomaterials improved angiogenic activity and osteogenic differentiation of nearby BMSCs in vitro [42].
We used hydrogels as cell carriers to evaluate the bone healing capacity of DMOG-treated ASCs. The hydrogel can self-assemble into a 3D nanofiber scaffold, which mimics the extracellular environment and supports cell migration, proliferation, and differentiation within it [43]. Their pore size and porosity are favorable for the transfer of growth factors and invasion of new microvasculars [44]. In addition, DMOG can be loaded in the hydrogels conveniently with sustained release kinetics. However, hydrogels have inherent disadvantages as bone repair materials due to their inadequate osteoconductivity. This study showed that peptide nanofiber hydrogels loaded with DMOG-treated ASCs were not enough to repair the bone defects completely. Many bone substitutes can provide more intrinsic stability and are more beneficial for bone regeneration than hydrogels. These scaffolds loaded with DMOG-treated ASCs may be more suitable in tissue engineering for repairing large bone defects and will be investigated in the future. The application of scaffolds loaded with DMOG, which can release DMOG in a sustained and controlled way in vivo, also deserves to be explored in bone tissue repair.
Conclusion
In summary, our results indicated that DMOG significantly enhanced VEGF production in ASCs and improved the osteogenic differentiation potential of ASCs in vitro by stabilizing the expression of HIF-1α in ASCs. DMOG-treated ASCs exhibited an increased bone healing capacity in critical defects in vivo, and promoted local revascularization. This study may serve as evidence for further study of the application of DMOG in cell tissue engineering to promote bone healing.
Footnotes
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81101368 and 81272003).
Author Disclosure Statement
No competing financial interests exist.