
Abstract
Mesenchymal stem cells (MSCs) are used in both human clinical trials and veterinary medicine for the treatment of inflammatory and immune-mediated diseases. MSCs modulate inflammation by decreasing the cells and products of the inflammatory response. Stimulated equine MSCs from bone marrow (BM), adipose tissue (AT), cord blood (CB), and umbilical cord tissue (CT) inhibit lymphocyte proliferation and decrease inflammatory cytokine production. We hypothesized that equine MSCs inhibit T cell proliferation through secreted mediators and that MSCs from different tissue sources decrease T cell proliferation through different mechanisms. To test our hypotheses, we inhibited interleukin-6 (IL-6), nitric oxide (NO), and prostaglandin E2 (PGE2) to determine their impact on stimulated T cell proliferation. We also determined how equine MSCs modulate lymphocyte proliferation either via cell cycle arrest or apoptosis. Inhibition of IL-6 or NO did not reverse the immunomodulatory effect of MSCs on activated T cells. In contrast, inhibition of PGE2 restored T cell proliferation, restored the secretion of tumor necrosis factor-α and interferon-γ, and increased IL-10 levels. MSCs from solid-tissue-derived sources, AT and CT, inhibited T cell proliferation through induction of lymphocyte apoptosis while blood-derived MSCs, BM and CB, induced lymphocyte cell cycle arrest. Equine MSCs from different tissue sources modulated immune cell function by both overlapping and unique mechanisms. MSC tissue source may determine immunomodulatory properties of MSCs and may have very practical implications for MSC selection in the application of MSC therapy.
Introduction
M
MSCs interact with both the innate and adaptive arms of the immune system. Once stimulated, they downregulate specific pro-inflammatory immune cell subsets (ie, cytotoxic CD8+ T cells, Th1 and Th17 T cells, NK cells, dendritic cells, and B-cells) and promote immunomodulatory subsets (ie, inhibitory dendritic cells and T regulatory cells) [14]. One aspect of MSC function is to inhibit T cell proliferation, described in humans, rodents, and many other veterinary species, including horses [12,14 –16]. Blocking prostaglandin E2 (PGE2) function restores human T cell proliferation and secretion of pro-inflammatory cytokines. Interleukin (IL)-10 has also been shown to participate in the inhibition of human lymphocyte proliferation [17] via the induction of T regulatory cells [14,18]. Nitric oxide (NO) production by stimulated MSCs in mice inhibits T cell proliferation; however, its role in the immune response is not fully elucidated [19]. In humans and rodents, mechanisms of MSC inhibition of T cell proliferation include arresting stimulated T cells in the G0/G1 phase of the cell cycle by downregulating cyclin D2 [20] and inhibiting T cell proliferation through apoptosis [21].
In horses, activated MSCs decrease stimulated T cell proliferation [16]. In our previous work, we identified key mediators secreted by equine MSCs in response to activation by stimulated T cells. However, the impact of these mediators, including IL-6, NO, and PGE2, in equine-MSC-based inhibition of T cell proliferation is not known. In addition, NO production varies by MSCs derived from different tissue sources [16], suggesting that MSC source may impact MSC immunomodulatory capability. A deeper understanding of how MSCs regulate T cell proliferation will promote more targeted stem-cell-based therapeutic options for inflammatory and immune-mediated diseases in horses and other species.
The objectives of this study were to determine the importance of key secreted mediators on MSC inhibition of T cell proliferation and to determine the mechanisms by which MSCs altered T cell proliferation. We hypothesized that equine MSCs inhibit T cell proliferation primarily through secreted mediators, similar to mice and humans. However, conflicting evidence between these two species suggests that each species may have unique mechanisms to regulate T cell proliferation. We also determined whether MSC-based inhibition of lymphocyte proliferation was a result of T cell cycle arrest or apoptosis. We hypothesized that MSCs from different tissue sources arrest T cell proliferation through different mechanisms.
Materials and Methods
Tissue collection
MSCs derived from AT, BM, CB, and CT were used in this study. Samples from each tissue type (n=5 each) were obtained from 2 sources: the Center for Equine Health, University of California, Davis (UCD), and the Regenerative Medicine Laboratory at the William R. Pritchard Veterinary Medical Teaching Hospital, UCD, as previously described [16]. Samples were obtained according to approved animal care and use protocols [6 –8,22 –24].
Isolation and culture of MSCs
AT, BM, CB, and CT were processed, cultured, expanded, and cryopreserved exactly as previously described [6,8, 23,24]. In brief, cells were cultured at 37°C, 5% CO2 in low-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco, Invitrogen) with 10% fetal bovine serum (Hyclone), 1% penicillin–streptomycin (Gibco), and 0.1% Fungizone (Gibco) [16].
Mixed leukocyte reaction
Mixed leukocyte reactions (MLRs) were performed exactly as previously described [16]. In brief, peripheral blood mononuclear cells (PBMCs) were obtained via jugular venipuncture followed by density gradient centrifugation. T cell enrichment was performed by column separation using 0.5 g nylon wool (Polysciences, Inc.). T cell-enriched PBMCs were resuspended in 5 mL of cold (4°C) medium. MSCs were irradiated with 10 Gy (Varian 2100C linear accelerator; Varian Medical Systems, Inc.) in suspension at a concentration of 1×106 cells/mL to prevent proliferation. Enriched T cells and MSCs were plated at ratio of 5 T cells:1 MSC and were incubated for 4 days. Cells were treated with 1 mM bromodeoxyuridine (BrdU; BD Biosciences) 24 h prior to analysis. Cells were collected and processed as per manufacturer's directions (FITC BrdU Flow Kit; BD Biosciences) and analyzed on a flow cytometer (Cytomics FC500; Beckman Coulter) exactly as previously described [16]. For cell cycle analysis, 7-aminoactinomycin D was added to cultures as per manufacturer's instructions (BrdU kit; BD Biosciences) and analyzed by flow cytometry on days 1–4.
To determine the role of secreted mediators on T cell proliferation, neutralizing antibodies to IL-6, indomethacin (chemical blockade of PGE2 production), or Nw-Nitro-L-arginine methyl ester hydrochloride (L-NAME; chemical blockage of NO production) were added to the MLR assays on the first day, at the following concentrations: anti-equine-IL-6, 10 μg/mL (R&D Systems); indomethacin, 10 μM (Sigma); and L-NAME, 1 mM (Sigma). See Supplementary Data (Supplementary Data are available online at
Mediator secretion assay
T cell-enriched PBMCs stimulated with phytohemagglutin (PHA; Sigma), with or without blocking agents, were incubated with MSCs at a 5:1 T cell:MSC ratio. Cells were incubated at 37°C, 5% CO2 for 4 days and then the media supernatant was removed. Any contaminating cells were pelleted (300 g, 10 min) and the supernatant was stored at −80°C as previously described [16]. ELISAs for PGE2 (Prostaglandin E2 Parameter Assay Kit; R&D Systems) [25], interferon-γ (IFN-γ; Equine IFN-γ Duoset; R&D Systems), tumor necrosis factor-α (TNF-α; Equine TNF-α Screening Set; Thermo Scientific) [26], and IL-10 (Equine IL-10 DuoSet; R&D Systems) were performed as previously described [16]. IL-6 was measured via ELISA exactly as previously described [27]. All ELISA plates were read spectrophotometrically on a microplate reader (Synergy HT Multi-Mode; Biotek) with Gen5 software (Biotek).
Assay for NO production
NO was measured as previously described using a Greiss reagent system [16]. Briefly, 1 volume of 1% sulfanilamide (Sigma) was combined with an equal volume of media and incubated for 5–10 min at room temperature. Following incubation, 1 volume of 0.1% napthylethylenediamine dihydrochloride (Sigma) was added and incubated for at least 5 min. All samples were read within 30 min at 540 nm on a microplate reader (Thermomax, Molecular Devices, Inc.) and compared to a standard curve.
Statistical analyses
All data are presented as mean±the standard error of the mean. Statistical significance was determined using nonparametric Mann–Whitney tests (α=0.05) based on the sample size (GraphPad InStat version 3.06 for Windows and StatXact 9; Cytel, Inc., Cambridge).
Results
MSC secretion of IL-6 does not inhibit T cell proliferation
As we have previously shown, T cell proliferation is significantly reduced after coincubation with equine MSCs from AT, BM, CT, or CB [16]. To determine the role of IL-6 in modulating T cell proliferation, we utilized an anti-IL-6 antibody that binds secreted IL-6. Titration of the antibody on an IL-6-replication-dependent B cell line demonstrated that the antibody effectively blocked IL-6 function (Fig. 1A). The addition of MSCs to stimulated T cells reduced T cell proliferation. Blocking secreted IL-6 did not reverse this inhibition of T cell proliferation, demonstrating that IL-6 is not responsible for the inhibition of T cell proliferation by equine MSCs (Fig. 1B).

Blocking interleukin (IL)-6 in mesenchymal stem cells (MSCs) does not prevent T cell proliferation. The addition of neutralizing IL-6 antibodies to an IL-6-dependent cell line prevents cellular replication, demonstrating the effectiveness of the antibodies
MSC secretion of NO does not inhibit T cell proliferation
Chemical blockage of NO with L-NAME was used to determine whether NO was involved in MSC inhibition of T cell proliferation. Equine AT and CT MSCs do not produce NO [16]. L-NAME significantly inhibited NO production from both CB- and BM-derived MSCs (Fig. 2A). Coincubation of CB or BM MSCs with stimulated T cells reduced T cell proliferation. The inhibition of NO synthesis by L-NAME did not significantly restore lymphocyte proliferation, indicating that NO is not solely responsible for the inhibition of T cell proliferation by MSCs (Fig. 2B).
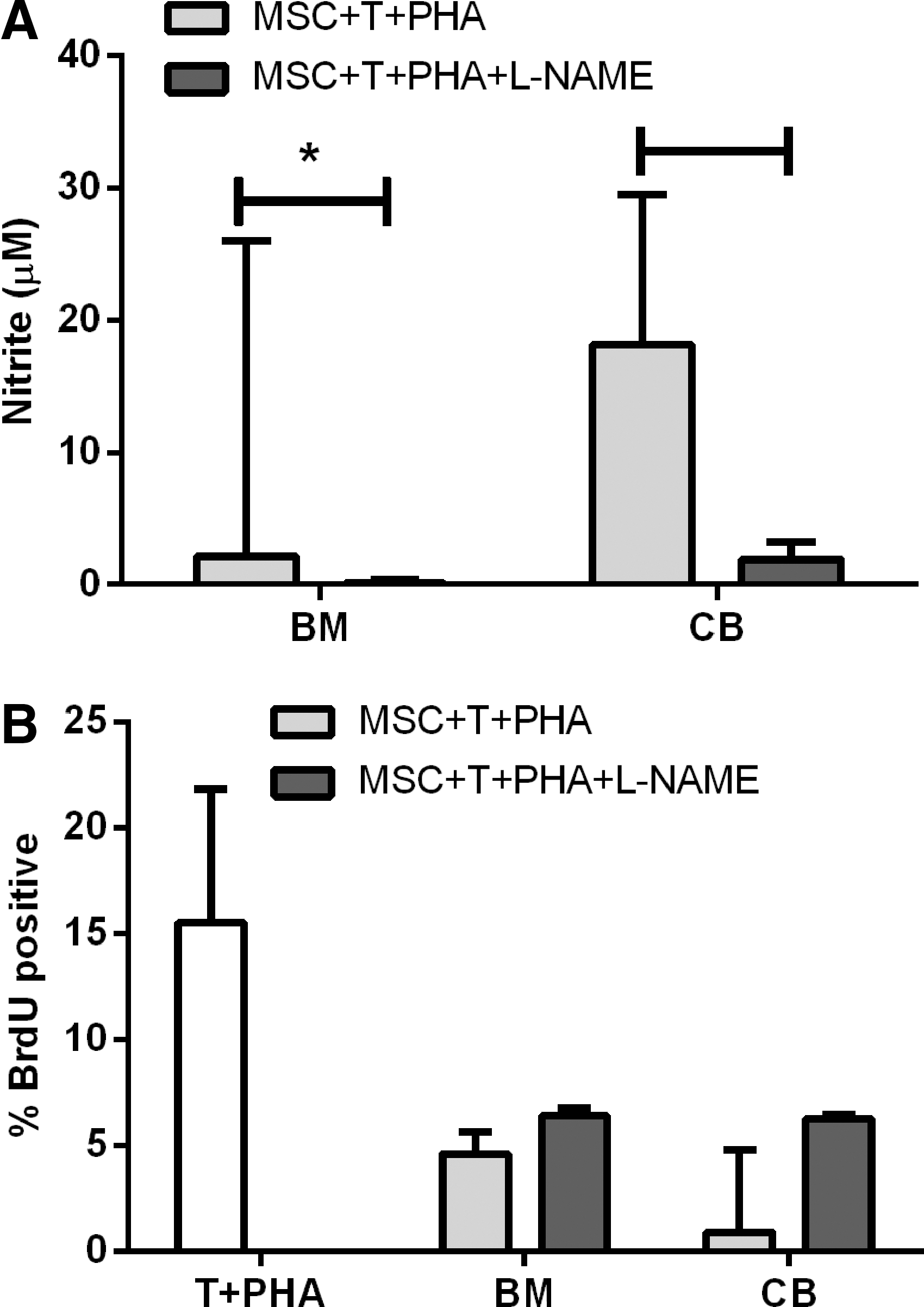
Blocking nitric oxide (NO) is not sufficient to control T cell proliferation. Addition of Nw-Nitro-L-arginine methyl ester hydrochloride (L-NAME) significantly inhibited NO production by stimulated MSCs
PGE2 regulates MSC-induced inhibition of T cell proliferation
Activated equine MSCs from AT, BM, CB, and CT secrete PGE2 in the presence of activated T cells [16]. Incubation with indomethacin resulted in the complete blockade of PGE2 accumulation from stimulated MSCs, regardless of the tissue of origin (Fig. 3A). Blocking PGE2 production significantly increased T cell proliferation (P<0.05) to levels comparable to untreated conditions (Fig. 3B). The restoration of T cell proliferation was accompanied by the restoration of inflammatory cytokine production by T cells, including TNF-α (Fig. 3C) and IFN-γ (Fig. 3D). IFN-γ concentration exceeded the detection threshold of the ELISA in PHA-stimulated T cells alone and in indomethacin-blocked samples. Blocking PGE2 also resulted in a significantly increased IL-10 concentration not found in any other condition when PGE2 is present (Fig. 3E).
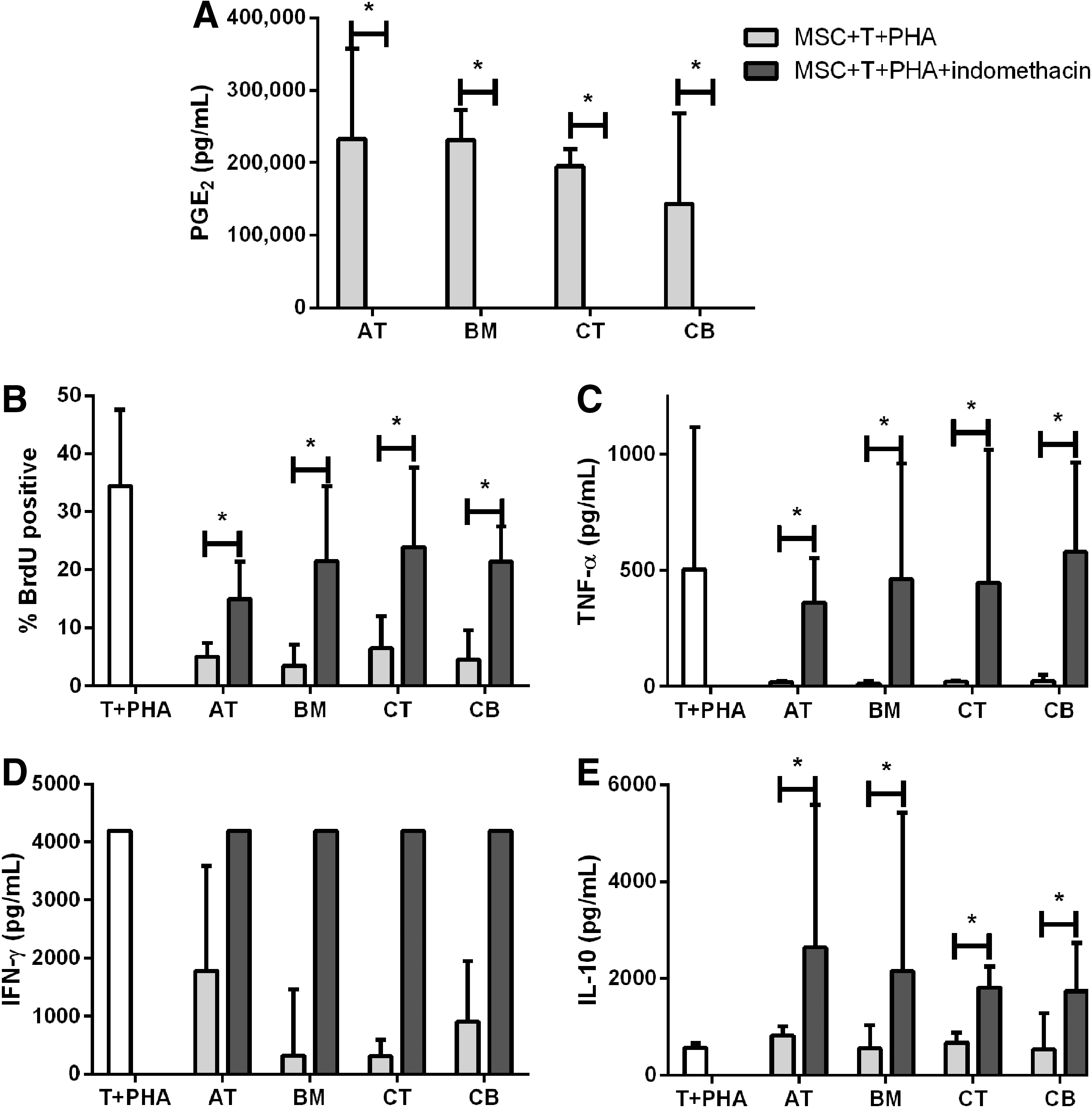
Blocking prostaglandin E2 (PGE2) production in MSCs restores T cell proliferation and cytokine production. Chemical blocking through indomethacin in MSC cultures prevents the production of PGE2
MSCs inhibit T cell proliferation through different mechanisms dependent on MSC tissue source
In addition to determining the chemical mediators responsible for equine-MSC-based inhibition of T cell proliferation, we examined the underlying mechanisms of this inhibition. MSC-induced inhibition of T cell proliferation can occur through multiple pathways, including decreased DNA synthesis in the S phase, cell cycle arrest in the G0/G1 phase, and induction of apoptosis. Stimulation of T cells by PHA results in activation-induced lymphocyte apoptosis over time (Fig. 4A–D, dashed lines). Equine AT and CT MSCs did not inhibit this activation-induced T cell apoptosis (Fig. 4A, B). However, CB and BM MSCs did significantly inhibit activation-induced T cell apoptosis at days 3 and 4 postculture (Fig. 4C, D). AT MSCs did not induce T cell arrest (Fig. 4E). CT did induce some cell cycle arrest by day 4 postculture (Fig. 4F); however, CB and BM MSCs induced a significant arrest of T cells in G0/G1 at days 2 (CB), 3, and 4 postculture (Fig. 4G, H). Regardless of the underlying mechanism (promotion of lymphocyte apoptosis or cell cycle arrest), the end result was a decrease in lymphocyte S phase (DNA synthesis) by days 3–4 of culture (Fig. 4I–L) compatible with the global decrease in T cell proliferation noted in earlier studies (Figs. 1B, 2B, and 3B) [18].

MSC immunomodulation varies by source of origin. Equine adipose tissue (AT) and cord tissue (CT) MSCs did not inhibit T cell apoptosis
Discussion
Overall, our findings demonstrate that MSCs modulate T cells through multiple mechanisms that vary by tissue source. Inhibition of PGE2 reversed MSC-based inhibition of T cell proliferation and restored the secretion of inflammatory cytokines, TNF-α and IFN-γ, as well as enhanced IL-10 production. Further, MSCs from solid-tissue-derived sources, AT and CT, inhibited T cell proliferation through apoptosis while blood-derived MSCs, BM and CB, inhibited T cell proliferation by the induction of cell cycle arrest.
Increased secretion of IL-6 by MSCs in response to allogeneic T cells has been demonstrated in several species, including humans and horses [14,15]. IL-6, in conjunction with TGF-β, regulates the Th17/T regulatory balance [28,29] but blocking IL-6 does not restore human lymphocyte proliferation [30]. Similar to findings in other species, we found little to no impact of IL-6 on T cell proliferation, demonstrating that it is neither necessary nor sufficient for MSC-based inhibition of T cell proliferation in vitro. However, IL-6 may play an important role in other MSC-based immunomodulatory abilities or may be more influential in vivo [31].
NO has been shown in murine, but not human, cells to be important in BM MSC inhibition of T cell proliferation [19,32,33]. NO is only generated by equine MSCs from hemic origins [16]. While NO was not sufficient to significantly restore T cell proliferation in presence of MSCs, there was an approximate 25%–50% increase in the percentage of proliferating T cells with the addition of L-NAME (Fig. 3). These data may suggest a partial restoration of lymphocyte proliferation when NO is blocked, but do not demonstrate that NO is sufficient to control T cell proliferation. In humans, NO is an upstream regulator of IL-10 and TGF-β, and a direct regulator of indoleamine 2,3,-dioxygenase (IDO), all of which regulate a number of immune and anti-inflammatory functions, including macrophage activation [34]. Horse MSCs do not secrete IDO [16], suggesting that NO may participate in upstream regulation of IL-10 or TGF-β as well as in its other functions, including vasodilation and the immune response to pathogens [34]. More research is needed in this area to fully elucidate the role of NO in MSC-based immunomodulation.
MSCs from multiple tissue sources and species have been shown to produce PGE2 as part of their immunomodulatory response [14 –16,35]. We observed similar results in equine MSCs from both blood and tissue sources and were able to abrogate the production of PGE2 using indomethacin [14]. Restoration of T cell proliferation has been shown following PGE2 blocking in MSCs from human BM [14] and CT [36]. However, to our knowledge, this is the first demonstration that PGE2 blocking restores T cell proliferation inhibited by MSCs from AT and CB sources. This is also the first demonstration that equine MSCs modulate T cell proliferation through a PGE2-dependent mechanism. T cell proliferation is one indicator that lymphocytes are activated but secretion of proinflammatory molecules is a critical part of this response. TNF-α and IFN-γ production were also restored with T cell proliferation, demonstrating that the release of proinflammatory cytokines is at least partially controlled by PGE2. Production of these cytokines is likely due to increased secretion by inflammatory T cells [14,37,38]. Our results demonstrate that the PGE2-based immunomodulatory pathway regulates T cell proliferation and cytokine secretion, and is conserved across equine MSCs isolated from multiple tissues (Fig. 3).
Interestingly, indomethacin treatment not only restored T cell proliferation but significantly upregulated IL-10 production. The source of IL-10 secretion has not been fully elucidated though its production is correlated in other publications to increased numbers of T regulatory cells [14,18]. IL-10 levels do not increase in PHA-stimulated T cell/MSC cocultures [17], which is consistent with our findings (Fig. 3E). IL-10 was only detected when PGE2 was blocked, suggesting that the absence of PGE2 may promote an alternate pathway of MSC immunomodulation that includes secretion of IL-10. This discovery further demonstrates the existence of multiple pathways in MSC-based immunomodulation.
MSCs in the presence of inflammatory stimulation significantly inhibited allogeneic T cell proliferation (Fig. 4) as has been previously shown [13,16,35,36]. MSC tissue source had no impact on this aspect of immunomodulation. However, MSCs from CB and BM inhibited activation-induced T cell apoptosis whereas MSCs from AT and CT had little effect on apoptosis (Fig. 4A–D). Our data suggest that both tissue source and animal species affect the mechanism through which MSCs modulate immune cell function. Induction of apoptosis in activated T cells has been previously demonstrated with human BM MSCs [21] but apoptosis of activated T cells does not occur with murine-BM-derived MSCs [39].
MSC inhibition of human T cell proliferation can occur through G0/G1 division arrest of activated T cells [20]. MSCs also inhibit the differentiation of monocytes into dendritic cells by arresting the cells in the G0/G1 phase [40]. We found that BM- and CB-derived MSCs induced T cell cycle arrest in the G0/G1 phase but AT- and CT-derived MSCs did not (Fig. 4E–H). Taken in context with the apoptosis data, we showed that MSCs from solid-tissue-derived sources inhibited T cell proliferation through apoptosis in contrast to blood-derived MSCs that inhibited T cell proliferation by cycle arrest. Direct comparison has elucidated unique mechanisms for the control of T cell proliferation by MSCs in a tissue-dependent manner. Preferential induction of apoptosis compared to cell cycle arrest may be an important indicator of the underlying mechanism of MSC-induced inhibition of lymphocyte proliferation. Indeed, IL-10 is well known to induce T cell anergy in humans [41], suggesting that blood-derived MSCs may preferentially regulate T cell proliferation through an IL-10-mediated pathway. Increased T cell apoptosis may be correlated to future T cell cycle arrest following traumatic injury [42]. Further, recent evidence has shown that MSC therapy may be beneficial for the treatment of HIV [43]. Increased T cell apoptosis is a hallmark of HIV [44,45] and administering MSC therapy that is known to induce T cell apoptosis may have unforeseen complications compared with MSC therapy that is known to induce T cell cycle arrest. To our knowledge, this is the first time that unique mechanisms for immunomodulation pathways have been demonstrated for MSCs originating from different tissues.
Footnotes
Acknowledgments
This project was supported by the Center for Equine Health (University of California, Davis) and a generous gift from Mr. Dick and Carolyn Randall. This work was presented as an abstract at the 3rd Annual North America Veterinary Regenerative Medicine Conference, Savannah, GA, 2012.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.