
Abstract
Intracellular Mg2+, which is implicated in arrhythmogenesis and transient cardiac ischemia, inhibits L-type Ca2+ calcium channel current (I CaL) of adult cardiomyocytes (CMs). We take the advantage of an in vitro model of CMs based on induced pluripotent stem cells to investigate the effects of intracellular Mg2+ on the phosphorylation or dephosphorylation processes of L-type Ca2+ channels (LTCCs) at early and late stages of cardiac cell differentiation. Using the whole-cell patch-clamp technique, we demonstrate that increasing intracellular Mg2+ concentration [Mg2+]i from 0.2 to 5 mM markedly reduced the peak of I CaL density, showing less effect on both the activation and inactivation properties in the late differentiation stage (LDS) of CMs more so than in the early differentiation stage (EDS). Increasing the [Mg2+]i from 0.2 to 2 mM in the presence of cAMP-dependent protein kinase A significantly decreased I CaL in LDS (70%) and in EDS (36%) CMs. In addition, the effect of forskolin was greatly attenuated in the presence of 2 mM [Mg2+]i in LDS but not in EDS CMs. The effect of forskolin was enhanced in the presence of ATP-γ-S in LDS CMs compared with EDS CMs. The exposure of both EDS and LDS CMs to 2 mM [Mg2+]i considerably reduced the effects of isobutylmethylxanthine (IBMX) and okadaic acid on I CaL. Our results provide evidence for differential regulation of LTCCs activities by cytosolic Mg2+ concentration in developing cardiac cells and confirm that Mg2+ acts under conditions that favor opening of the LTCCs caused by channel phosphorylation.
Introduction
I
Intracellular Mg2+ concentration [Mg2+]i in adult CMs is estimated to be between 0.6 and 1.3 mM. Modulation of [Mg2+]i within this concentration interval markedly affects the activity of LTCCs, the release of Ca2+ from the sarcoplasmic reticulum (SR) via Ryanodine receptor, and the sensitivity of the contractile apparatus of the cell [10]. Extracellular applied Mg2+, for instance, can decrease the current amplitude by entering the cell and blocking the Ca2+ channel [11]. As a competitive agent of the Ca2+ channel, extracellular applied Mag2+ decreases the permeability for Ca2+ ions. Previous studies have revealed both the inhibition and stimulation of cardiac L-type Ca2+ channel current (I CaL) under different [Mg2+]i with regard to experimental conditions [8,10,12,13]. These effects of [Mg2+]i on I CaL have been shown to be mainly dependent on the phosphorylation state of the channel [12,14,15]. However, the exact mechanism by which [Mg2+]i affects LTCCs channel phosphorylation in adult CMs is unknown. Two mechanisms may explain how [Mg2+]i modulates I CaL: (i) alteration of the activity of Mg2+-dependent kinases and phosphorylation [10,16,17] and (ii) modulation of the effect of channel phosphorylation on gating kinetics [15]. Nevertheless, information regarding the modulation of I CaL by [Mg2+]i at different developmental stages of cardiac cells is lacking. During development, cardiac cells undergo tremendous and normal physiological changes (eg, morphological changes and the generation of spontaneous electrical activity). These developmental changes have been shown to be associated with a variation in ion channel gene expression and function that not only participates in the intrinsic maturation processes but also contributes to cardiac cell development [18]. Therefore, the properties of ion channels that are intrinsic in developing cardiac cells and the intracellular concentration of each ion differ markedly from those of adult cells. Moreover, the source and pathways of Ca2+ influx required to produce contraction relaxation are highly different between both developing and adult CMs [19 –21]. In developing CMs, Ca2+ ions handling the facilitator's t-tubule system and the SR are also structurally and functionally underdeveloped compared with those in adult CMs. In this study, we aimed at investigating whether and how [Mg2+]i modulates the phosphorylation processes and participates in the [Mg2+]i-dependent regulation of I CaL during cardiac development. Using the whole-cell patch-clamp technique to record I CaL, we examined the [Mg2+]i effects on I CaL under different phosphorylation conditions at different stages of induced pluripotent stem (iPS) cell-derived CMs that have been previously well characterized [22 –24]. Our data demonstrate that LTCCs was markedly regulated by [Mg2+]i under conditions that activate channel phosphorylation in late differentiation stage (LDS) compared with early differentiation stage (EDS), suggesting important developmental changes in terms of receptors (ie, sensitivity to agonists and antagonists), signaling pathways, and interactions.
Materials and Methods
iPS cell line and cell culture
The iPS cell line TiB7-4 was generated from murine embryonic fibroblasts by Alexander Meissner and Marius Werning at the laboratory of Rudolf Jaenisch at the Whitehead Institute of Technology, MA, USA [25]. The cell line harbors a cardiac-specific αMHC-promoter driving puromycin resistance and enhanced green fluorescent protein [26], yielding highly purified cultures under selection pressure. Undifferentiated iPS cells were cultured on inactive murine embryonic feeder (MEF) layers in the presence of LIF. MEFs used for the propagation of undifferentiated iPS cells were prepared from HIMOF1 outbred mice at embryonic day 14.5 and inactivated by mitomycin C treatment (SERVA Electrophoresis GmbH). The propagation medium was composed of Iscove's modified Dulbecco's medium that was supplemented with 15% fetal calf serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% nonessential amino acids (all purchased from Invitrogen GmbH), 0.1 mM β-mercaptoethanol, and 1 U/μL LIF (ESGRO®; Millipore). Adherent confluent cells were passaged every 2 or 3 days by enzymatic dissociation with 0.25% trypsin/0.05% EDTA for 5 min at 37°C.
Embryoid body formation and cardiac differentiation
To investigate the maturation process toward the formation of growing cell clusters, cardiac differentiation of the iPS cells was performed in mass culture. Briefly, 1×105 cells were suspended in 12 mL Iscove's modified Dulbecco medium, which was supplemented with 20% fetal calf serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 1% nonessential amino acids, and 0.1 mM β-mercaptoethanol and placed on a horizontal shaker inside an incubator (37°C) to allow embryoid body (EB) formation. Subsequently, on day 2, EBs were diluted to 1,000 EBs/dish in suspension culture. On day 5 after dilution, EBs were plated onto 0.1% gelatin-coated bacterial dishes for further differentiation. Beating EBs were generally observed at 2 days after plating. The rhythmically beating areas began appearing in EBs at 9 or 10 days postdifferentiation, suggesting the appearance of CM differentiation of iPS cells. For immunostaining and patch-clamping experiments, the beating areas were mechanically dissected, dissociated by trypsinization, and then plated on 0.1% gelatin-coated glass coverslips. CMs isolated from beating EBs at days 9–11 (eg, the day at which the majority of EBs started to beat) were defined as the EDS, and those isolated from beating EBs on days 15–18 were defined as the LDS. Isolated cells were incubated 2–3 days before the experiments.
Immunocytochemistry
Beating EBs (EDS, LDS) were dissociated into single cells and small cell clusters, plated on 0.1% gelatin-coated cover slips and cultured for up to 3 days in incubator. Thereafter, samples were fixed in Prolong Gold Antifade Reagent (Invitrogen) to detect α-actinin, which is characteristic of heart cells. Mouse monoclonal IgG1 anti-sarcomeric α-actinin antibody (Sigma-Aldrich) was used at 1:800 dilutions. The secondary antibody was mouse anti-mouse Alexa 555 (Invitrogen) used at a 1:1,000 dilution. Cell nuclei were stained with Hoechst 33432 and were analyzed with an inverted microscope (Zeiss Axiovert 200/ApoTome) and Zeiss Axiovision 4.5 software (Zeiss).
Electrophysiological recording
Standard voltage-clamp recordings were performed with the classic whole-cell patch-clamp configuration [27]. An EPC-9 amplifier (Heka Electronics) was used for signal enhancement, and the PULSE/PULSEFIT program (Heka) was used for data acquisition and analysis. Leak subtraction was applied in all experiments. Pipettes (at 3–5 MΩ resistance when filled with standard intracellular solution) were made from thin-walled borosilicate glass capillary tubes (World Precision Instruments) on a Zeitz DMZ Universal Puller (DMZ). Glass coverslips with attached single cells were transferred into a recording chamber and placed on the stage of an Axiovert 135 TV inverted microscope (Zeiss). During measurements, cells were constantly superfused using a gravitational perfusion system with a perfusion rate of ∼2 mL per minute. The bath temperature was maintained at 37°C±1°C. After establishment of the gigaseal, the membrane capacitance Cm and the series resistance Rseries were compensated to minimize the capacitive transient. A P/4 protocol was used in all experiments to subtract linear leak and capacitance. Only cells showing stable values were included in the analysis. I CaL were recorded in the voltage-clamp mode. The compositions of the different solutions used during the experiment are as follows. The standard extracellular solution contained (in mM): 120 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 20 TEA-Cl, and 10 HEPES, adjusted to pH 7.4 with CsOH. To avoid contamination by Na+ channels, 10 μM Tetrodotoxin (TTX citrate) was administered. The intracellular solution used to fill the patch pipettes contained the following (in mM): 120 CsCl, 10 EGTA, 1 MgCl2, 5 MgATP, and 5 HEPES, titrated to pH 7.4 with CsOH, thus making Cs+ the main intracellular cation. The internal and external solutions contained Cs+ and tetraethylammonium, respectively, to effectively block K+ currents. The intracellular Mg2+ concentration was adjusted by adding the appropriate amounts of MgCl2. Free [Mg2+]i were calculated as previously described [28]. Experiments involving the use of nifedipine (10 μM in DMSO) were performed in the dark. Unless stated otherwise, all substances were obtained from Sigma-Aldrich.
Investigation of the phosphorylation-dependent modulation of ICaL by intracellular Mg2+ concentration during cardiac development
The effect of cytosolic/intracellular Mg2+ concentration on I CaL was studied during CMs development. For this purpose, I CaL was measured in the presence of different agents that were applied extra- or intracellularly. The functional expression of I CaL at different stages of development was tested by the extracellular application of nifedipine (10 μM). The function of adenylyl cyclase (AC) was tested using its potent agonist forskolin (1 mM). The function of phosphodiesterases (PDEs) was tested by using the nonselective inhibitor isobutylmethylxanthine (IBMX, 100 μM), as previously described [29]. Receptor-mediated phosphorylation was investigated by the coapplication of 1 mM forskolin and the thiophosphorylating compound ATP-γ-S (2 mM via patch pipette). The effect of Ca2+ channel phosphorylation was tested by cell perfusion with the catalytic subunit of 7 mM PKA. ATP-γ-S (1 mM) was added to the pipette solution and to the catalytic subunit of PKA to make channel phosphorylation irreversible. We further examined the role of PP1/PP2A on the calcium-dependent processes and the signal transduction mechanisms in CMs using inhibitor okadaic acid (OA, 10 mM).
The current-voltage (I-V) relationship of I CaL was determined by applying test pulses (T, 100 ms) from −60 to +50 mV in 10 mV increments at a frequency of 0.2 Hz. Prepulses from a holding potential of −80 to −40 mV (P, duration: 50 ms) along with TTX (10 μM) were applied to inactivate Na+ currents (I Na). The prestep to −40 mV was also applied to inactivate the T-type Ca2+ current (I CaT).
Steady-state activation was determined by stepping to various prepulse voltages (10 mV increments) levels from −60 to +20 mV for 100 ms from an HP of −80 mV before repolarizing to a fixed test pulse at −50 mV to evoke the tail currents. To evaluate the steady-state activation, we calculated peak conductance (G) as follows: G=I/(Vm – Vr), where Vm is membrane voltage test level and Vr is reversal potential, derived from I-V curve extrapolation for each cell. Peak currents obtained at all voltages were normalized to the maximal value. Steady-state inactivation was determined by stepping to various prepulse voltages (−60 to +40 mV, 10 mV increments) for 5 s before depolarization to a fixed test potential of +10 mV for 100 ms to evoke channel opening. There was a 5 ms interpulse interval that enabled the resetting of the activation gate between the end of the prepulse and the beginning of the test pulse. Both steady-state activation and inactivation curves were fitted with a Boltzmann function (G=A2+(A1−A2)/(1+exp((V 1/2−x)/k) that describes conductance (G) as a function of the potential (x). V 1/2 is half-activating or half-inactivating potential, and k is the slope factor.
Statistical data analysis
The experimental values are expressed as means±standard error of the mean (SEM) for the number of cells indicated. Significance was determined using analysis of variance and Student's t-test. A P value less than 0.05 was considered statistically significant.
Results
Murine iPS cells differentiate toward spontaneously beating CMs
The murine iPS cell line TiB7.4 derived from adult fibroblasts was differentiated to CMs as previously reported [22,23,30]. On inducing differentiation, portions of the iPSC-derived EBs from day 8 or 9 exhibiting GFP-positive areas (Fig. 1A, left panel) contained spontaneously contracting CMs. For all experiments performed, the maximum number (n=3, ∼70%) of beating EBs was obtained between days 15 and 20 postdifferentiation. The functional, structural, and molecular properties of iPS-CMs used in this study were characterized extensively and will be reported elsewhere (Fatima A., et al., manuscript in preparation). Immunocytochemical analysis revealed that iPS-CMs at day 11 of differentiation expressed sarcomeric α-actinin and displayed the typical pattern of cross-striations indicative of normal architecture organization (Fig. 1A, right panel), which are important in maintaining the full function of CMs.
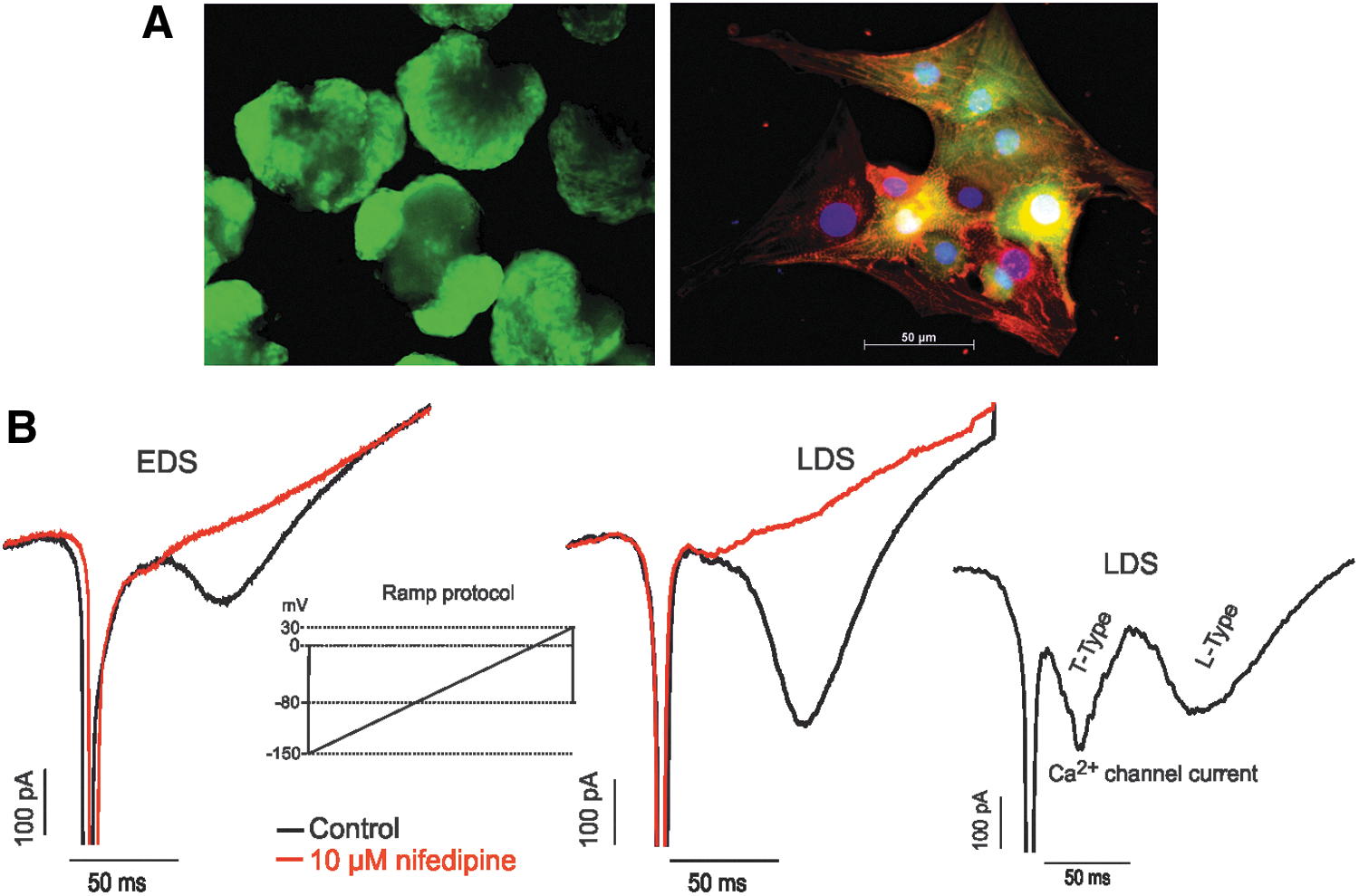
Murine iPS cell-derived cardiomyocytes (iPS-CMs) and functional expression of Ca2+ channel.
Identification of ICaL and its modulation by extracellular Mg2+
Given the positive α-actinin staining and beating activity, we further determined whether these EDS and LDS cells express functional L-type Ca2+ channels. An inward current with two components was recorded in EDS and LDS cells when the cells were depolarized from −60 to 50 mV with a ramp protocol (Fig. 1B, left insert). However, cells showing both components (Fig. 1B, right insert) were detected in large numbers at LDS compared with EDS (data not shown). As previously described [31,32], the first component of the current corresponds to the T-type current (ICaT ), and the second component corresponds to the L-type current (ICaL ). ICaL was detected in all cells at both EDS and LDS but was predominant in LDS, which expressed a robust ICaL . However, ICaT varied from cell to cell with large amplitudes in LDS relative to EDS cells. In LDS cells expressing ICaT , the amplitude of the current was larger than or comparable with that of LDS cells expressing ICaL ; whereas in a few EDS cells in which ICaT was recorded, the amplitude was always small compared with the amplitude of ICaL (data not shown). To further confirm the identity of the L-type Ca2+ channels of murine iPS-CMs at EDS and LDS, we used nifedipine as a specific L-type Ca2+ channel blocker. The extracellular application of 10 μM nifedipine completely blocked the current of EDS and LDS cells. These effects developed rapidly and were partially reversible on washout (Fig. 1B).
As a first approach for our experiments, we determined whether [Mg2+]i modulates L-type Ca2+-channels in murine iPS cells. Concentration-dependent [Mg2+]i effects were monitored between 0.2 and 5 mM in CMs of EDS and LDS. Figure 2A shows the effect of changing [Mg2+]i on the I-V relationship of I CaL obtained from EDS and LDS CMs. The traces indicate that I CaL was activated at different potentials in a concentration- and developmental stage-dependent manner. In EDS cells, increased [Mg2+]i, from 0.2 mM (control condition) to 2 mM, revealed no significant effect on the peak I CaL density (−11.7±1.7 pA/pF, n=8 vs. −9.5±1.0 pA/pF, n=12; P>0.05); whereas in the presence of 5 mM [Mg2+]i, the I CaL of EDS declined significantly to −7.8±1.09 pA/pF (n=7, P<0.05). In LDS cells, the mean I CaL density recorded in the presence of 2 mM [Mg2+]i declined significantly from −20.8±3.11 pA/pF (control, n=10) to −12.5±2.09 pA/pF (n=13, P<0.05). With 5 mM [Mg2+]i, the effect was even more pronounced, declining from −20.8±3.11 pA/pF (control, n=13) to −9.22±1.33 pA/pF (n=10), P<0.01) in mean I CaL density of LDS (Fig. 2B). Thus, increasing [Mg2+]i from 0.2 to 5 mM not only reduced the amplitude of the mean peak I CaL density but also significantly shifted the I-V curve to more negative potentials from 10 mV (−20.8±3.11 pA/pF) to 0 mV (−9.22±1.33 pA/pF) in LDS (n=10, P<0.05) but not in EDS (Fig. 2A).
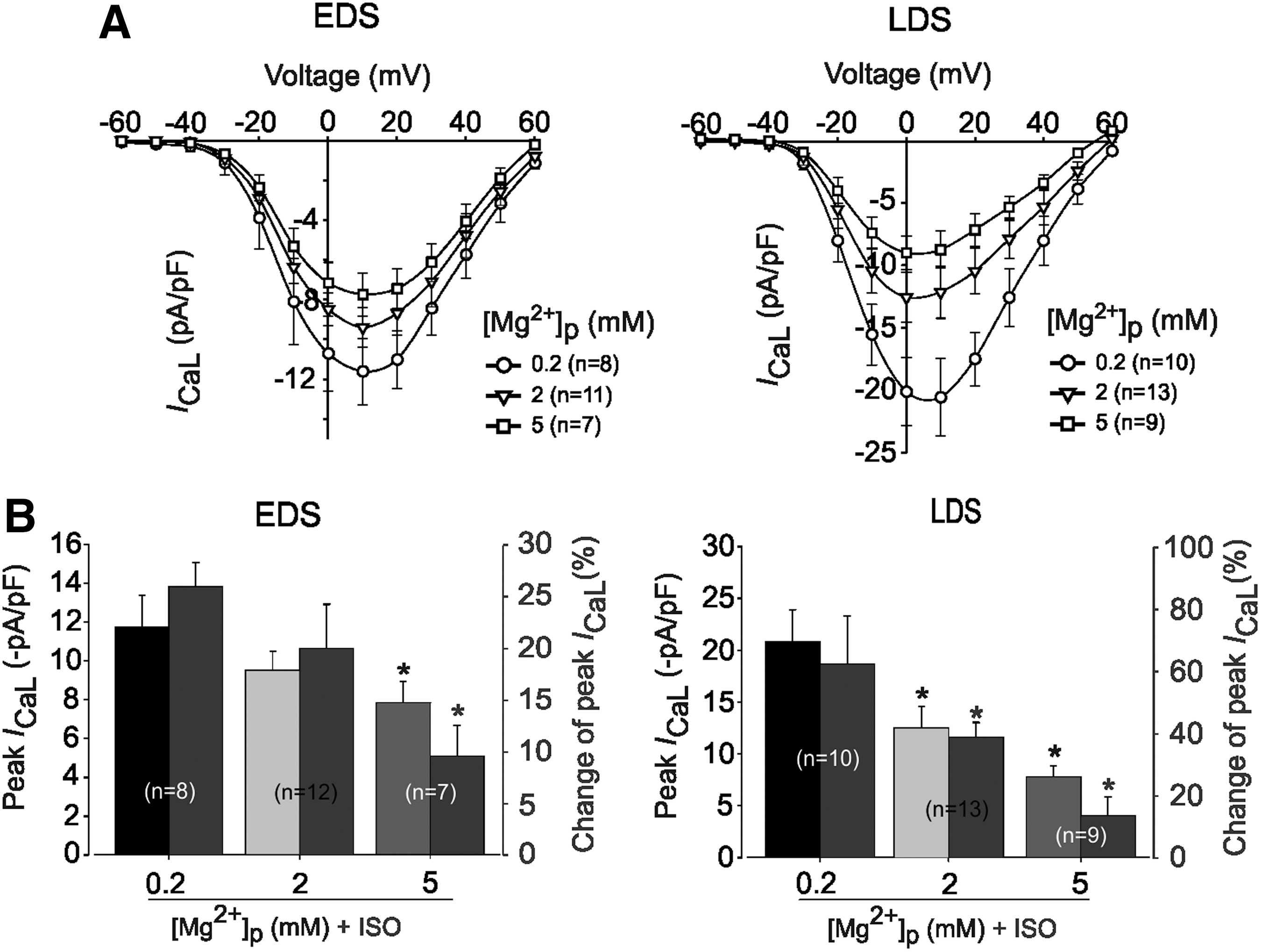
Effects of different concentrations of intracellular Mg2+ on L-type Ca2+ channel current (I
CaL) density in murine iPS cell-derived CMs at different stage of differentiation.
As previously revealed [8], the exposure of adult CMs to higher [Mg2+]i attenuated the response of the β-adrenergic receptors (β-ARs) stimulation of I CaL. However, the extent to which changes in [Mg2+]i affect β-ARs stimulation at an early stage of heart formation or embryonic CMs has not yet been investigated and remains elusive to date. Increasing [Mg2+]i from 0.2 to 2 mM in EDS (Fig. 2B, left) did not significantly reduce the stimulation effect of Iso (1 μM) on the I CaL density [25.9%±2.4% (n=8) vs. 21.0%±4.3% (n=12), P>0.05]. However, the same manipulation in LDS cells (Fig. 2B, right) significantly reduced the Iso (1 μM) stimulation of I CaL, from 62.3%±15.6% (n=10) to 38.9%±4.8% (n=13, P<0.05). In both EDS and LDS cells, an additional increase of [Mg2+]i to 5 mM did not completely block Iso (1 μM) stimulation of I CaL but caused pronounced inhibition.
Since [Mg2+]i may affect the potential drop across the membrane [10], we next examined the possible effects of changing [Mg2+]i on the voltage-dependent activation and steady-state inactivation of I CaL in EDS and LDS CMs. The data shown in Figure 2A were used to determine both the voltage dependence of activation and steady-state inactivation of I CaL as previously described [33]. Consistent with the left shift in the peak of the I-V relationship of I CaL, as indicated by the statistical analysis in Table 1, the increase of [Mg2+]i from 0.2 to 2 mM also induced a significant negative shift of activation curves in LDS; a minor effect was observed in EDS CMs (Fig. 3A). Moreover, the data from the voltage dependence of steady-state inactivation (Fig. 3B) also revealed a left shift V 1/2 of LDS cells, from −21.8±0.9 mV (n=9) in control cells (0.2 mM) to −28.4±1.0 mV (n=6) in the presence of 5 mM [Mg2+]i (P<0.05). These results suggest that [Mg2+]i can differentially modulate the cardiac I CaL in a developmental stage-dependent manner, as LDS CMs showed increased sensitivity to higher [Mg2+]i compared with EDS cells.
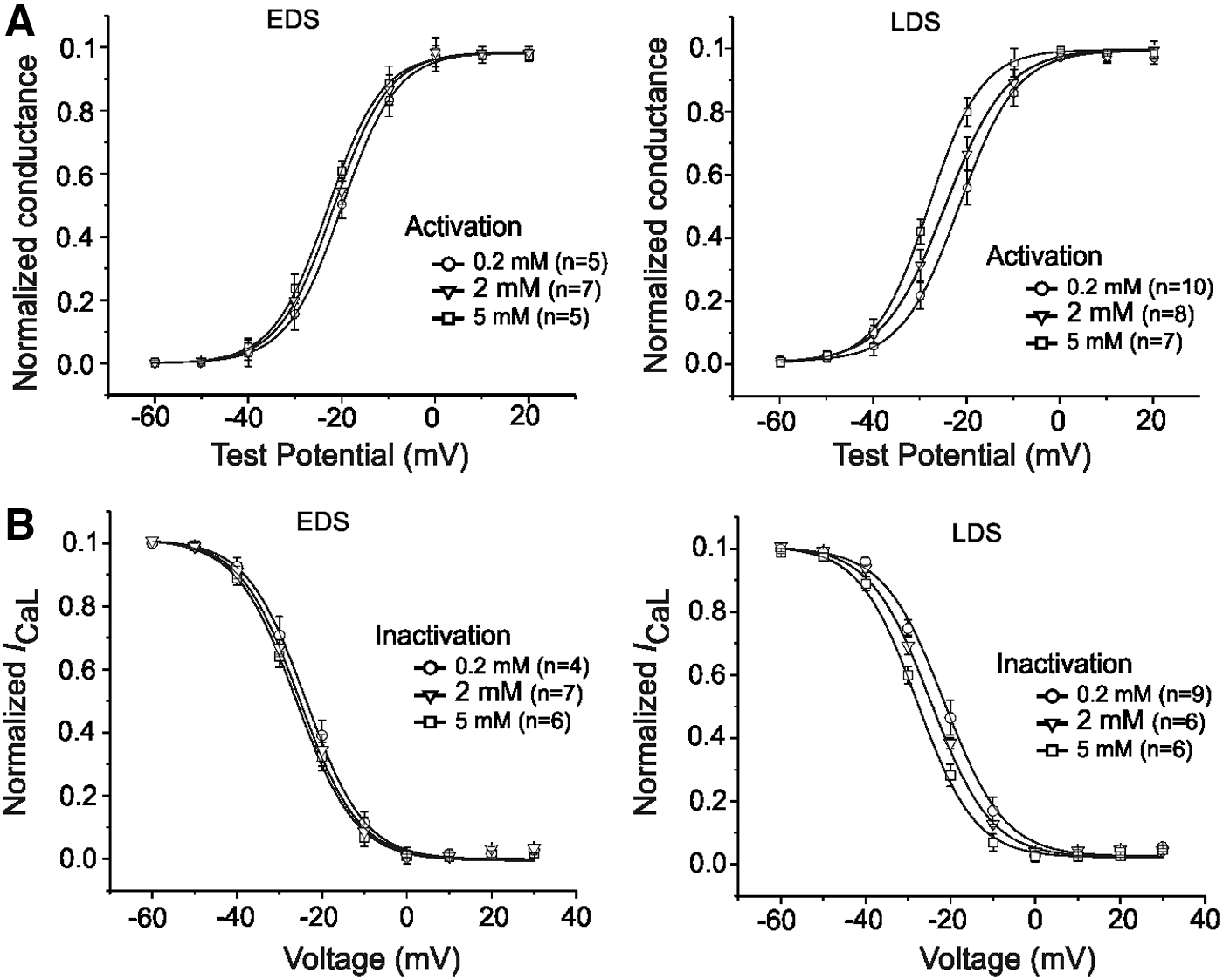
Effects of different concentrations of intracellular Mg2+ on activation and steady-state inactivation properties of I
CaL recorded in murine iPS cell-derived CMs at different stages of differentiation.
Significant difference versus 0.2 mM [Mg2+]p.
EDS, early differentiation stage; LDS, late differentiation stage.
Effects of [Mg2+]i on ICaL recorded under channel phosphorylation and dephosphorylation states
To determine whether the effects of [Mg2+]i on I CaL of EDS and LDS cells are dependent on channel phosphorylation state, as previously shown in adult cardiac cells [5,8,10,12,15,16,34], we performed a series of experiments recording the I CaL of EDS and LDS cells in the presence of low (0.2 mM, control) and high (2 mM) [Mg2+]i.
We first stimulated the phosphorylation pathways by adding PKA (7 μM) to the intracellular solution via pipette. This manipulation induced a significant increase of I CaL density by ∼30% in EDS CMs (Fig. 4A, left); the effect was more pronounced in LDS, with an ∼51% (Fig. 4A, right) increase of I CaL density. Under these conditions, an increase in [Mg2+]i from 0.2 to 2 mM blocked the stimulation effect of PKA in a developmental stage-dependent manner (Fig. 4B) and induced a left shift in the I-V curves (from 10 mV; −29.05±3.0 pA/pF to 0 mV; −8.9±1.0 pA/pF) of I CaL in LDS cells (Fig. 4B, right) but not in EDS cells (Fig. 4B, left).

Effects of changing intracellular Mg2+ concentration in the presence of PKA on I
CaL recorded in EDS and LDS CMs.
To increase phosphorylation, we also treated EDS and LDS CMs with the AC activator forskolin (1 μM) (Fig. 5). With 0.2 mM [Mg2+]i, the mean peak I CaL density was slightly stimulated by forskolin in EDS cells (increased by 10%, Fig. 5A, left) compared with LDS cells (Fig. 5A, left), where it induced an increase of ∼30% (from −20.8±2.3 pA/pF (n=10) to −27.2±1.7 pA/pF (n=6); Fig. 5B). Increasing [Mg2+]i from 0.2 to 2 mM decreased the mean peak I CaL density from −27.2±1.7 pA/pF (n=6) to −9.3±1.3 pA/pF (n=6, P<0.05) in LDS (Fig. 5B, left) and induced a left shift from 10 to 0 mV in the I-V relationship of I CaL. This reduction of I CaL density was not statistically significant in EDS cells (Fig. 5B, right).
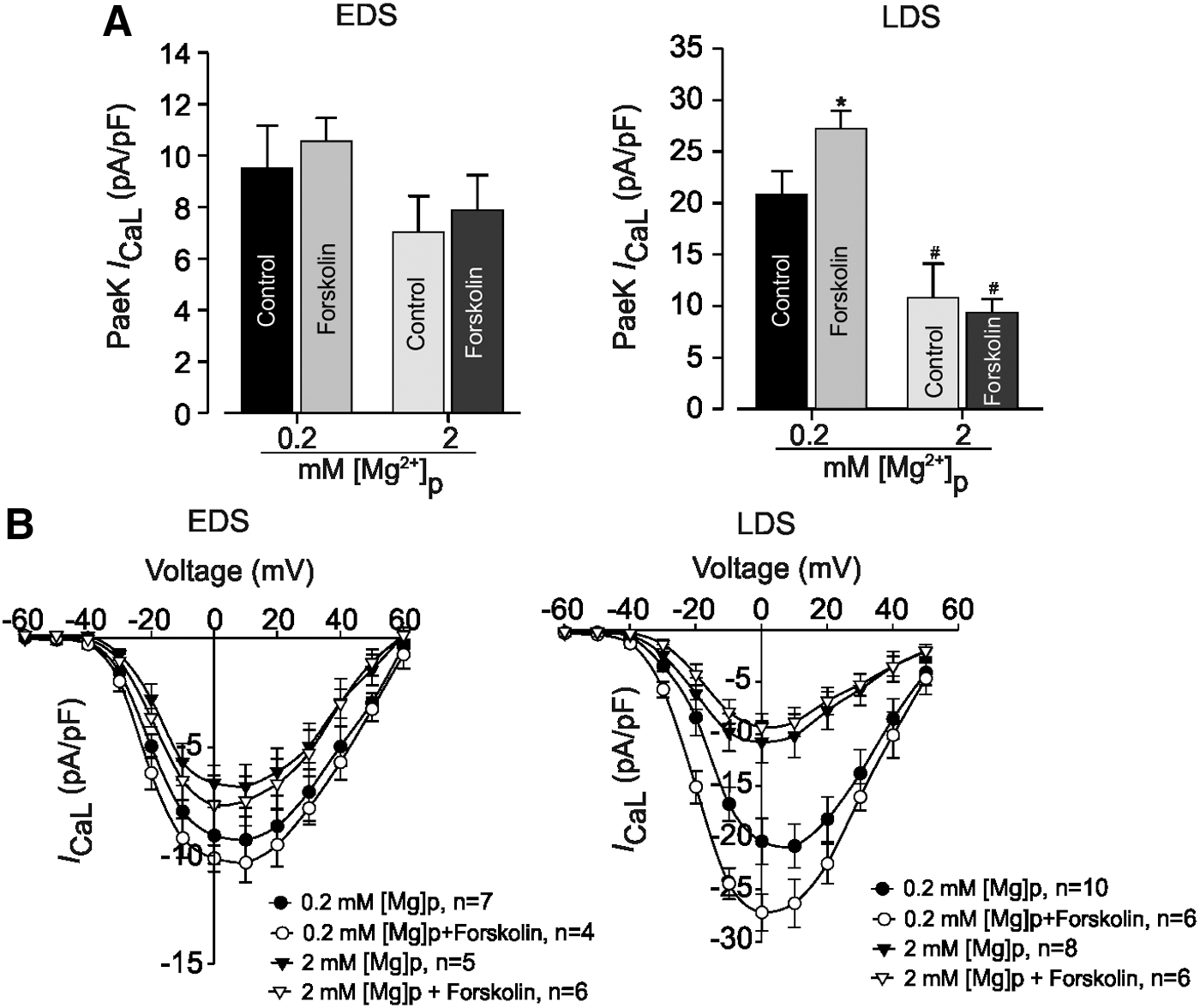
Effect of intracellular Mg2+ concentration on adenylate cyclase stimulation of I
CaL in EDS and LDS CMs.
As recently revealed, increased PDE activity under higher [Mg2+]i leads to decreased cAMP levels [8]. The application of IBMX (100 μM), a nonselective PDE inhibitor, induced an increase of I CaL density in EDS cells (Fig. 6A, B, left). However, this effect was more pronounced in LDS cells (Fig. 6A, B, right). Consistent with previous results in adult CMs, increasing [Mg2+]i decreased the effect of IBMX on I CaL in both EDS and LDS CMs, with a relative left shift of the I-V relationship curve of I CaL. The phosphorylation dependence of [Mg2+]i effects on the I CaL of EDS and LDS cells, as described earlier, can be explained by the modification in the channel phosphorylation levels and the modulation of the effects of channel phosphorylation on gating kinetics (opened/closed probability). To discriminate between these two mechanisms, the I CaL of EDS and LDS CMs were measured with pipette solutions containing the thiophosphorylating agent ATP-γ-S. The addition of 2 mM ATP-γ-S resulted in a large increase of the basal I CaL density in EDS cells, whereas it slightly augmented I CaL in LDS cells (Fig. 7). Under these conditions, changing [Mg2+]i from 0.2 to 2 mM significantly reduced the mean peak I CaL density from 66.6%±2.1% (n=7) to 11.7%±5.6% (n=4) in EDS cells; in LDS cells, a minor effect was observed (Fig. 7B, left).
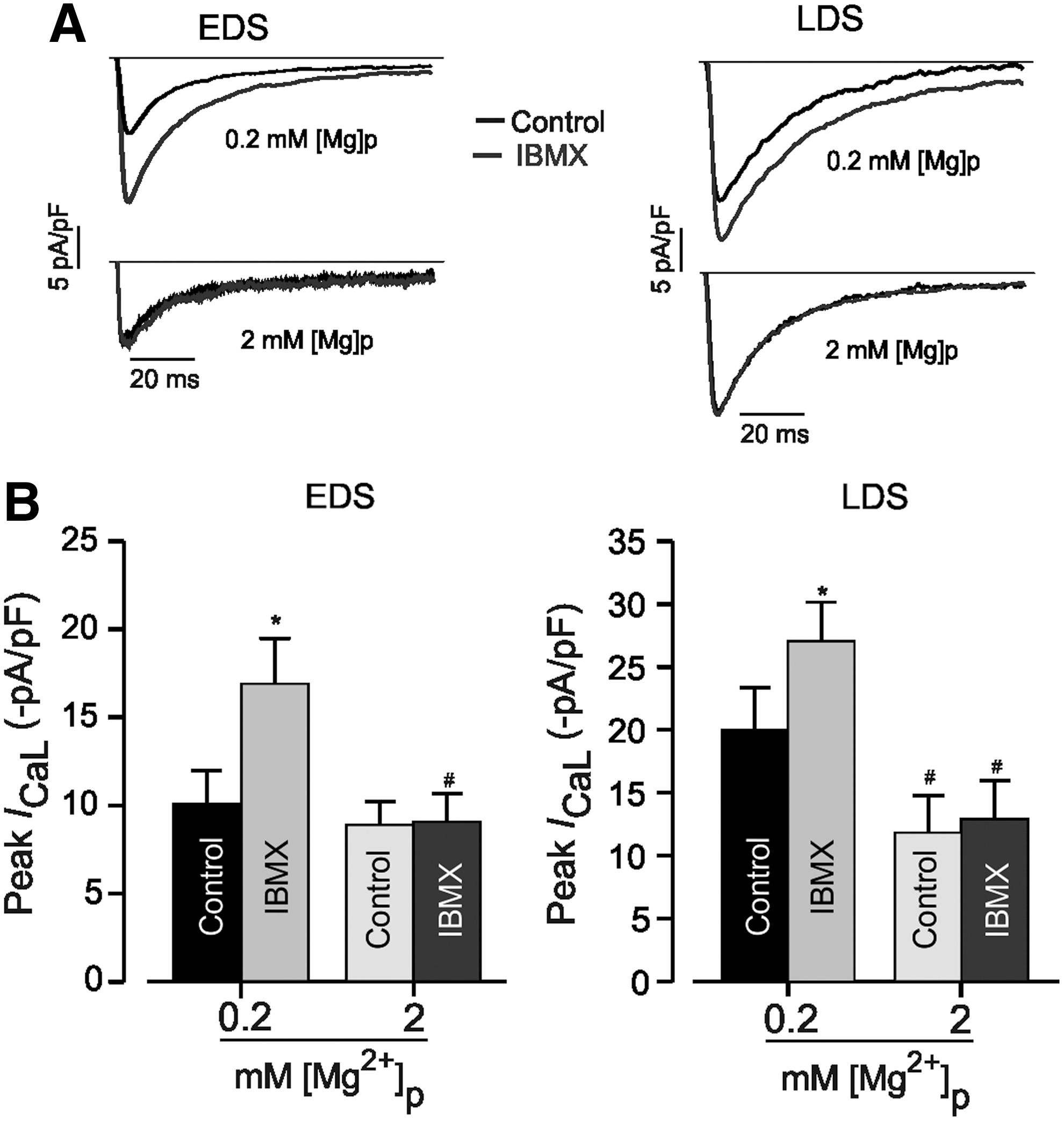
Effects of changing intracellular Mg2+ concentration in the presence of phosphodiesterase inhibitor, IBMX on I
CaL in EDS and LDS of murine iPS cell-derived CMs.

Effects of changing intracellular Mg2+ concentration in the presence of the thiophosphorylating compound ATP-γ-S on I
CaL in EDS and LDS of murine iPS cell-derived CMs.
We further investigated the receptor-mediated phosphorylation by the coapplication of forskolin and ATP-γ-S via pipette solution. This manipulation induced a strong increase of peak I CaL density in EDS (by 75.3%±21.4%, n=7) and LDS (by 111.0%±25.6%, n=10) cells. In the presence of higher [Mg2+]i (2 mM), the I CaL density decreased in EDS cells by 40.7%±12.6% (n=6) and in LDS cells by 25.7%±5.2% (n=7) (Fig. 7B, right). The statistical analysis also revealed that increasing [Mg2+]i from 0.2 to 2 mM induced a larger inhibition of peak I CaL density in LDS (by ∼85%) than in EDS (by ∼35%) and caused a left shift in the peak of the I-V relationship (data not shown).
High intrinsic phosphatase activity was further evaluated in EDS and LDS cells by examining the effect of OA, an inhibitor of type 1 and type 2A phosphatases, on I CaL density under 0.2 and 2 mM [Mg2+]i (Fig. 8). The exposure of cells to OA (10 mM) resulted in an increase of peak I CaL amplitude in EDS cells by 67.0%±9.6% (n=6) and in LDS cells by 69.3%±8.7% (n=7). Increasing [Mg2+]i from 0.2 to 2 mM significantly decreased the peak I CaL density that had been prestimulated with OA in EDS CMs to 22.5%±2.8% (n=4) and in LDS cells to 27.2%±6.5% (n=6) (Fig. 8B). Thus, the most important interpretation of our data presented here is that increasing [Mg2+]i from 0.2 to 2 mM markedly caused the inhibition of I CaL density under conditions that stimulated channel phosphorylation in a developmental stage-dependent manner. In addition, several mechanisms could explain the [Mg2+]i modulation of LTCCs. For example, various interactions involving the Ca2+ channel, β-AR, Gs, AC, and cAMP-PKA and leading to channel phosphorylation or dephosphorylation are [Mg2+]i dependent. We predict that the differences observed in the [Mg2+]i modulation of I CaL may depend on the species, cell type, and changes that happen during development with regard to the intracellular concentration of both Ca2+ and Mg2+ and their interactions with other ions.
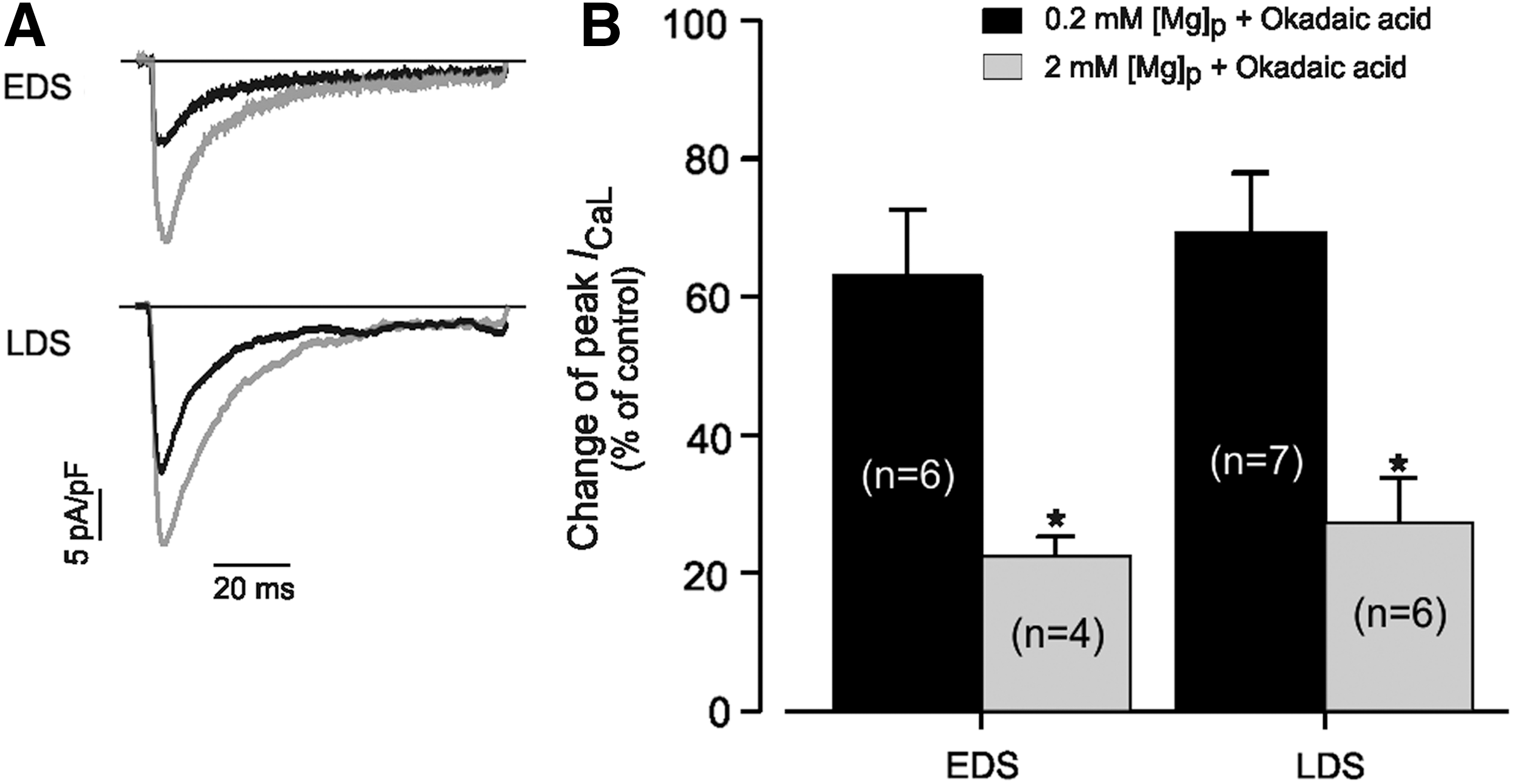
Effect of changing intracellular Mg2+ concentration on I
CaL of EDS and LDS of murine iPS cell-derived CMs measured in the presence of 10 μM okadaic acid.
Discussion
In the present study, using different conditions that mediated the stimulation or inhibition of I CaL as a functional assay, we have compared the effects of intracellular Mg2+ concentration ([Mg2+]i) on I CaL in patch-clamped murine iPS cell-derived CMs at different stages of maturation. Such studies are of major importance, because I CaL density has been shown to significantly increase during cardiac development [7,29]. Moreover, differential changes of proteins and signaling pathways, which are modulated by or require Mg2+ and ATP, occur during cell lineage and organ specification [35,36]. Alterations of their respective intra- or extracellular concentrations could affect the function of several proteins [37]. In addition, in ischemia, the concentration of free [Mg 2+]i is known to increase nearly threefold, thereby increasing the myogenic tone [8]. Although the effects of this divalent cation on I CaL have long been recognized in mature CMs, the mechanisms by which Mg2+ modulates I CaL in the early stages of cardiac cell formation have not yet been studied. Since considerable electrophysiological changes occur during cardiac cell formation and the diseased adult heart recapitulates the gene profiles of embryonic cardiac cells [7,38], these studies may help elucidate some of the pathophysiological effects of Mg2+ deficiency that also occur in the adult heart.
Increasing [Mg2+]i inhibited ICaL density in LDS but not in EDS CMs
The present data revealed that changing [Mg2+]i from 0.2 to 2 mM induced significant inhibitory effects on the I CaL density and altered both the activation and inactivation properties of I CaL at LDS. However, the effects were less pronounced in EDS cells. These effects were accompanied by the acceleration and shift of the steady-state activation and inactivation kinetics to more negative membrane potentials in LDS cells. These observations are in agreement with previous work on adult CMs [8,10,16,34]. The modulation in peak I CaL density and amplitude may be explained by the change in the channel gating state (ie, opened or closed) and/or the movement rates of ions across the LDS cell membrane. Previous studies suggested that the effect of Mg2+ (applied intracellularly) on the Ca2+ channel is dependent on the channel phosphorylation state in adult CMs [10,12,15,39]. Moreover, β-AR stimulation has been shown to strongly increase the I CaL density in mature CMs compared with embryonic and fetal cardiac cells [7,29]. In fact, in mature cardiac cells, signal transduction consists of several pathways of interaction between receptors and the Ca2+ channel. Several works have provided extensive evidence demonstrating robust up-regulation of I CaL by the β-AR/cAMP/PKA cascade during cardiomyogenesis.
The binding of an agonist such as isoprotorenol (ISO) to the β-AR activates a guanine nucleotide-binding (G) protein, which initially induces the activation of AC and thereafter increases the level of cytosolic cAMP. Then, the Ca2+ channel is phosphorylated via activation of the cAMP-dependent PKA, leading to an increase in both the probability of the channel being open and the channel availability and, in turn, an increase in I CaL density [29,40]. In line with previous reports [7,29,41,42], the exposure of EDS CMs to β-AR agonist isoproterenol did not significantly affect the I CaL density, suggesting that significant changes in expression levels and/or pathway interactions occur during the course of cardiac cells' development/differentiation. Our results revealed that the increase of [Mg2+]i from 0.2 to 2 mM attenuated β-AR agonist ISO stimulation of I CaL in LDS, as previously reported in adult mammalian ventricular cells [8]; however, no effect was observed in EDS cells under the same condition. The important changes in the functional expression of the components of the β-AR signaling cascade occurring during cardiomyogenesis may explain this observation. Furthermore, a low level of Gs protein modulation observed in EDS CMs compared with LDS cells, as previously reported [29,35], may also account for a smaller ISO effect observed at this early stage of cell differentiation.
Calcium channel phosphorylation/dephosphorylation and [Mg2+]i effects
In the presence of PKA (applied intracellularly), which was shown to enhance channel phosphorylation [43,44], the change in [Mg2+]i from 0.2 to 2 mM increased the I CaL density in both EDS and LDS CMs. Low concentrations of [Mg2+]i (0.2 mM) significantly inhibited the I CaL density of EDS and LDS CMs under conditions that impaired channel phosphorylation; whereas under normal conditions, low [Mg2+]i did not affect the channel. Therefore, changing the concentration to 2 mM in the presence of intracellular PKA induces moderate effects on I CaL. This observation suggests that [Mg2+]i reduced the I CaL density of CMs at EDS and LDS under PKA, a high phosphorylation condition, in a dose-dependent manner, which was consistent with previous data observed in adult cardiac cells [15,45]. Interestingly, in the presence of a selective PKA inhibitor, PKI (a condition that impairs channel phosphorylation), increasing [Mg2+]i from 0.2 to 2 mM had no significant effect on the peak I CaL for both EDS and LDS cells. However, the effect was relatively more pronounced in EDS CMs than in LDS CMs. This observation suggests that [Mg2+]i modulates the Ca2+ channel by phosphorylation via cAMP-dependent PKA, indicating similar properties between the L-type Ca2+ channel and a closely related protein during cardiomyogenesis with regard to channel phosphorylation sites, as previously suggested [9].
Moreover, in the presence of 0.2 mM [Mg2+]i, manipulation of the phosphorylation conditions significantly affected the peak I CaL density under forskolin in LDS CMs compared with EDS CMs. Interestingly, the strong modulation of I CaL by 0.2 mM [Mg2+]i was detected after the coapplication of forskolin and ATP-γ-S in EDS cells, suggesting that phosphatases are involved in the modulation of I CaL by [Mg2+]i at this stage of cardiac cell development. This observation was further confirmed by the fact that the application of ATP-γ-S alone led to a significant modulation of I CaL by [Mg2+]i in EDS when compared with LDS cells. Since the regulation of I CaL during cardiac heart development was shown to induce high intrinsic activity for most PDE subtypes, such as PDE2-4 at the EDS cell stage [9,43], we further tested whether the intrinsic PDE activity is involved in the modulation of I CaL by [Mg2+]i during cardiac cell development. In agreement with previous studies [8,12,46], in the presence of low [Mg2+]i (0.2 mM), the treatment of CMs with the PDE inhibitor IBMX strongly increased the peak I CaL density at both early and LDS. However, this effect was more pronounced in EDS than in LDS cells, confirming the role and high PDE activity in EDS compared with LDS CMs, as previously suggested [9,43]. In addition, the use of a wide variety of phosphatase inhibitors such as OA (type 1 and type 2A inhibitor) showed the importance of dephosphorylation mechanisms in the control of I CaL under different conditions [47 –50]. We, therefore, tested whether the effect of [Mg2+]i on I CaL was mediated via the activation of PP1 and PP2A. Using OA to block both phosphatases, we found that at 0.2 mM [Mg2+]i, the exposure of EDS CMs to OA induced a substantial increase in I CaL; the same effect was observed in LDS CMs. These observations suggest that the functional intrinsic AC activity and expression of PKA lead to the phosphorylation of Ca2+ channels and take place in both developmental stages. In contrast, the same manipulation conditions mentioned earlier (in the presence of forskolin, ATP-γ-S forskolin+ATP-γ-S, IBMX, and OA) with 2 mM [Mg2+]i had less effect on the peak I CaL density in both EDS and LDS. According to previous observations, several intracellular domain structures of the L-type Ca2+ channel, such as the C-terminus of the α-subunit, have been shown to be critical for channel function (eg, activation and inactivation) [34,51 –53]. In the C-terminus, the EF-hand motif was suggested as a potential binding site at which Mg2+ may regulate channel gating [8,15]. Moreover, other domains, including the calmodulin binding domain and the Ser-1928 residue (which is thought to be phosphorylated in response to elevated cAMP in mammalian CMs), are not excluded in adult CMs.
Taken together with the results discussed earlier, our data suggest that [Mg2+]i modulation of I CaL is boosted by conditions which induce the phosphorylation of specific sites within the α-subunit of the LTCCs. We also previously revealed the expression of all major LTCCs subunit genes in embryonic stem (ES) and iPS cell-derived CMs during the differentiation process [24], suggesting that different response of LTCCs to similar [Mg2+]i observed is not due to the subunit composition of the Ca2+ channel. Moreover, since the T-tubule is absent at EDS and LDS of CMs even at fetal developmental stages [7,54], we assume that Mg2+ may at least, in part, modulate the Ca2+ channel located in the sarcolemma membrane of EDS and LDS cells. Since any modulation of the Ca2+ pathways results in coordinated interplay between the activities of kinases and phosphatases, even in the absence of stimulation [55], our data also suggest that in EDS CMs, all signaling components of Ca2+ channels may be in an inactive state or may not be fully functional (eg, developmental changes in the coupling between β-AR and G proteins in cardiac-specific genes and ionic currents). These observations, at least in part, provide an explanation for the relatively low modulation of I CaL by [Mg2+]i under low or high phosphorylation conditions at this stage of development, suggesting that significant modifications in the channel phosphorylation levels or pathways modulate the channel gating kinetics (opened/closed probability). However, given the complexity of Ca2+-gating properties and the modulation of others ion channels such as sodium and potassium by [Mg2+]i, great caution should be taken when defining the structural basis and experimental protocol regarding [Mg2+]i actions on CMs. Further studies will be required to assess the exact [Mg2+]i in mouse ES or iPS cell-derived CMs at different stages of differentiation.
Footnotes
Acknowledgments
The authors acknowledge the skillful technical assistance of Susan Rohani, Annette Köster, and Cornelia Böttinger. They also thank Suzanne Wood and Elke Lieske for the secretarial assistance and the electronic and mechanic workshop, especially Frank Stassen and Harald Metzner for their technical support.
This study was supported by EU FP7 (STEMCAM) and the BMBF (FKZ 0313926B).
Author Disclosure Statement
No competing financial interests exist.