
Abstract
Pluripotent stem cells (PSCs) offer unprecedented biomedical potential not only in relation to humans but also companion animals, particularly the horse. Despite this, attempts to generate bona fide equine embryonic stem cells have been unsuccessful. A very limited number of induced PSC lines have so far been generated from equine fibroblasts but their potential for directed differentiation into clinically relevant tissues has not been explored. In this study, we used retroviral vectors to generate induced pluripotent stem cells (iPSCs) with comparatively high efficiency from equine keratinocytes. Expression of endogenous PSC markers (OCT4, SOX2, LIN28, NANOG, DNMT3B, and REX1) was effectively restored in these cells, which could also form in vivo several tissue derivatives of the three germ layers, including functional neurons, keratinized epithelium, cartilage, bone, muscle, and respiratory and gastric epithelia. Comparative analysis of different reprogrammed cell lines revealed an association between the ability of iPSCs to form well-differentiated teratomas and the distinct endogenous expression of OCT4 and REX1 and reduced expression of viral transgenes. Importantly, unlike in previous studies, equine iPSCs were successfully expanded using simplified feeder-free culture conditions, constituting significant progress toward future biomedical applications. Further, under appropriate conditions equine iPSCs generated cells with features of cholinergic motor neurons including the ability to generate action potentials, providing the first report of functional cells derived from equine iPSCs. The ability to derive electrically active neurons in vitro from a large animal reveals highly conserved pathways of differentiation across species and opens the way for new and exciting applications in veterinary regenerative medicine.
Introduction
T
Although fibroblasts have been the cell source of choice for generating iPSCs, studies in humans showed that other cell types such as keratinocytes were more amenable to reprogramming and generated iPSCs with much higher efficiency [14 –16]; whether this may be also the case in other species has not been investigated. Moreover, iPSC lines reported so far from domestic species, including the horse, depended on complex culture conditions for growth, including feeder layers and media supplementation with several growth factors and/or signaling inhibitors, which impose significant limitations in regard to potential future biomedical applications of these cells.
iPSC technology has shown particular potential for the study of human neuron disease, with recent advances leading to the successful generation of different neuronal types and in vitro neuron disease models [17 –19]. In sharp contrast, to our knowledge the generation of neurons from domestic species in vitro has not been reported. Yet, neuropathies and neurodegenerative conditions in general are very poorly understood in veterinary species, and affected animals often end up being euthanized. This is the case of equine grass sickness, a highly prevalent disease in certain parts of the world, particularly the United Kingdom [20], and equine motor neuron diseases similar to ALS or progressive muscular atrophy in humans [21]. Therefore, alongside the desire for better sources of stem cells from horses, there is a real need to develop models to understand neurogenesis and neuropathogenesis in this species. Such models could be useful to better understand mammalian neurophysiology and to reduce the current dependence on murine models [22,23].
In this study, we describe the generation of iPSCs from equine keratinocytes under feeder-free conditions and their robust differentiation into motor neuron-like cells. These novel cell populations will allow insight into development pathways and mechanisms that are otherwise difficult to study in large animals. Our results may also pave the way for new and exciting veterinary applications including disease modeling [24], improved drug development [25,26], and even targeted cell replacement.
Materials and Methods
Cell reprogramming and culture
Both the derivation of primary equine keratinocyte cultures from a 5-month-old filly [27] and the preparation of MMLV constructs have been described in earlier studies [11]. Primary keratinocytes at passage 3 [27] were plated on gelatine-coated six-well plates at a density of 105 cells/well and transduced 48 h later with viruses coding for murine Oct4, Sox2, Klf4, and Myc sequences or carrying the pCLXSN-GFP vector, as previously described for equine fibroblasts [11]; the latter was used to determine transfection efficiency. Three days after transduction, keratinocytes were seeded on irradiated mouse fibroblast STO cell line transformed with neomycin resistance and murine LIF gene (SNL) feeder cells (Cell Bio Labs) in 10 cm dishes at a density of 5×104 cells/dish and kept at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) with 10% fetal bovine serum (FBS) gold (PAA), 0.1 mM β-mercaptoethanol (Invitrogen), 0.1 mM minimum essential medium (MEM) nonessential amino acids, and 1% penicillin-streptomycin supplemented with 4 ng/mL human basic fibroblast growth factor (bFGF) (Peprotech) and/or 1,000 U/mL human leukemia inhibitory factor (LIF) (Millipore). Putative iPSC colonies were mechanically harvested 7 to 15 after transduction and replated in 96-well plates, and they were subsequently passaged individually in 12-well plates. Unless indicated, cell colonies were always harvested using StemPro® accutase (Life Technologies).
iPSCs were later maintained in feeder-free conditions using plates coated with Matrigel (BD) and conditioned media supplemented with 1,000 U/mL human LIF (Millipore). Conditioned media was obtained from mitomycin C-treated equine fetal fibroblasts cultured in Knockout (KO) DMEM (Invitrogen) containing 20% KO serum replacement (Invitrogen), 0.1 mM β-mercaptoethanol (Invitrogen), 200 mM Glutamax-1 (Invitrogen), 0.1 mM MEM nonessential amino acids (Invitrogen), and 1% penicillin-streptomycin. Media was harvested every 24 h for 5 days, filtered and stored at −20°C.
Mouse ES cells (Bruce 4) [11] were cultured in media containing DMEM, 10% FBS gold, 0.1 mM β-mercaptoethanol, 0.1 mM MEM nonessential amino acids, 1% penicillin-streptomycin, and 1,000 U/mL human LIF.
Cell growth analysis
Equine iPSCs, mouse ESCs (Bruce 4) and primary equine keratinocytes were seeded at a density of 104 cells per well for electrical impedance analyses using the XCelligence Real-Time Cell Analysis system (version 1.2.1002; Roche). A normalized Cell Index was obtained from 6 replicate wells, log-transformed and used to calculate Cell Index Doubling Time as per manufacturer instructions.
Spontaneous differentiation of iPSCs in vitro
iPSCs were allowed to grow in suspension in low attachment culture dishes (106 cells/dish) for 5 days in KO DMEM containing 20% FBS (nonheat inactivated; Sigma), 1% non-essential amino acid (NEAA), 200 mM Glutamax-1, and 0.1 mM B-mercaptoethanol. Embryoid bodies (EBs) were transferred onto gelatine-coated coverslips or chamber slides in the same medium and allowed to differentiate for 15–20 days after which they were fixed in 4% paraformaldehyde (PFA) prior to immunostaining for appropriate germ layer-specific markers.
Differentiation of iPSCs in vivo
iPSCs grown in feeder-free conditions (5×106 cells, passage 20) were suspended in 100 μL DMEM containing HEPES, 10%FBS, and 200 nM L-glutamine, and 20 μL were transferred under the kidney capsule of 8-week-old male non obese diabetic/ severe combined immunodeficiency (NOD-SCID) gamma mice (
Neuronal culture and differentiation
iPSC were placed in suspension culture with slow rocking and maintained for 72 h in media containing KO DMEM/F12 (Invitrogen) supplemented with LIF 500 U/mL, 0.1 M B-mercaptoethanol, Glutamax-1, 0.1 mM NEAA, and 10% KO serum replacement. Neurospheres were then treated for 15 to 30 s with accutase and plated on poly (d) lysine and Mg-coated glass coverslips with the same media. After 24 h (day 0), media was either left unchanged or changed to neuronal maturation media consisting of KO DMEM/F12 with Glutamax-1 and 0.1 mM NEAA supplemented with 1:100 StemPro® neural supplements (Invitrogen), and cells were cultured for a further 21 days. Samples were harvested on days 0, 3, 7, and 14 of culture and further analyzed to determine neuronal maturation. All experiments were conducted in triplicate.
Acetylcholine assay
Culture media collected from triplicate wells was analyzed using Amplex Red Acetylcholine assay kit (Invitrogen), and a Synergy HT plate reader (Bio-TEK instruments), as per manufacturer's instructions.
Electrophysiological recordings
Whole-cell current-clamp recordings were obtained from neurons using thick-walled borosilicate glass electrodes (∼4–7 MΩ) filled with 155 mM K-gluconate, 2 mM MgCl2, 10 mM Na-HEPES, 10 mM Na-PiCreatine, 2 mM Mg2-ATP, and 0.3 mM Na3-GTP, pH 7.3. Coverslips of cultured neurons were placed in a recording chamber perfused with an extracellular solution composed of 152 mM NaCl, 2.8 mM KCl, 10 mM HEPES, 2 mM CaCl2, and 10 mM glucose, pH 7.3 that was supplemented with the blockers of fast synaptic transmission, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 5 μM), D-2-amino-5-phosphonopentanoic (D-AP5; 50 μM), picrotoxin (50 μM), and strychnine (50 μM) to prevent any potential synaptic transmitter activity [28]. Recordings were performed at room temperature at a holding potential of −74 mV (including liquid junction potential) using an Axon Multiclamp 700B amplifier (Molecular Devices). Voltage measurements were low-pass filtered online at 2 kHz and digitized at 10 kHz.
Quantitative polymerase chain reaction
See Supplementary Data (Supplementary Data are available online at
Southern blotting
See Supplementary Data.
Statistical analyses
See Supplementary Data.
Results
Generation of iPSC lines
Keratinocyte monolayers were derived from epidermal explants [27] (Fig. 1A, B) and were transduced with individual retroviral constructs coding for mouse Oct4, Sox2, Klf4, and Myc sequences. Transduction efficiency was about 75%, as indicated by fluorescence-activated cell sorting analyses of cells separately transduced with pCLXSN-GFP. Cells were plated on irradiated SNLs with bFGF and LIF, and within 5 days distinct colonies began to appear that were flat, had well-defined borders, contained cells with a high nuclear to cytoplasmic ratio, and stained positively for alkaline phosphatase (ALP; Fig. 1C); these features are similar to those of human iPSCs [15] and previously reported equine iPSCs [12]. A previous study reported the generation of equine iPSCs without Myc [13]. Therefore, in an additional experiment we transduced cells with Oct4, Sox2, and Klf4 only, however, using this approach we could generate colonies with very low efficiency and that could not be maintained beyond a few passages.
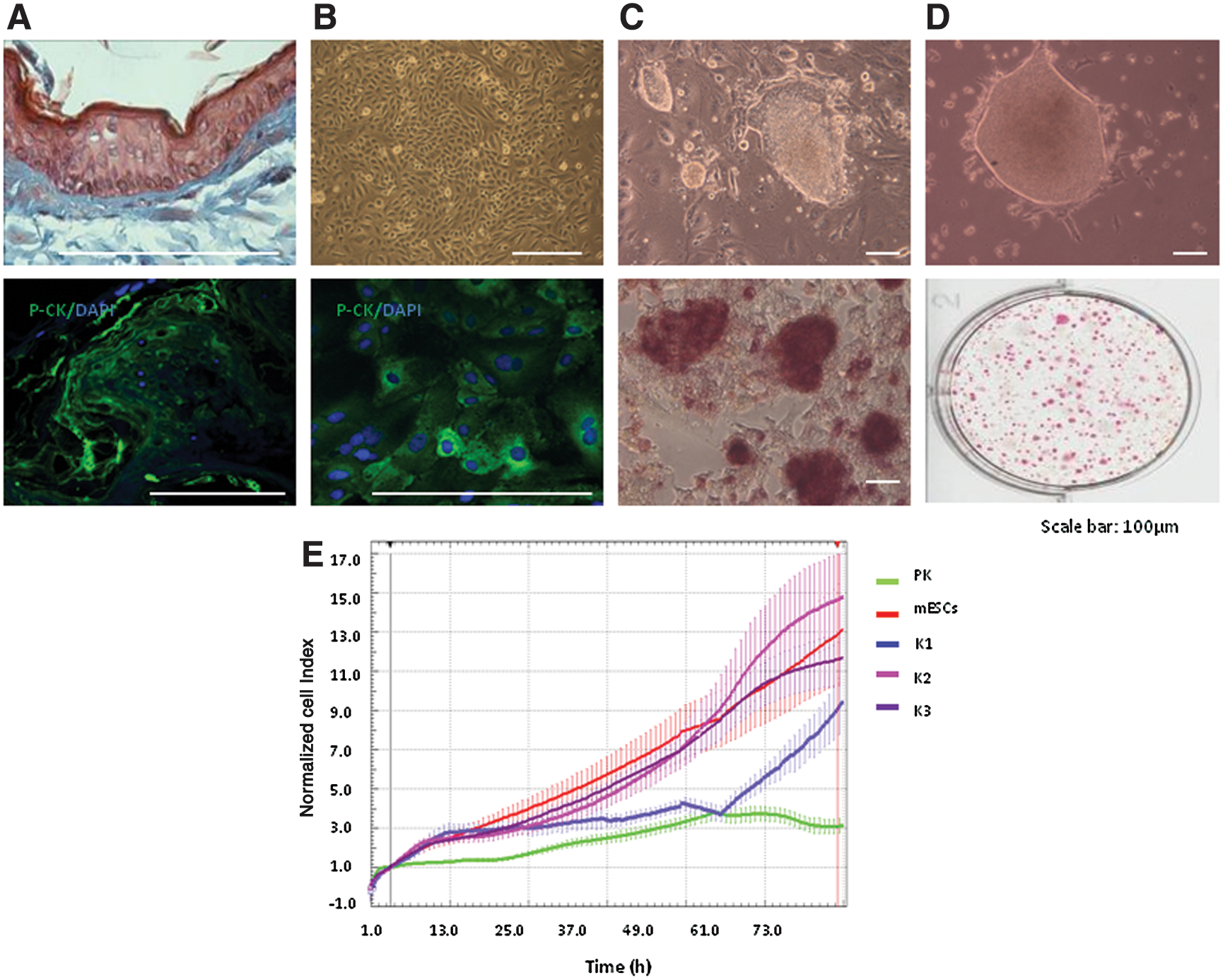
Reprogramming of equine keratinocytes.
A total of 92 ALP-positive colonies were harvested between 7 and 15 days postviral transduction. The efficiency of reprogramming was determined from the number of ALP-positive colonies generated from 50,000 transduced cells plated on SNLs and was equivalent to 0.18%. Sequential passaging of these colonies proved to be unsuitable for long-term maintenance, as they began to show signs of differentiation (Supplementary Fig. S1A). Therefore, cells from newly harvested colonies were gradually adapted to feeder-free and serum-free conditions using equine fetal fibroblast-conditioned media containing knockout serum replacement (Fig. 1D). Cells grown over serial passages under these conditions in the presence of LIF strongly stained for ALP (Fig. 1D), whereas supplementation with bFGF or both bFGF and LIF led to a loss of typical iPSC colony morphology and ALP staining (Supplementary Fig. S1B), indicating dependence of equine iPSC self-renewal on LIF and a pro-differentiating effect of FGF on these cells, as reported for mouse iPSCs [29]. We found that equine fetal fibroblasts were a superior source of conditioned media than mouse fibroblasts, an observation that is in line with the reported benefit of using homologous fetal feeders to maintain equine iPSCs [12]. Six putative iPSC lines selected on the basis of ALP activity were maintained under these conditions for over 40 passages. Three of these lines (K1, K2, and K3) were further characterized.
Growth of these equine iPSC lines was analyzed using the XCelligence RTCA system. Normalized cell index and doubling time were compared between iPSCs, parental keratinocytes, and mouse ESCs (Fig. 1E and Supplementary Fig. S1D). Growth profiles were comparable between K2, K3, and mouse ESCs, and were clearly distinct in parental primary keratinocytes, which became confluent and stopped growing within 48 h of culture. K1 cells had distinct profiles from other iPSC lines or mouse ESCs.
Analyses of pluripotency markers
Immunofluorescence and quantitative polymerase chain reaction (QPCR) analyses showed detectable expression of the endogenous pluripotency markers, OCT4, SOX2, NANOG, LIN28, DNMT3B, and REX1 in all or some of the three cell lines analyzed (Figs. 2 and 3A), as reported in equine fibroblast-derived iPSCs and equine embryonic cells [9,11,12]. Specifically, relative to parental keratinocytes, significant expression of OCT4 and REX1 was detected in line K2 but not in the other two lines, whereas SOX2, NANOG, DNMT3B, and LIN28 were expressed, albeit at variable levels, in the three lines (Fig. 3A). Thus, reprogramming with four factors clearly resulted in reactivation of the endogenous pluripotency machinery, although at a variable extent highlighting the stochastic nature of the reprogramming process [30,31].

Pluripotency marker immunostaining of reprogrammed cell lines. Representative confocal images showing immunostaining of equine iPSCs (line K2) for OCT4, SOX2, LIN28, and SSEA1 (upper row). The lower row illustrates lack of immunostaining in parental keratinocytes. Color images available online at
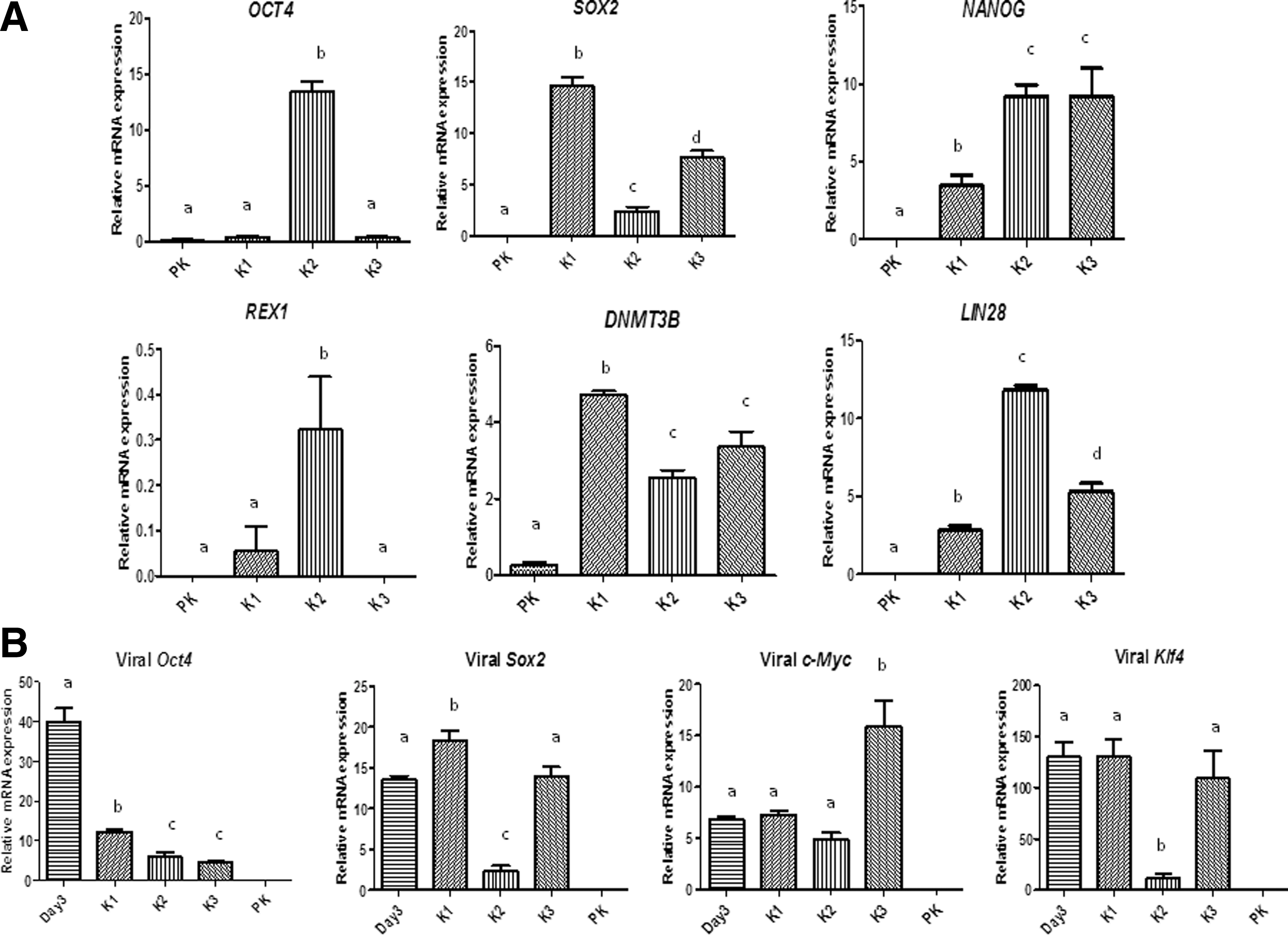
QPCR analysis of pluripotency markers in reprogrammed cell lines. Transcript levels of
PCR and southern blotting revealed effective integration of viral transgenes in the reprogrammed cell lines (Supplementary Fig. S2A, B); moreover, the integration patterns of each of the Oct4 and Klf4 transgenes were clearly distinct among the three lines (Supplementary Fig. S2B), indicating their different clonal origins. Mean transcript levels of viral Oct4 were significantly lower (>3-fold) in each of the reprogrammed cell lines than in the transduced parental keratinocytes (Fig. 3B). The expression of Sox2 and Klf4 was also reduced (6 to 13-fold) in K2 but not in the other two lines. Thus, overall expression of viral transgenes was distinctly attenuated in K2 compared with the other two cell lines.
Multilineage differentiation in vitro and in vivo
After 5 days on low-adherence culture dishes, iPSCs readily formed EBs (Supplementary Fig. S1E). These were allowed to differentiate further on gelatine-coated surfaces soon after which they produced heterogeneous populations containing cells that positively stained for AFP and GATA4 (endoderm), VIM and SMA (mesoderm), and NES and TUBB3 (ectoderm) (Fig. 4). In addition, expression of OCT4 and SOX2 was no longer detected in these cells.
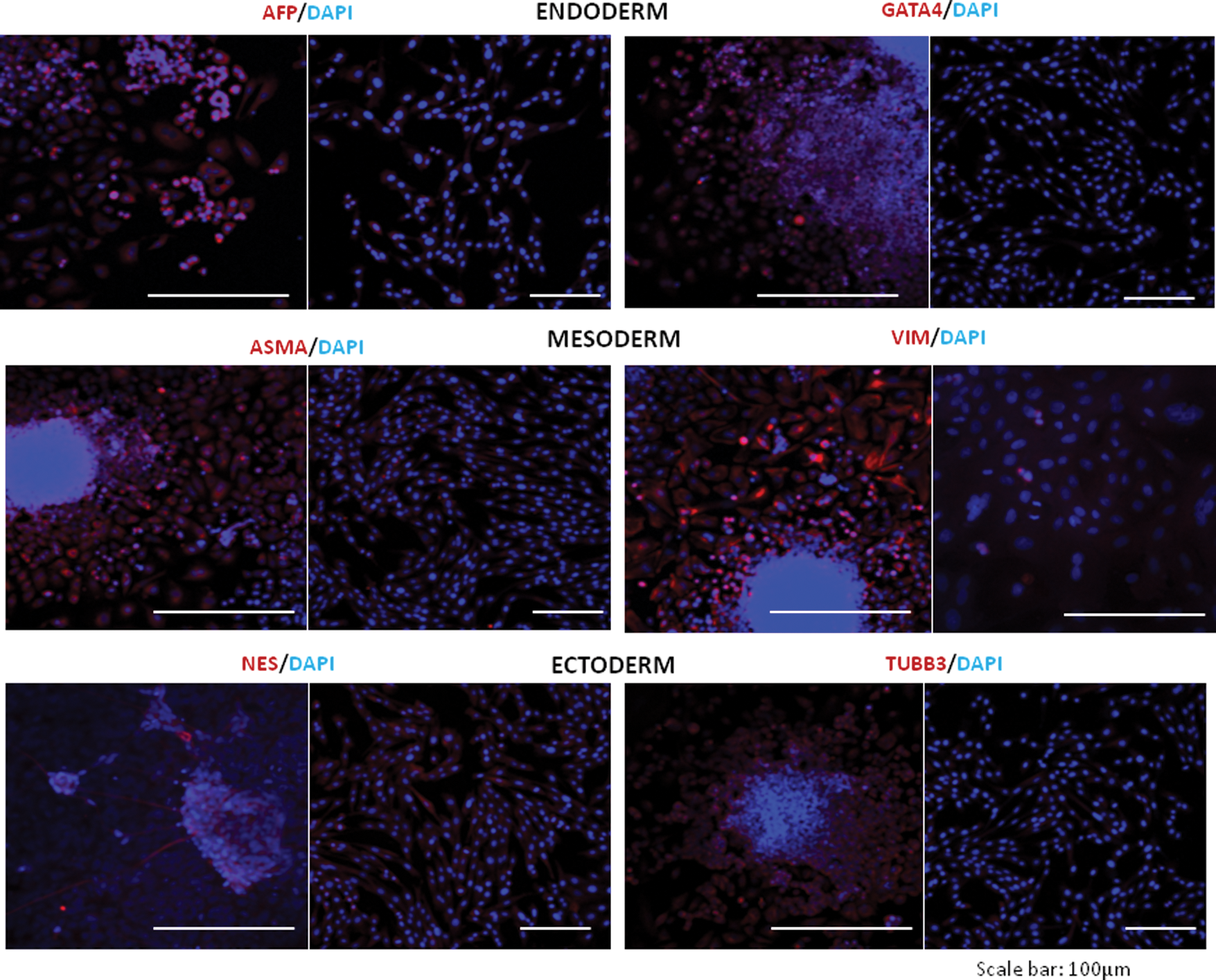
In vitro differentiation of reprogrammed cell lines. Representative confocal images showing immunofluorescence staining for markers of ectoderm, mesoderm, and endoderm. For each marker, the left image shows embryoid body cultures derived from line K2 and the right image shows parental keratinocytes (negative control). Color images available online at
Ability for multilineage differentiation in vivo was determined by injecting iPSCs into NOD-SCID mice. Two to three months after injection, animals that developed large tumors were euthanized. Histological analyses showed that tumors derived from K2 line were best differentiated with unambiguous evidence of all three germ layer derivatives, including well-developed respiratory and gastric epithelia, cartilage, bone, muscle, keratinized epithelium, and mature neurons (Fig. 5A); similar features were evident in teratomas from mice injected with ESC line JV09 (Fig. 5C). In contrast, lines K1 and K3 resulted in tumors predominately containing embryonic carcinoma-like cells with some keratinized epithelium (Fig. 5B). Thus, despite all three iPSC lines could efficiently differentiate in vitro, particularly K2 and K3, only K2 was able to produce differentiated teratomas, highlighting the importance of in vivo testing to fully characterize iPSC populations.
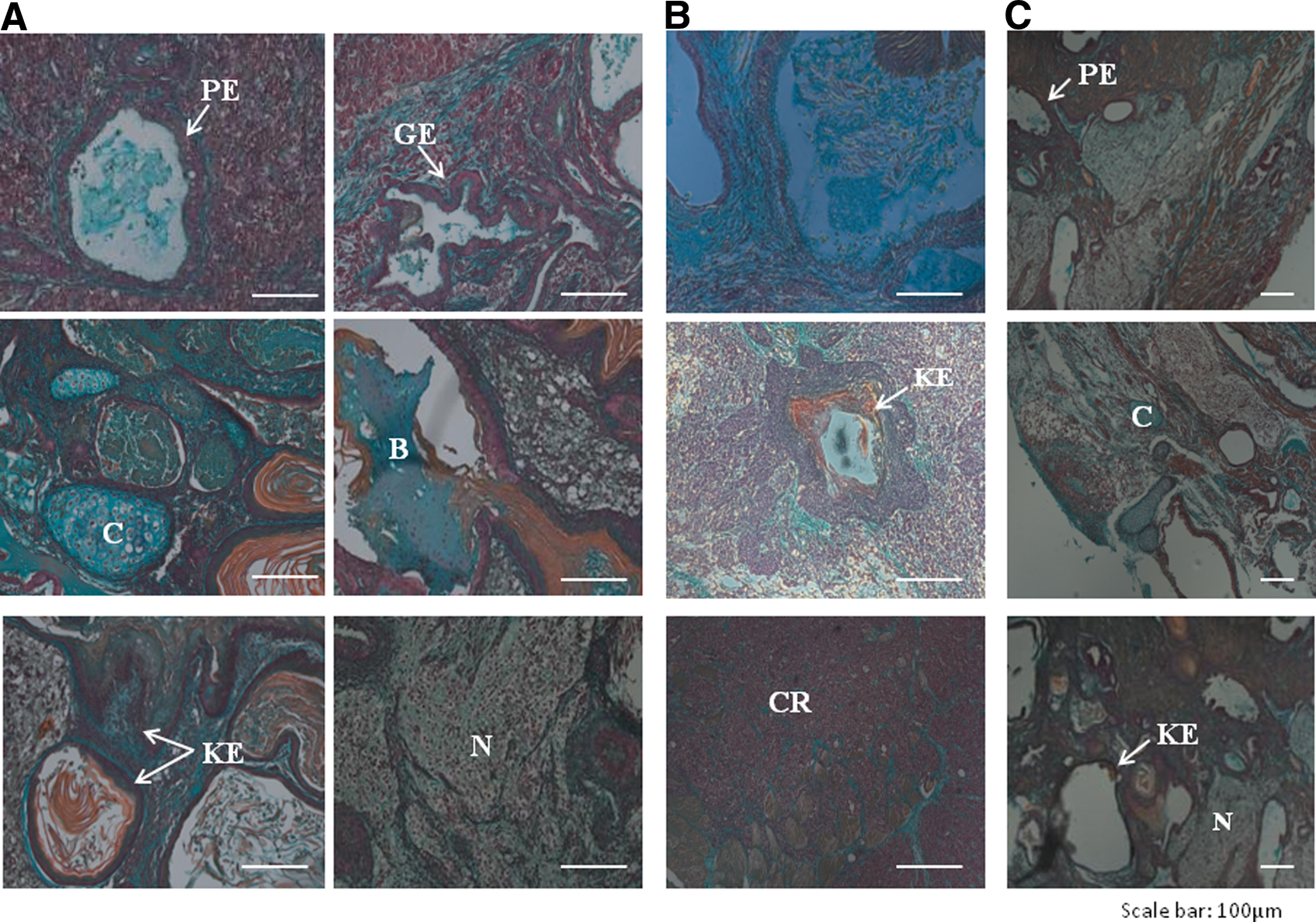
In vivo differentiation of reprogrammed cell lines.
Directed neuronal differentiation of equine iPSCs
To determine whether equine iPSCs could be directed toward neural lineages we followed a two-step protocol involving generation of neurospheres for 72 h followed by specification to mature neurons in serum-free neural medium over 3 weeks (Supplementary Fig. S3). Notably, retinoic acid, FGF, and/or epidermal growth factor provided no obvious advantage over LIF in their ability to induce neurosphere formation during step 1. Indeed, supplementation with retinoic acid resulted in the formation of large EBs that were difficult to dissociate enzymatically and contained dark core areas indicative of necrosis. Compared with neurosphere cultures maintained in LIF (Supplementary Fig. S3D, E), transfer of these cultures to media with StemPro® neural supplement in step 2 supported highly efficient specification of a more mature cell type as evidenced by extensive axonal growth (Supplementary Fig. S3B, C and Supplementary Video S1); failure to remove LIF from culture media at this stage significantly reduced cell differentiation.
Characterization of iPSC-derived equine neurons
Neuronal populations from iPSC line K2, the line that produced well-differentiated teratomas, were examined in detail. Neurospheres and developing neurites positive for TUBB3 were observed after 3 days of culture in neural supplement, and TUBB3 expression distinctly increased up to day 14 (Figs. 6A and 7). Progressive neurogenesis was also associated with distinct staining for MAP2, and the presynaptic marker, SYN1 (Fig. 6A), [32,33]. To further characterize these neuronal populations, we determined the expression of several cholinergic and motor neuron markers. Expression of acetylcholinesterase (ACHE) increased during culture in neural supplement (Figs. 6A and 7). At day 14, neurons also expressed the acetylcholine-synthesising enzyme, CHAT (Fig. 6A), with about 35% cells simultaneously staining positively for both ACHE and CHAT. Consistent with these results, cells cultured in neural supplement produced increasing levels of acetylcholine (Fig. 6B). Furthermore, ISL1 and HOXA2, two genes involved in motor neuron specification [34 –36], distinctly increased in cells cultured with StemPro® neural supplement (Fig. 7), however, the lack of suitable antibodies prevented the detection of these markers at the protein level. To examine whether the neurons were functional, whole-cell current-clamp recordings were performed at the end of culture with neural supplement (day 14: Fig. 6C). All cells examined (n=6) had the ability to fire action potentials (APs) in response to a 500 ms current stimulation (range: 10–40 pA) and such activity was blocked with the voltage-gated sodium ion channel blocker tetrodotoxin (300 nM). Four neurones were observed to fire multiple APs (between two and five APs per 500 ms stimulus), with a mean (±standard error) of 2.7±0.8 APs per stimulus.

Immunostaining and functional characterization of neurons derived from equine iPSCs.
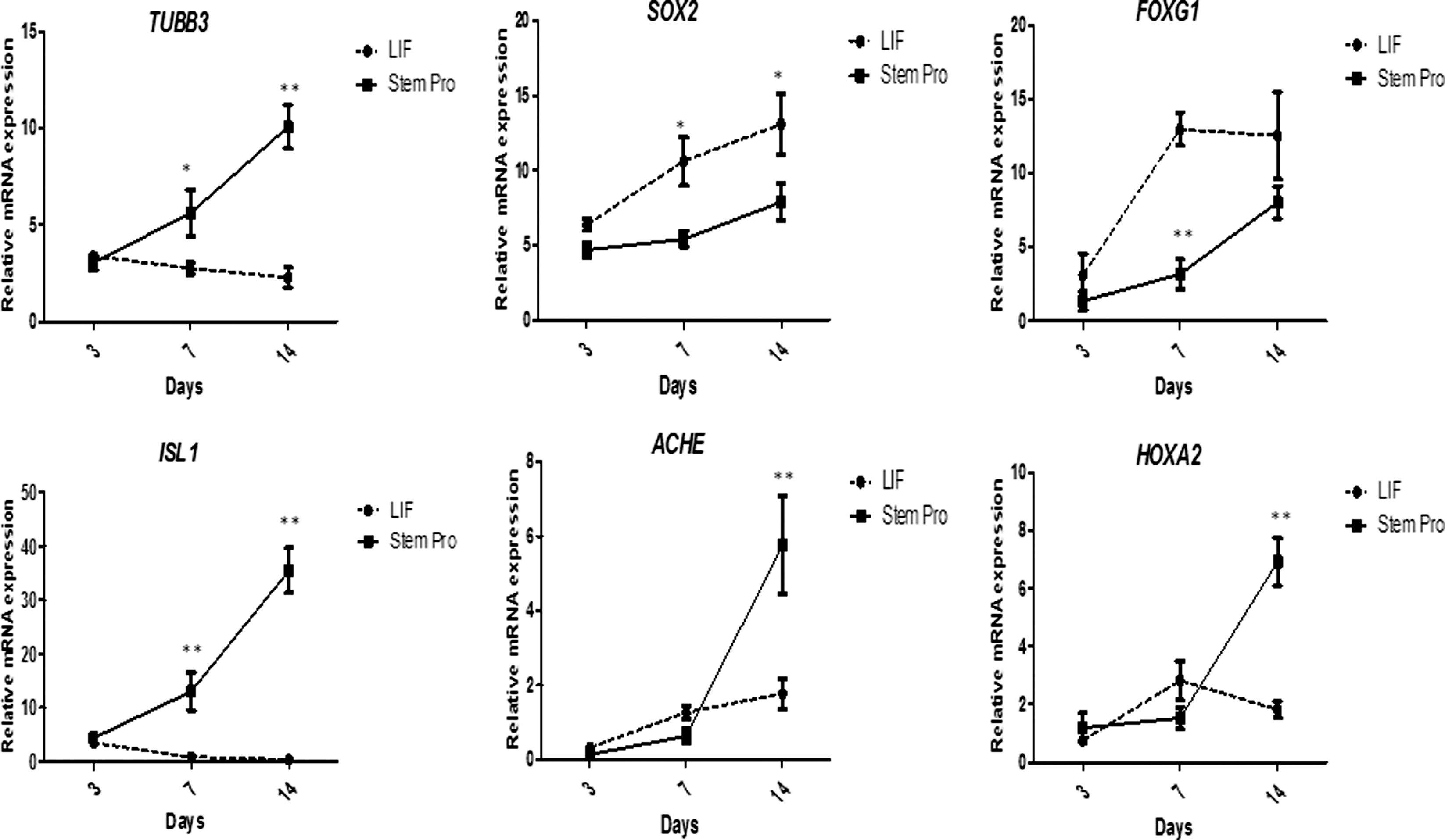
Gene expression analysis during induced neuronal differentiation of equine iPSCs. Transcript levels (mean±SE, normalized to GAPDH levels) of different neuronal markers in iPSC-derived neurosphere cultures maintained for 14 days in media containing either LIF or StemPro® neural supplement (day 0=day of addition of neuronal maturation media to neurospheres plated in glass coverslips). All experiments were conducted in triplicate. Differences between group mean within days are shown by stars (*P<0.05;**P<0.001).
Finally, given the apparent high levels of neurogenesis reached in cultures containing neural supplement compared to supplementation with LIF only, we sought to examine in these two cell populations the relative expression of SOX2 and FOXG1, two transcriptional regulators involved in neural stem cell self-renewal and differentiation [37 –39]. In contrast to markers of neuronal maturation, both SOX2 and FOXG1 were expressed at significantly higher levels by cells maintained in LIF-containing media (Fig. 7), which was indicative of a more abundant progenitor neuronal cell population under these conditions and reflects upon a default mechanism of neurogenesis [40,41].
Thus, results of immunostaining, QPCR, acetylcholine analyses, and electrophysiological analyses indicated that equine iPSCs could be differentiated into electrically active cells with motor neuron-like properties.
Discussion
The veterinary potential of PSCs is highest in the horse, both in relation to clinical applications and for developmental/disease modeling and toxicology. Although three different studies reported the generation of ES-like cells by transgene-mediated reprogramming of equine fibroblasts [11 –13], compared with human iPSCs, very little effort has been made to optimize the generation and culture conditions of equine iPSCs; critically, the biomedical potential of these cells remains unexplored. This study reports the comparatively efficient and robust generation of equine iPSCs with several novel features in relation to previous cell lines generated from horses or other domestic species, namely, (i) the use of cells other than fibroblasts as parental population, (ii) the successful expansion and maintenance of equine iPSCs in feeder-free conditions, and, importantly, (iii) their successful differentiation into functional cells, neurons. These achievements, particularly the latter two, constitute substantial progress in the field and represent an important step toward future veterinary biomedical applications of iPSCs.
Reports in humans [14 –16] showed that keratinocytes could be reprogrammed at a significantly faster rate and with considerably higher efficiency than fibroblasts, the only source of iPSCs so far reported in domestic species; these differences were attributed to the fact that mesenchymal-to-epithelial transition, a limiting step during fibroblast reprogramming, would not be required during reprogramming of keratinocytes and, in addition, keratinocytes may already express high endogenous levels of the reprogramming factors, MYC and KLF4. Consistent with this, in the present study well-defined iPSC-like colonies could be observed within just 5 days after viral transduction of equine keratinocytes; this compares with a 2 to 3-week interval required for colony formation from equine fibroblasts [11 –13]. Furthermore, the efficiency of equine keratinocyte reprogramming, 0.18%, was almost 10-fold higher than that reported using fibroblasts [12]. Thus, in the horse as in humans, compared with fibroblasts, keratinocytes appear to be a superior source for cell reprogramming.
Teratoma induction in mice is currently the most stringent test of pluripotency for equine cells. The levels of tissue differentiation displayed by tumors derived from iPSC line K2 were comparable to those obtained from mouse ESCs in this study and were much higher than those resulting from lines K1 and K3 or from equine iPSCs in previous studies [11,13], thus identifying K2 as bona fide equine iPSCs. Of note, this cell line displayed significant expression of endogenous OCT4 and REX1; these were absent from the other two lines. Transcriptional re-activation of endogenous OCT4 is a recognized hallmark of full reprogramming [42,43], whereas REX1, a marker of naïve PSCs [44,45], is expressed by teratoma-competent iPSCs of different species [46 –48] including, as previously shown by us, the horse [11]. Further, in an earlier report, expression of REX1, but also DNMT3B, were shown to be a distinguishing feature of bona fide human iPSCs, whereas NANOG expression was not [49]. Although a strict association of endogenous OCT4 and REX1 protein with line K2 could not be confirmed in this study due to the unavailability of species-specific antibodies, the present results strongly indicate that, as in other species, those two transcription factors are critically important for pluripotency in the horse. In addition to its distinct endogenous gene expression profile, the expression of viral transgenes was differentially attenuated in line K2; this feature may have contributed to the unique ability of this cell line to produce fully differentiated teratomas, a conclusion consistent with the established notion that significant downregulation or silencing of viral transgenes is a hallmark of successful cell reprogramming [30,50]. Taken together, the distinct features of equine iPSC line K2 in this study support the notion that the mechanisms of induced pluripotency are conserved across species.
A major challenge toward future biomedical applications remains the derivation of iPSCs that can be robustly maintained in defined, feeder-free conditions. Although significant advances have been done in that regard in humans, very little progress has been reported in other species [51]. Thus, available iPSC lines from domestic species including the horse were derived using heterologous or homologous fetal feeders in media containing bFGF with or without LIF [11 –13,46 –48], more complex mixtures of growth factors and kinase inhibitors [12,13], or LIF in the presence of MEK and GSK3β inhibitors [52]. In this study, we developed a comparatively much simpler culture system by successfully adapting keratinocyte-derived iPSCs to feeder-free and serum-free conditioned media supplemented only with LIF, without the need to use kinase inhibitors. Although not a fully defined media, the fact that bona fide equine iPSCs could be maintained for over 40 passages under these culture conditions represents a very important step toward a future veterinary application of these cells.
Further advances in the iPSC field will also require the development of robust protocols for specific and efficient differentiation into clinically relevant cell types. Under standard iPSC culture conditions, the equine reprogrammed cell lines showed a clear although variable tendency to spontaneously form neuron-like structures, an observation we did not make with fibroblast-derived iPSCs [11] and that may be related to the ectodermic origin of the parental cells in the present study [53]. Transferring iPSC aggregates to reduced LIF readily induced formation of neurospheres that upon continuous culture with LIF did not go on to form mature neurons but showed a distinct increase in expression of SOX2 and FOXG1, two transcription factors expressed during early neurogenesis in embryos and PSCs [37 –39], and which are necessary for neural stem/progenitor cell self-renewal [39,54]. This result is consistent with the reported “default” differentiation of PSCs into LIF-dependent primitive neural stem cells that remain undifferentiated upon continuous exposure to LIF [40,41,55], reflecting upon a mechanism of equine neurogenesis presumably similar to that identified in other species. In contrast, transferring neurospheres to media containing neural supplement led to formation of cells phenotypically similar to mature neurons that expressed, among other markers, distinctly high transcript levels of ISL1and HOXA2, two highly conserved genes essential for multiple aspects of motor neurogenesis [34 –36,56,57], and which suggests the acquisition of a distinct motor neuron subtype identity. Expression of these markers could not be characterized further due to the lack of suitable antibodies. However, consistent with those results, equine iPSC-derived neurons concomitantly acquired the ability to produce acetylcholine and to fire multiple APs, attesting to their functionality acquired using a relatively short in vitro differentiation protocol.
Summary
In summary, this study reports for the first time the generation of keratinocyte-derived iPSCs from a domestic species. Compared with previous reports, equine iPSCs were generated with high efficiency and, more importantly, could be maintained using simplified, feeder-free culture conditions. To our knowledge, this is also the first time that differentiated functional cells have been obtained from equine iPSCs, representing an important step toward future applications in veterinary biomedicine. Neurodegenerative conditions such as grass sickness and equine motor neuron disease, which are nontreatable and usually fatal, could be eventually targeted using equine iPSCs. Further, the ready availability in vitro of equine neurons, otherwise difficult to obtain and culture from in vivo sources, will potentially lead to a better understanding of neurogenesis in large animals and might in turn help reduce the dependence of neuroscience research on mouse models [23]. Horses suffer up to 90 genetic diseases that resemble human disorders, including amyotrophic lateral sclerosis [58]. Thus, the availability of disease models from large animal iPSCs could be of significant importance [59,60] especially when neurons derived from PSCs are being considered for human clinical trials [61].
Footnotes
Acknowledgments
We thank Mr. Robert Fleming for help with bioimaging. Financial support was provided by the Petplan Charitable Trust and the Horserace Betting Levy Board (to F.X.D.), and by the University of Edinburgh Staff Scholarship (to R.S.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.