
Abstract
The aim of this study was to investigate the effects of systemically administered bone-marrow-derived mesenchymal stromal cells (MSCs) on the early acute phase of inflammation in the alkali-burned eye. Mice with damaged eyes were either untreated or treated 24 h after the injury with an intravenous administration of fluorescent-dye-labeled MSCs that were unstimulated or pretreated with interleukin-1α (IL-1α), transforming growth factor-β (TGF-β), or interferon-γ (IFN-γ). Analysis of cell suspensions prepared from the eyes of treated mice on day 3 after the alkali burn revealed that MSCs specifically migrated to the damaged eye and that the number of labeled MSCs was more than 30-times higher in damaged eyes compared with control eyes. The study of the composition of the leukocyte populations within the damaged eyes showed that all types of tested MSCs slightly decreased the number of infiltrating lymphoid and myeloid cells, but only MSCs pretreated with IFN-γ significantly decreased the percentage of eye-infiltrating cells with a more profound effect on myeloid cells. Determining cytokine and NO production in the damaged eyes confirmed that the most effective immunomodulation was achieved with MSCs pretreated with IFN-γ, which significantly decreased the levels of the proinflammatory molecules IL-1α, IL-6, and NO. Taken together, the results show that systemically administered MSCs specifically migrate to the damaged eye and that IFN-γ-pretreated MSCs are superior in inhibiting the acute phase of inflammation, decreasing leukocyte infiltration, and attenuating the early inflammatory environment.
Introduction
A
More recently, mesenchymal stromal cells (MSCs) have been suggested and tested as a promising therapeutic tool for the treatment of many disorders involving corneal defects. These cells have the potential to differentiate into various cell types [6], including epithelial cells [7 –9], and thus they have been used for ocular surface reconstruction. Moreover, MSCs possess potent immunomodulatory properties and can influence various functions of immune cells, including dendritic cells, naive and effector T lymphocytes, and natural killer (NK) cells [10]. The immunomodulatory properties of MSCs have been documented in numerous in vitro and in vivo studies that demonstrate the ability of MSCs to prolong allograft survival [11,12], ameliorate experimental autoimmune disorders [13], or attenuate severe acute graft-versus-host disease [14].
The beneficial effects of MSCs consist not only in their ability to replace injured cells, but also in their modulation of the local proinflammatory microenvironment by the production of numerous immunomodulatory and trophic factors. It has been shown that the curative effect of MSCs on corneal injury can consist partly in the epithelial transdifferentiation of MSCs [7,15] and in the suppression of corneal inflammation [16 –19].
This study was focused on monitoring and evaluating the effects of systemically administered MSCs on the early acute phase of inflammation in the alkali-burned eye using an experimental murine model. Since the immunomodulatory properties of MSCs can be modified by proinflammatory cytokines [20 –22], we compared the effect of unstimulated MSCs and MSCs pretreated with interleukin (IL)-1α, interferon (IFN)-γ, or transforming growth factor (TGF)-β on the inflammatory environment in the eye.
Materials and Methods
Mice
Female BALB/c mice at the age of 8–12 weeks were obtained from the breeding unit of the Institute of Molecular Genetics (Prague, Czech Republic). The use of animals was approved by the local Animal Ethics Committee.
Isolation, culture, and purification of MSCs
Bone marrow for the cultivation of MSCs was isolated from the femurs and tibias of female BALB/c mice. The bone marrow was flushed out, a single-cell suspension was prepared using a tissue homogenizer, and the cells were seeded at a concentration of 2×106 cells/mL in Dulbecco's modified Eagle's medium (DMEM; Sigma, St. Louis, MO) containing 10% fetal calf serum (FCS; Gibco BRL, Grand Island, NY), antibiotics (100 U/mL of penicillin and 100 μg/mL of streptomycin), and 10 mM HEPES buffer (hereafter referred to as complete DMEM) in 75-cm2 tissue culture flasks (TPP, Trasadingen, Switzerland). Nonadherent cells were washed out after 72 h of cultivation, and the remaining adherent cells were cultured for an additional 3 weeks (two passages) at 37°C in an atmosphere of 5% CO2. Plastic-adherent cells were harvested by incubating the cells with 8 mL of 10 mM EDTA for 5 min and subsequent gentle scraping. The resulting cell suspension was incubated for 15 min with CD11b MicroBeads and CD45 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. The cell suspension was then immunodepleted of CD11b+ and CD45+ contaminating cells using a magnetic activated cell sorter (AutoMACS; Miltenyi Biotec). The remaining CD11b− and CD45− cells were evaluated in terms of their purity and differentiation potential.
Phenotypic characterization of MSCs by flow cytometry
Unstimulated and cytokine-pretreated MSCs were washed in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) and then incubated for 30 min on ice with the following anti-mouse monoclonal antibodies (mAbs): allophycocyanine (APC)–labeled anti-CD44 (clone IM7; BD PharMingen, San Jose, CA), phycoerythrin (PE)–labeled anti-CD105 (clone MJ7/18; eBioscience, San Diego, CA), APC-labeled anti-CD11b (clone M1/70; BioLegend, San Diego, CA), or fluorescein isothiocyanate (FITC)–labeled anti-CD45 (clone 30-F11; BioLegend). Dead cells were stained using Hoechst 33258 fluorescent dye (Invitrogen, Carlsbad, CA) added to the samples 10 min before flow cytometry analysis. Data were collected using an LSRII cytometer (BD Biosciences, Franklin Lakes, NJ) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Labeling of MSCs with PKH26 fluorescent dye
MSCs were labeled with a fluorescent dye (PKH26 Red Fluorescent Cell Linker Kit; Sigma) in order to monitor their fate after intravenous administration. MSCs were labeled according to the manufacturer's instructions with modifications introduced in the protocol to achieve optimal labeling for this type of cells. Particularly, a final concentration of 2 μM of PKH26 for 1×106 MSCs/mL and a 5-min incubation were determined to be the optimal conditions. The fluorescence intensity and homogeneity of the staining were tested by fluorescent microscopy (Inverted fluorescent microscope Olympus IX71, Center Valley, PA) and flow cytometry.
A model of corneal damage
Female BALB/c mice were deeply anesthetized by an intramuscular injection of a mixture of xylazine and ketamine (Rometar, Spofa, Prague, Czech Republic). The surface (corneal and limbal region) of the left eye was damaged by the application of 3-mm-diameter filter paper soaked with 8 μL of 1 N NaOH for 30 s. The eye was then thoroughly rinsed with 10 mL of PBS.
In vitro stimulation and the intravenous administration of MSCs
MSCs were labeled with PKH26 dye and incubated at a concentration of 0.5×106 cells/mL in a volume of 1.5 mL of complete DMEM in a 12-well tissue culture plate (Nunc, Roskilde, Denmark) for 24 h. MSCs were cultured either unstimulated or were pretreated with 10 ng/mL of mouse recombinant IL-1α (Immunotools, Friesoyte, Germany), 10 ng/mL of IFN-γ (Immunotools), or 2 ng/mL of human TGF-β (PeproTech, Rocky Hill, NJ). Stimulated MSCs were then harvested and centrifuged in an excess of serum-free DMEM in order to remove the rest of the serum and added cytokines. For intravenous application, 0.5×106 MSCs were resuspended in 200 μL of serum-free DMEM. The cell suspension was administered to mice through the tail vein using a 30G Omnican 100 syringe (B.Braun, Melsungen, Germany) 24 h after corneal damage.
Preparation of single-cell suspension from the eye and other organs
Single-cell suspensions from the whole eyeballs of both damaged and control eyes were prepared for flow cytometry analysis and cell culturing. The eyballs were cleaned of redundant tissue, cut into pieces, and centrifuged in 600 μL of HBSS to obtain a tissue extract for ELISA analysis. The pelleted tissue was then digested with 1 mg/mL of collagenase I (Sigma) in HBSS for 50 min at 37°C. To monitor the fate of PKH26-labeled MSCs after their intravenous administration, selected tissues and organs were obtained and digested to prepare single-cell suspensions for flow cytometry. Eyes, lung, and liver were digested in 1 mg/mL of collagenase I in HBSS for 50 min at 37°C. Lymph nodes (inguinal, brachial, cervical, and submandibular) and spleen were digested in 1 mg/mL of collagenase II (Sigma) in HBSS for 60 min at 37°C. A cell suspension from the bone marrow was prepared in the same way as in the protocol for the culture of MSCs.
Monitoring the migration of PKH26-labeled MSCs in the body
To determine the distribution of PKH26-labeled MSCs in the body 48 h after their intravenous administration, single-cell suspensions were prepared from several tissues and organs (eyes, lung, liver, spleen, lymph nodes, and bone marrow). For flow cytometry analysis, the concentration of the cells was adjusted to 1×106/mL in PBS containing 0.5% BSA. The number of PKH26-labeled cells was determined using an LSRII cytometer, and the data were analyzed by FlowJo software. To determine more precise location of PKH26+ cells in damaged eye, single-cell suspensions were prepared from anterior segment, vitreous humor, and posterior segment of damaged eyes from mice treated with unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ. The cell suspensions were incubated with APC-labeled anti-CD44 mAb (BD PharMingen, San Jose, CA) and a total number of PKH26+CD44+ MSCs in particular segments was assessed by flow cytometry.
Immunofluorescent staining of frozen sections of damaged eyes
For frozen sectioning, damaged eyes of mice that were untreated or were treated with unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ were enucleated and immersion fixed in 4% paraformaldehyde for 1 h, followed by overnight cryoprotection in 15% sucrose. The eyes were embedded in optical cutting temperature medium and frozen sections at a thickness of 7 μm were prepared using a Leica CM 3050 cryostat (Leica, Wetzlar, Germany). The sections were refixed by 4% paraformaldehyde for 10 min, washed in PBS, blocked by 10% BSA in PBS containing 0.5% Triton X-100 for 30 min, and then incubated with APC-labeled anti-CD45 mAb (clone 30-F11; BioLegend) in blocking solution for 2 h at room temperature. After washing three times with PBS, slides were mounted with DAPI (Vectashield; Vector Labs, Burlingame, CA). CD45+ leukocyte and PKH26+ MSC infiltration were analyzed using fluorescent microscope Olympus Cell-R.
Detection of gene expression by real-time polymerase chain reaction
Total RNA was extracted from unstimulated and cytokine-pretreated MSCs using TRI Reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer's instructions. One microgram of total RNA was treated with deoxyribonuclease I (Promega, Madison, WI) and used for subsequent reverse transcription. The first-strand cDNA was synthesized using random hexamers (Promega) in a total reaction volume of 25 μL using M-MLV Reverse Transcriptase (Promega). Quantitative real-time polymerase chain reaction (PCR) was performed in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) as we have previously described [23,24]. The sequences of primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IL-6, TGF-β, indoleamine 2,3-dioxygenase (IDO), inducible nitric oxide synthase (iNOS), hepatocyte growth factor (HGF), and cyclooxygenase 2 (COX2) used for amplification are presented in Table 1. The PCR parameters included denaturation at 95°C for 3 min, 40 cycles at 95°C for 20 s, annealing at 60°C for 30 s, and elongation at 72°C for 30 s. Fluorescence data were collected at each cycle after an elongation step at 80°C for 5 s and were analyzed using StepOne Software version 2.2.2 (Applied Biosystems).
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin; TGF-β, transforming growth factor-β; IDO, indoleamine 2,3-dioxygenase; iNOS, inducible nitric oxide synthase; HGF, hepatocyte growth factor; COX2, cyclooxygenase 2.
Samples for testing changes in the expression of genes for cytokines and iNOS in control and damaged eyes were prepared from fresh single-cell suspensions (IL-6) or obtained after culturing suspensions for 48 h (IL-1α, IL-10, or iNOS). In brief, cells (1×106/mL) from control and damaged eyes were cultured in a volume of 800 μL of RPMI 1640 medium (Sigma) containing 10% FCS (Gibco BRL), antibiotics (100 U/mL of penicillin and 100 μg/mL of streptomycin), and 10 mM HEPES buffer (hereafter referred to as complete RPMI) in 48-well tissue culture plates (Corning, Corning, NY) in the presence of 1.5 μg/mL of concanavalin A (ConA; Sigma) and 1.5 μg/mL of lipopolysaccharide (LPS; Difco Laboratories, Detroit, MI) for a 48-h incubation period. The sequences of primers for GAPDH, IL-1α, IL-6, IL-10, and iNOS used for amplification are shown in Table 1.
Flow cytometry characterization of leukocyte populations infiltrating the damaged eyes
Single-cell suspensions prepared from control and damaged eyes were washed in PBS containing 0.5% BSA and incubated for 30 min on ice with the following anti-mouse mAb (all purchased from BioLegend): peridin-chlorophyll protein/Cyanine5.5 (PerCP/Cy5.5)–labeled anti-CD45 (clone 30-F11), APC-labeled anti-CD3 (clone 17A2), FITC-labeled anti-CD4 (clone GK1.5), PE-labeled anti-CD8a (clone 53–6.7), PE-labeled anti-F4/80 (clone BM8), APC-labeled anti-CD80 (clone 16-10A1), PE-labeled anti-CD14 (clone Sa14-2), APC-labeled anti-CD11b (clone M1/70), FITC-labeled anti-Ly6G/Ly-6C (Gr-1) (clone RB6-8C5), and FITC-labeled anti-CD19 (clone 6D5). Dead cells were stained using Hoechst 33258 fluorescent dye (Invitrogen) added to the samples 10 min before flow cytometry analysis. Data were collected using an LSRII cytometer and analyzed using FlowJo software. One hundred thousand events from each sample were measured. These events were gated for CD45+ leukocytes after the exclusion of cell debris and dead cells and analyzed for particular markers.
Cytokine and NO measurement
Cytokines and NO were measured in tissue extracts and culture supernatants from control and damaged eyes. Tissue extracts were obtained during the preparation of single-cell suspensions from eyeballs. Supernatants were obtained after culturing cell suspensions from control and damaged eyes. Cells (1×106/mL) were cultured in a volume of 800 μL of complete RPMI medium (Sigma) in the presence of 1.5 μg/mL of ConA and 1.5 μg/mL of LPS for a 48-h incubation period. The production of IL-1α, IL-2, IL-4, IL-6, IL-10, IL-17, and IFN-γ was quantified by ELISA. The production of IL-2, IFN-γ, and IL-6 was measured using cytokine-specific capture and detection of mAbs purchased from BD Pharmingen (San Diego, CA). IL-1α, IL-4, IL-6, IL-10, and IL-17 were measured using ELISA kits purchased from R&D Systems (Minneapolis, MN). Only IL-1α, IL-6, and IL-10 were produced in significant concentrations and with enhanced production in the damaged eyes. The concentrations of NO in the supernatants were determined using the Griess reaction [25]. A mixture of 50 μL of 1% sulfanilamide and 50 μL of 0.3% N-1-naphthylethylendiamine dihydrochloride (both in 3% H3PO4) was incubated with 100 μL of the tested supernatant. Nitrite was quantified by spectrophotometry at 540 nm using sodium nitrite as a standard.
Statistical analysis
The statistical significance of differences between individual groups was calculated using the Student's t-test. A value of P<0.05 was considered statistically significant.
Results
Characterization of MSCs
The purity and phenotypic markers of MACS-separated MSCs pretreated with cytokines were assessed with flow cytometry. The results showed that unstimulated and cytokine-treated MSCs were positive with a corresponding intensity for CD44 and CD105, which are the markers attributed to murine MSCs (Fig. 1A). On the other hand, <1% of the cells were CD11b+ and <3% were CD45+ (Fig. 1A); therefore, the population of bone marrow cells was depleted of contaminating CD11b+ and CD45+ cells with a high efficiency. In addition, both unstimulated and cytokine-treated MSCs were able to undergo adipogenic and osteogenic differentiation (data not shown). Further, PKH26-labeled MSCs possessed a sufficient, detectable, and relatively homogenous fluorescent signal that could be detected using a fluorescent microscopy and flow cytometry even after cultivation (Fig. 1B, C).
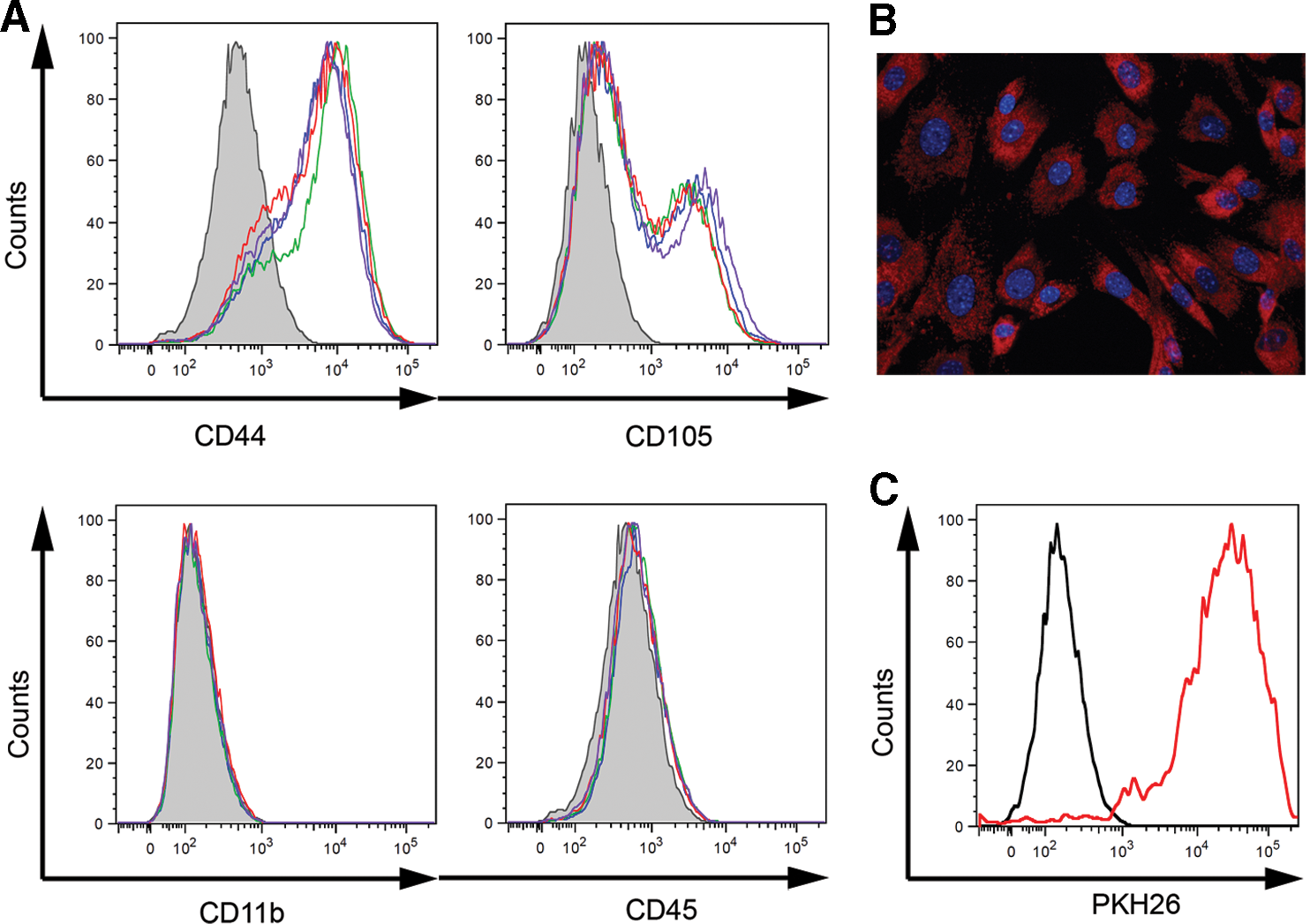
Characterization of unstimulated mesenchymal stromal cells (MSCs) and MSCs pretreated with interleukin-1α (IL-1α), transforming growth factor-β (TGF-β), or interferon-γ (IFN-γ).
Real-time PCR analysis of the expression of genes for immunomodulatory molecules was performed for further characterization of unstimulated MSCs and MSCs pretreated with IL-1α, TGF-β, or IFN-γ. As shown in Fig. 2, both unstimulated and pretreated MSCs expressed significant but variable levels of genes for TGF-β, HGF, and COX2. Moreover, MSCs pretreated with IL-1α and IFN-γ expressed a significant level of mRNA for iNOS. Only MSCs pretreated with IL-1α expressed the IL-6 gene and, on the other hand, only MSCs pretreated with IFN-γ expressed the IDO gene (Fig. 2).
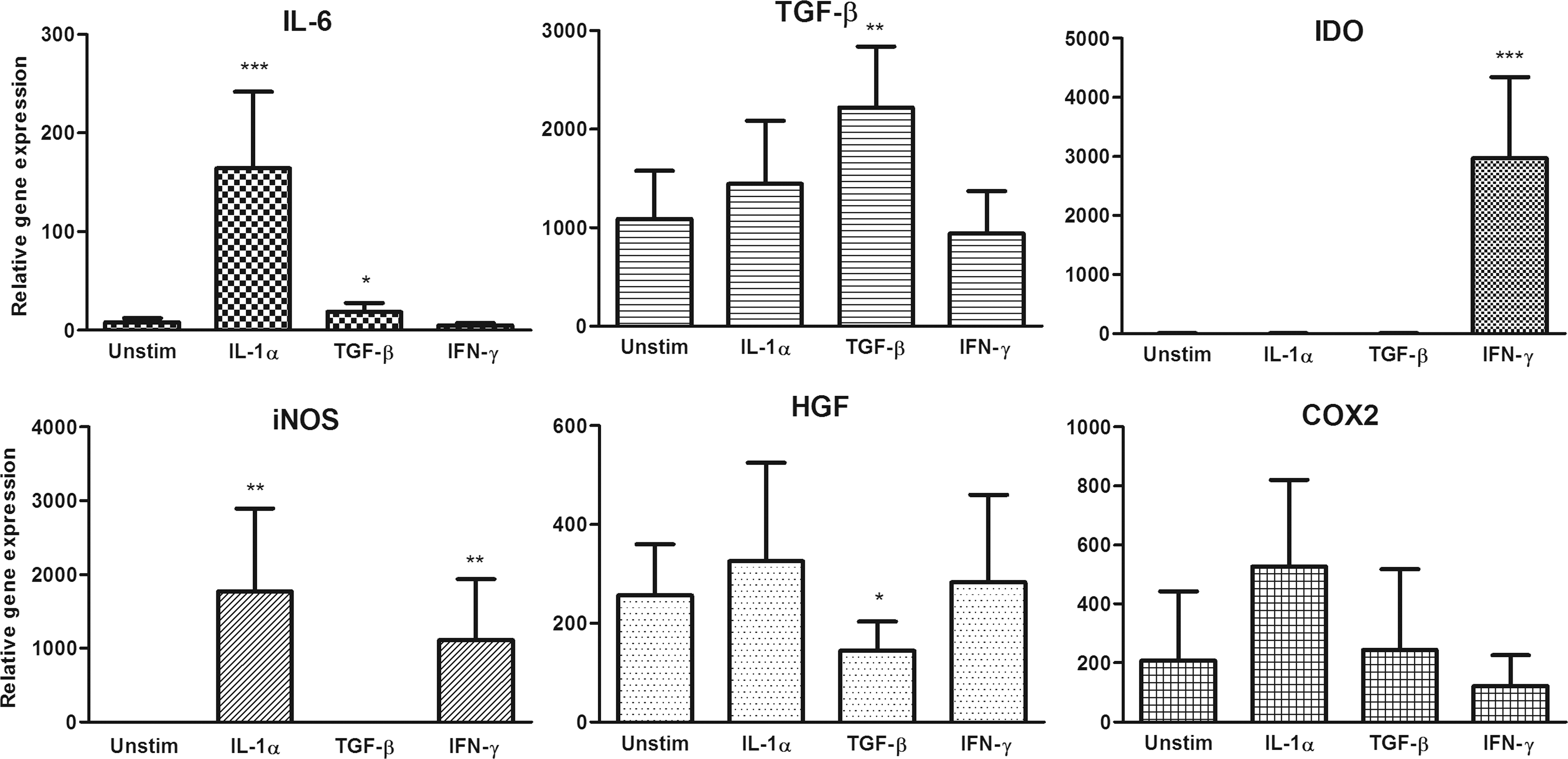
Expression profile of genes for immunomodulatory molecules in unstimulated MSCs and MSCs pretreated with IL-1α, TGF-β, or IFN-γ. MSCs were cultured for 24 h unstimulated or were pretreated with 10 ng/mL of IL-1α or IFN-γ or 2 ng/mL of TGF-β. The expression of genes for IL-6, TGF-β, indoleamine 2,3-dioxygenase (IDO), inducible nitric oxide synthase (iNOS), hepatocyte growth factor (HGF), and cyclooxygenase 2 (COX2) was detected by real-time PCR. Values with asterisks are significantly different (*P<0.05, **P<0.01, ***P<0.001) from the control value (unstimulated MSCs). Each bar represents the mean±SD from six independent experiments.
Monitoring of the distribution of PKH26-labeled MSCs in the body
The distribution of PKH26+ MSCs in the body 48 h after their systemic administration and possible differences in the migration of unstimulated and cytokine-pretreated MSCs were assessed using flow cytometry. Single-cell suspensions from the eye, lung, liver, and spleen; the inguinal, brachial, cervical, and submandibular lymph nodes; and the bone marrow were analyzed. A low number of PKH26+ MSCs was detected in the lymph nodes, while higher numbers of PKH26+ cells migrated to the spleen and bone marrow and the highest number of labeled MSCs was trapped in the lung and liver (data not shown). The analysis of suspensions prepared from damaged and control eyes showed that both unstimulated and cytokine-pretreated MSCs migrated preferentially into the damaged eye and that the number of PKH26+ MSCs was more than 30-times higher in the damaged eye compared with the control eye (Fig. 3A). No significant differences were revealed between the migratory properties of unstimulated MSCs and MSCs pretreated with IL-1α, TGF-β, or IFN-γ (Fig. 3B). Flow cytometry analysis of single-cell suspensions from anterior segment, vitreous humor, and posterior segment of damaged eyes from mice treated with unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ revealed that ∼20% of PKH26+CD44+ cells detected in eye was present in anterior segment, 10% of labeled cells was detected in vitreous humor, and 70% of cells was present in posterior segment of the damaged eye (Fig. 3B).
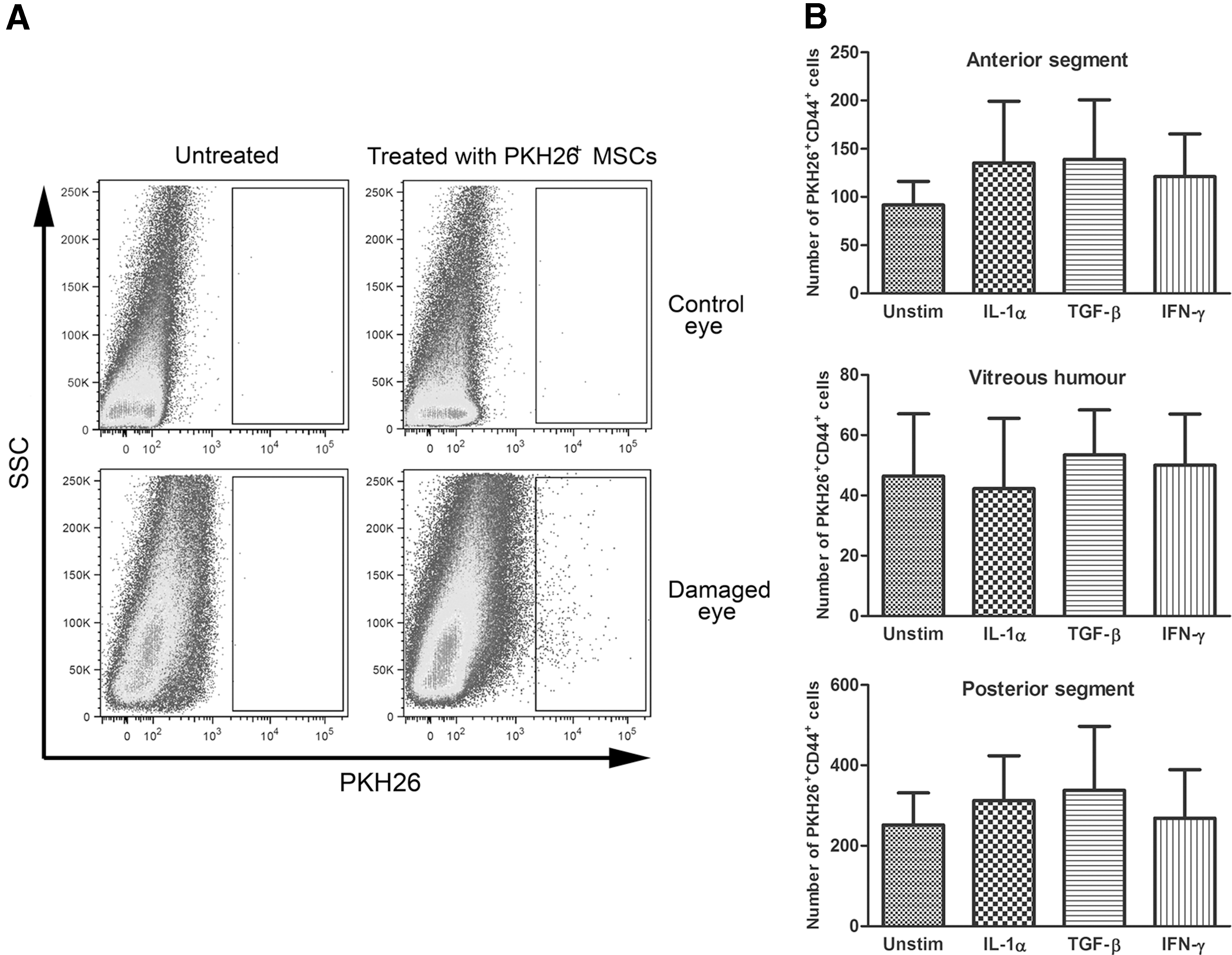
Monitoring of the migration of PKH26-labeled MSCs into damaged and control eyes.
Determination of leukocyte populations infiltrating damaged eyes after treatment with MSCs
Since both unstimulated and cytokine-pretreated MSCs preferentially migrated to the damaged eye, the potential effect of MSCs on the local environment was tested in the next experiments. Cell suspensions from control and damaged eyes from untreated mice or mice treated with systemically administered MSCs were labeled with selected combinations of mAbs and analyzed by flow cytometry. To test the lymphoid lineages, the percentage of CD3+CD4+ (T lymphocytes and NK-T cells), CD3+CD8+ (T lymphocytes and NK-T cells), and CD19+CD22+ (B lymphocytes) cells was monitored. Figure 4 shows that the presence of all of the tested types of MSCs slightly decreased the percentage of damaged-eye-infiltrating lymphoid populations, but only MSCs pretreated with IFN-γ decreased the number of CD3+CD4+, CD3+CD8+, and CD19+CD22+ cells significantly. The effect of MSCs on the myeloid lineage was tested by assessing the percentage of infiltrating CD80+, CD14+, F4/80+, and CD11b+Gr-1+ (granulocytes and macrophages) cells. As demonstrated in Fig. 4, both unstimulated and cytokine-treated MSCs slightly decreased the percentages of CD80+ and CD11b+Gr-1+ cells, but only MSCs pretreated with IFN-γ significantly decreased the percentages of all myeloid populations, while MSCs pretreated with TGF-β significantly decreased the percentage of CD11b+ Gr-1+ cells. In general, the effect of IFN-γ-pretreated MSCs was more profound on myeloid than lymphoid populations.
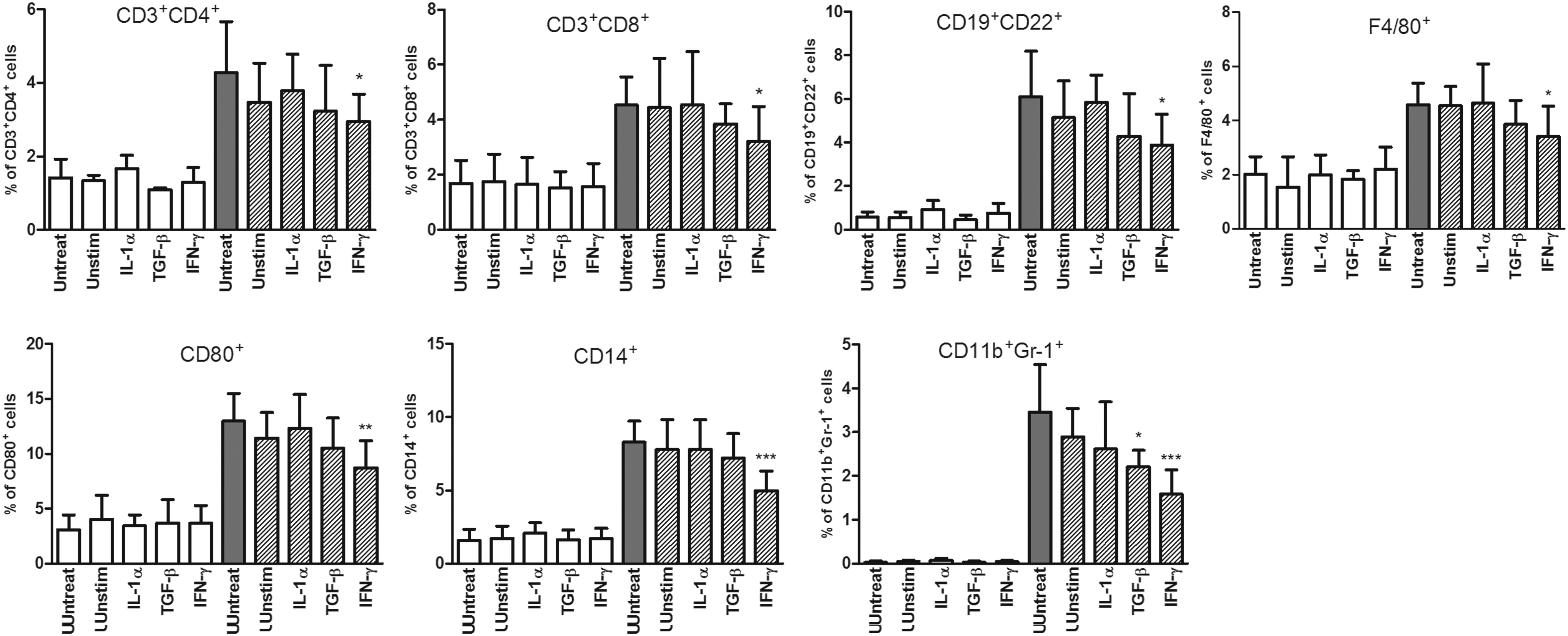
Flow cytometry analysis of leukocyte populations infiltrating control (white columns) and damaged (dashed and gray columns) eyes of untreated mice and mice treated with the systemic administration of unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ (in the graphs labeled as Unstim, IL-1α, TGF-β, and IFN-γ). One hundred thousand events from each sample were measured. These events were gated for CD45+ leukocytes after the exclusion of cell debris and dead cells and analyzed for the percentage of CD3+CD4+, CD3+CD8+, CD19+CD22+, CD80+, CD14+, F4/80+, or CD11b+Gr-1+ cells. Values with asterisks are significantly different (*P<0.05, **P<0.01, ***P<0.001) from the control values (damaged eyes of untreated mice). Each bar represents the mean±SD from 10 independent experiments.
Microscopical analysis of frozen sections of damaged eyes confirmed the results from flow cytometry. As demonstrated in Figure 5, a strong infiltration with CD45+ leukocytes was observed in damaged eyes from untreated mice, and this infiltration was decreased in eyes from mice treated with MSCs.
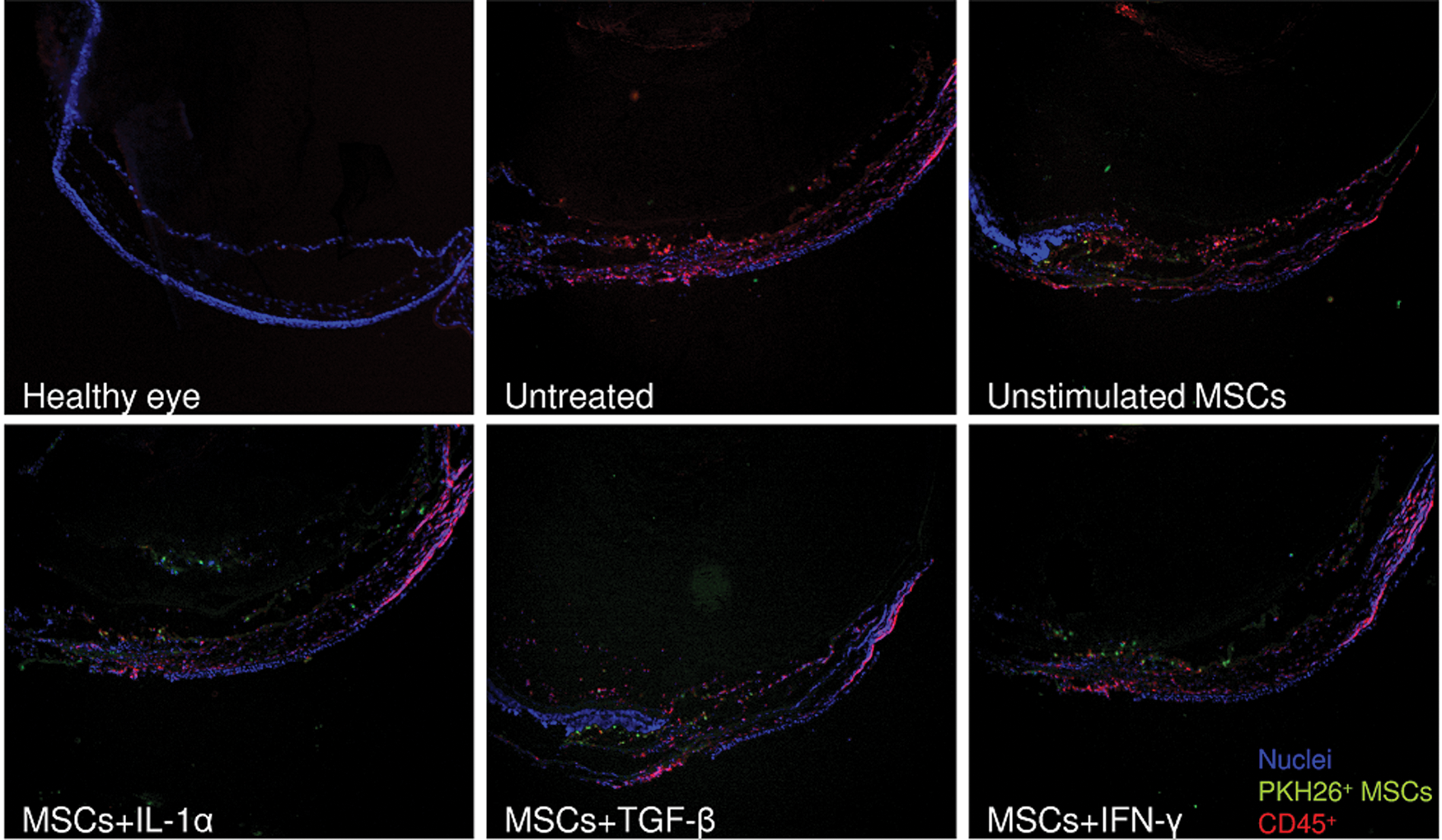
Immunofluorescent staining of frozen sections of eyes from untreated mice and mice treated with unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ. Representative pictures show part of anterior segment (right upper part of image) and lateral segment of control healthy eye and damaged eye from untreated mouse and from mouse treated with unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ. The infiltration of the eye with MSCs labeled with PKH26 fluorescent dye (green) and CD45+ leukocytes (red) is shown. The nuclei are blue (DAPI staining); original magnification is 40×. Color images available online at
Effect of MSCs on cytokine and NO production in control and damaged eyes
The flow cytometry data showed a significantly decreased infiltration of both lymphoid and myeloid cells in damaged eyes from mice injected with IFN-γ-pretreated MSCs. To extend this observation, we determined the production of the proinflammatory cytokines IL-1α and IL-6 and the secretion of NO in damaged eyes from mice treated with unstimulated or cytokine-pretreated MSCs. As demonstrated in Figure 6, systemic treatment with IFN-γ-primed MSCs significantly inhibited the local production of IL-1α, IL-6, and NO in the damaged eye. In contrast, the production of the anti-inflammatory cytokine IL-10 was not inhibited by MSCs (Fig. 6). These results were confirmed by real-time PCR analysis. The expression of genes for IL-1α and IL-6 in the damaged eyes of mice injected with IFN-γ-pretreated MSCs was significantly inhibited. Similarly, the expression of the gene for iNOS was significantly decreased in mice treated with IFN-γ-pretreated MSCs. In agreement with the results from ELISA, the expression of the IL-10 gene was not inhibited by MSCs (Fig. 7).
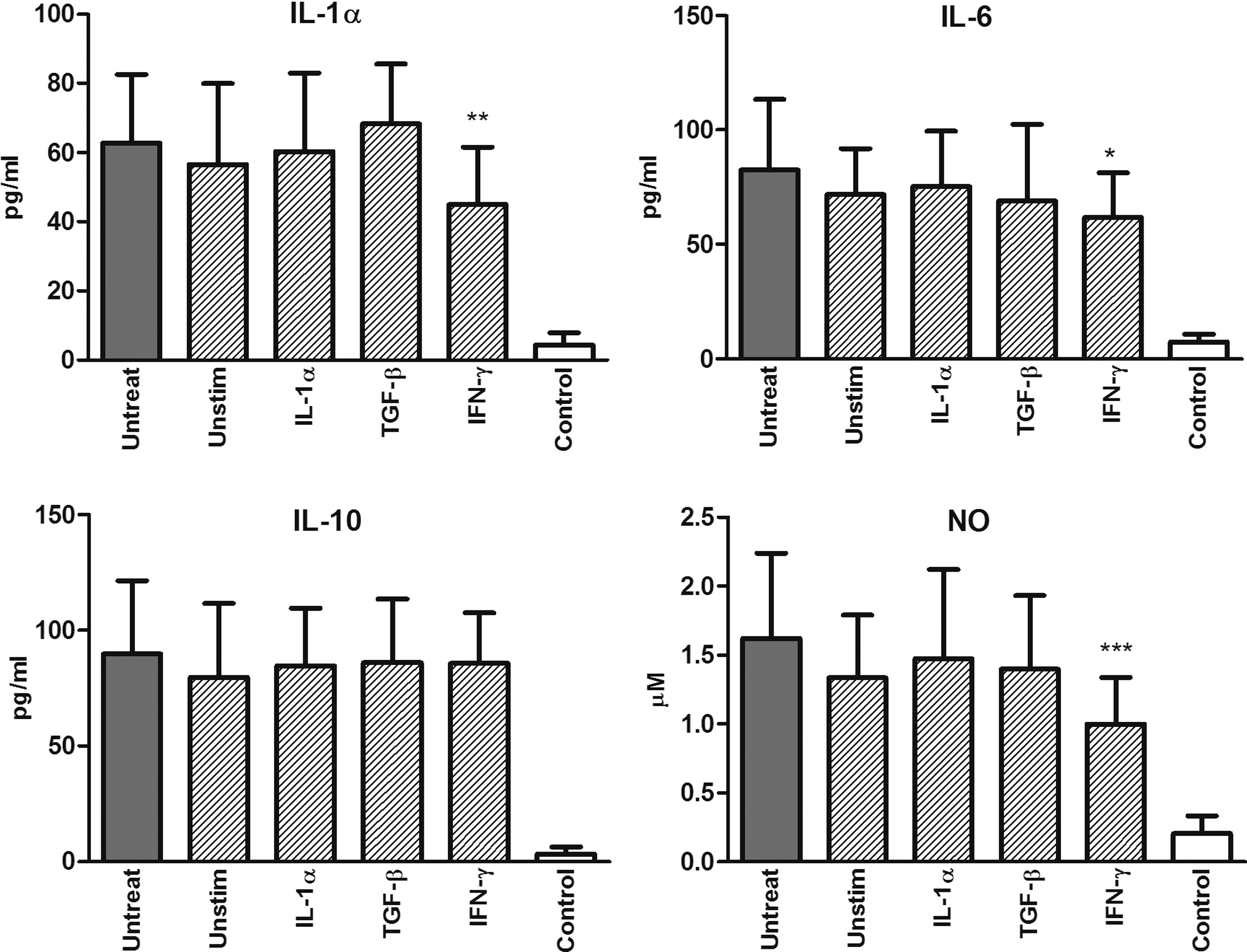
The effect of MSCs on cytokine and NO production. Cytokine and NO production in control (white column) and damaged (dashed and gray columns) eyes of untreated mice and mice treated with the systemic administration of unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ (in the graphs labeled as Unstim, IL-1α, TGF-β, and IFN-γ). The level of IL-6 was measured by ELISA in tissue extracts obtained from freshly isolated eyes. The production of IL-1α and IL-10 was determined by ELISA in supernatants from cultures of control and damaged eyes after stimulation with concanavalin A (ConA; 1.5 μg/mL) and lipopolysaccharide (LPS; 1.5 μg/mL) for 48 h. The production of NO was measured in culture supernatants using the Griess reaction. Values with asterisks are significantly different (*P<0.05, **P<0.01, ***P<0.001) from the control values (damaged eyes of untreated mice). Each bar represents the mean±SD from 12 independent experiments.

The effect of MSCs on the expression of genes for cytokines and iNOS. The expression of genes for IL-1α, IL-6, IL-10, and iNOS in control (white column) and damaged (dashed and gray columns) eyes of untreated mice and mice treated with the systemic administration of unstimulated MSCs or MSCs pretreated with IL-1α, TGF-β, or IFN-γ (in the graphs labeled as Unstim, IL-1α, TGF-β, and IFN-γ) was measured by real-time PCR. The expression of the IL-6 gene was assessed in cells from freshly isolated eyes. The expression of genes for IL-1α, IL-10, and iNOS was determined in cells from cultures of control and damaged eyes after stimulation with ConA (1.5 μg/mL) and LPS (1.5 μg/mL) for 48 h. Values with asterisks are significantly different (*P<0.05, **P<0.01, ***P<0.001) from the control values (damaged eyes of untreated mice). Each bar represents the mean±SD from eight independent experiments.
Discussion
The present study was designed to evaluate the effects of MSCs on immunological processes occurring in the eye in the early period after the ocular surface damage. We focused on the eye infiltration by different leukocyte populations and on the local cytokine microenvironment, but not on characterization of the eye surface regeneration, which occurs in the later phase after ocular surface damage. Majority of published studies investigated the effects of MSCs on the ocular surface regeneration, but not on immunological processes occurring in the eye within the first 3 days after ocular damage.
Since previous studies have demonstrated that cytokines can modulate the immunoregulatory properties of MSCs [20 –22], we compared the therapeutic effects of unstimulated MSCs and MSCs pretreated with IL-1α, IFN-γ, or TGF-β. MSCs were administered intravenously 24 h after corneal damage, at the time when the number of eye-infiltrating myeloid and lymphoid cells markedly increases. The immunomodulatory effects of MSCs were evaluated 72 h after the injury.
Initially, we tested the expression of cell surface markers and the migratory properties of unstimulated and cytokine-pretreated MSCs. These cells were positive for CD44 and CD105 and no significant differences in the intensity of expression of the tested markers were detected. A similar conclusion was reached by Najar et al. [20], who tested a panel of endothelial, stromal, and adhesive markers in unstimulated MSCs and MSCs stimulated with a cocktail of proinflammatory cytokines; no differences were detected. In addition, we did not find any variation in the migration of unstimulated and cytokine-pretreated MSCs, and a comparable distribution of injected MSCs was detected within the tested organs and tissues. In another study, Hemeda et al. [26] assessed the effect of IFN-γ and TNF-α on MSCs, and TNF-α was recognized as the predominant regulator of MSC migration. On the other hand, pretreatment of MSCs with IFN-γ increased their migration to the inflammed intestine in an animal model of colitis [27].
Our results confirmed that systemically administered MSCs are able to specifically migrate to the damaged eye. We found a more than 30-times higher number of injected MSCs in the damaged eye compared with the control contralateral eye. These findings are in agreement with the observation of Lan et al. [28], who detected systemically administered MSCs on day 3 in the cauterized cornea but not in the contralateral cornea. Systemically administered MSCs were also detected in the cornea 14 days after their injection in a rabbit eye alkali-burn model [29] but not in the case of xenogeneic (human) MSCs administered systemically in order to heal a corneal injury in the rat [19].
Since both unstimulated and cytokine-pretreated MSCs preferentially migrated into the injured eye, we tested the effects of the engrafted MSCs on the infiltration of the damaged eye by populations of lymphoid and myeloid cells. All types of MSCs slightly decreased infiltration by the lymphoid populations, but only MSCs pretreated with IFN-γ decreased the number of CD3+CD4+, CD3+CD8+, and CD19+CD22+ cells significantly. To date, a decreased expression of CD45 has been detected in damaged rat eyes transplanted with MSCs on an amniotic membrane [17], and a reduced infiltration of CD4+ cells was observed in injured corneas of mice treated with MSCs [16]. Further, we found that MSCs pretreated with IFN-γ suppressed the infiltration of the damaged eye by CD14+, CD80+, and CD11b+Gr-1+ myeloid populations even more effectively than they inhibited infiltration by lymphoid cells. These results are in agreement with the findings of Yao et al. [18], who showed that the number of CD68+ cells infiltrating the site of injury on day 7 was significantly lower in a group treated with MSCs [18]. In addition, the systemic injection of human MSCs significantly reduced the infiltration of neutrophils into the cornea on days 1 and 3 after injury in a rat model of a chemically burned eye [19]. These effects of MSCs on neutrophil infiltration are consistent with our observation that MSCs pretreated with IFN-γ strongly decreased the infiltration of the damaged eye by myeloid cell populations.
We next tested the effects of systemically administered MSCs on the early cytokine environment of the damaged eye. Although we tested a wide range of cytokines, that is, IL-1α, IL-2, IL-4, IL-6, IL-10, IL-17, and IFN-γ, only IL-1α, IL-6, and IL-10 were produced in this phase of inflammation in significant concentrations. We found that systemically administered MSCs stimulated with IFN-γ significantly decreased the production of IL-6 and IL-1α with the most profound inhibition of IL-1α production. This observation is in accordance with the findings of Roddy et al. [19], who showed in a rat model that the production of IL-1 on days 1 and 3 after injury was significantly decreased in corneas treated with human MSCs. On the other hand, Oh et al. [16] observed an increased production of IL-6 in rat corneas 3 weeks after injury and treatment with MSCs. This discrepancy could be explained by the different kinetics of IL-1 and IL-6 production. The peak of IL-1 production occurs at day 3 after injury, while the production of IL-6 culminates later [30].
Further, we detected a highly significant inhibition of iNOS expression and NO production in the damaged eye after treatment with MSCs prestimulated with IFN-γ. Since NO is a toxic and immunomodulatory molecule, its inhibition may represent another mechanism of the therapeutic action of MSCs.
In contrast to Roddy et al. [19], who found a significant increase in IL-10 production 3 weeks after the treatment of injured corneas with MSCs, we detected only a slight enhancement of IL-10 production in the damaged eyes of MSC-treated mice. This discrepancy can be due to the different time points used for IL-10 detection in our and Roddy's studies.
Since MSCs preincubated with various cytokines differ in their immunomodulatory effects, we tested the expression of genes for immunoregulatory molecules in unstimulated MSCs and in MSCs pretreated with IL-1α, IFN-γ, or TGF-β. Both unstimulated MSCs and cytokine-pretreated MSCs expressed significant levels of genes for TGF-β, HGF, and COX2. It has been already shown that these immunomodulatory molecules are constitutively expressed by MSCs [31]. On the other hand, we did not observe an increase in COX2 expression after stimulation with IFN-γ. English et al. [22] detected an increased expression of COX2 after the stimulation of murine MSCs with IFN-γ, but in their study a 20-times higher concentration of IFN-γ was used. We found that MSCs stimulated with IL-1α or IFN-γ expressed a significant level of the iNOS gene. Accordingly, the treatment of mouse MSCs with IFN-γ and any of three other proinflammatory cytokines (TNF-α, IL-1α, or IL-1β) induced the expression of several chemokines and iNOS [21]. In agreement with other studies [22,31,32], a significant increase in the expression of the IDO gene was observed in MSCs pretreated with IFN-γ.
Finally, MSCs stimulated with IFN-γ expressed significant levels of the genes for TGF-β, IDO, iNOS, HGF, and COX2; there were differences mainly in the expression of iNOS and IDO in comparison with the other types of MSC stimulation. Although iNOS and IDO are important immunomodulatory molecules [33 –35], other immunoregulatory mechanisms are probably also involved in MSC-mediated immunosuppression. Nevertheless, our results clearly show that systemically administered MSCs rapidly migrate into the site of injury and attenuate the early phase of the inflammatory reaction. Infiltration by both myeloid and lymphoid cells and the local production of proinflammatory cytokines are decreased by MSCs, and MSCs pretreated with IFN-γ are superior in the inhibition of this early inflammatory microenvironment.
Footnotes
Acknowledgments
This work was supported by grants 668012, 889113, and 546613 from the Grant Agency of Charles University; grants P304/11/0653, P301/11/1568, and 14-12580S from the Grant Agency of the Czech Republic; grant NT/14102 from the Grant Agency of the Ministry of Health of the Czech Republic; and the Charles University grant SVV 260083.
Author Disclosure Statement
No competing financial interests exist.