
Abstract
We aimed to study the relationship between glucosamine and FoxO1/Notch in gluconeogenesis and maintenance of mouse embryonic stem cell (mESC) self-renewal. Glucosamine (GlcN) increased glucose production and gluconeogenic enzyme (G6Pase and PEPCK) expression. GlcN also increased the percentage of cells in S phase, number of cells, and the protein expression of cell cycle regulatory proteins that were blocked by 3-mercaptopicolinic acid (gluconeogenesis inhibitor) or glucose transporter (GLUT) 1 neutralizing antibody. GlcN increased the O-GlcNAc transferase (OGT)–dependent protein O-GlcNAc level. Moreover, inhibition of OGT (by ST045849) decreased glucose production. GlcN enhanced the expression of OGT-dependent O-GlcNAcylated Notch1 and then increased the translocation of cleaved Notch1 to the nucleus. Moreover, GlcN stimulated the translocation of O-GlcNAcylated FoxO1 to the nucleus. GlcN increased the binding between cleaved Notch1 and FoxO1 with CSL, a transcription factor, which was blocked by L-685,458 (γ-secretase inhibitor) or ST045849, respectively. Simultaneous blockage of cleaved Notch1 and FoxO1 also decreased the expression of G6Pase and PEPCK more significantly than that by inhibition of cleaved Notch1 alone or FoxO1 alone. In addition, GlcN maintained the undifferentiation status while depletion of Notch1 and FoxO1 for 3 days decreased Oct4 and SSEA-1 expression and alkaline phosphatase activity or increased the mRNA expression of GATA4, Tbx5, Cdx2, and Fgf5. In conclusion, GlcN-induced OGT activation mediated glucose production through cleaved Notch1 and FoxO1, which contributed to the regulation of maintenance of self-renewal in mESCs.
Introduction
G
Forkhead box (Fox)–containing transcription factors of the FoxO subfamily are one of the key effectors of GlcN action in glucose production [14,15]. FoxOs are O-GlcNAcylated following increased oxidative stress, and this correlates with FoxO activation [16]. Hepatic FoxO1 promotes transcription of genes encoding G6Pase and PEPCK [17,18]. Several very specific downstream effects have illustrated the diversity in FoxO-regulated gene programs. These functions are probably elicited by cell-type-specific upstream regulators and cofactors and also through crosstalk between them and other pathways. Previous reports demonstrated that FoxO1 and Rbp-Jk directly interact, leading to corepressor clearance from and coactivator recruitment to promoters of Notch target genes, which is considered to be a key switch that regulates cell fate and plays a pivotal role in the regulation of stem cell maintenance [19,20]. It was reported that Notch signaling has various functions depending on the cell type, cell context, and crosstalk with other signaling systems [19,21]. Therefore, further studies on the regulation of Notch and FoxO are needed for a better understanding of the roles of O-GlcNAc modification in gluconeogenesis and maintenance of ESCs. In this study, we aimed to examine the relationship between glucosamine and FoxO1/Notch in gluconeogenesis and maintenance of mESC self-renewal.
Materials and Methods
Materials
The mESC line ES-E14TG2a was obtained from the American Type Culture Collection. The fetal bovine serum (FBS) was purchased from HyClone. The D-(+)-Glucosamine hydrochloride (GlcN), N-acetylcysteine (NAC), L-685,458, PD98059, fluorescein isothiocyanate (FITC)–goat anti-mouse IgM, and tetramethylrhodamine isothiocyanate (TRITC)–goat anti-rabbit IgM were supplied by Sigma Chemical Company. The ST045849 was purchased from TimTec. The leukemia inhibitory factor (LIF), 3-mercaptopicolinic acid, primary antibody (Ab) against G6Pase, PEPCK, glucose transporter (GLUT) 1, cyclinD1, cyclin-dependent kinase (CDK)4, cyclinE, CDK2, normal rabbit IgG, O-GlcNAc transferase (OGT), CTD110.6 (O-linked N-acetylglucosamine), RL2 (O-GlcNAc), ALG1 (N-linked glycosylation; asparagines-linked glycosylation 1), β-actin, Lamin A/C, Notch1, p-JNK, JNK, p-ERK, ERK, p-p38, CBF1/RBP-Jκ/Suppressor of Hairless/LAG-1 (CSL), Oct4, SSEA-1, and secondary Ab against horseradish peroxidase (HRP)–conjugated goat anti-rabbit IgG and goat anti-mouse IgG were purchased from Santa Cruz Biotechnology. The primary Ab against cleaved Notch1, FoxO1, and p38 was obtained from Cell Signaling Technology. The SP600125 and SB203580 were purchased from Biomol. All other reagents were of the highest purity commercially available and were used as received. To confirm the specificity, efficiency, and cytotoxicity of inhibitors, we treated inhibitors with various concentrations (nontreated, higher concentration, used concentration, and lower concentration) and examined the specificity, efficiency using western blotting, and cytotoxicity using MTT assay. Used concentration in this study inhibited target proteins or enzymes efficiently and had no cytotoxic effect (Supplementary Figs. S1 and S2; Supplementary Data are available online at
mESC culture
Cells were cultured in Dulbecco's modified Eagle's media (DMEM; Gibco-BRL) supplemented with 3.7 g/L sodium bicarbonate, 1% penicillin and streptomycin, 1.7 mM L-glutamine, 0.1 mM β-mercaptoethanol, 5 ng/mL mouse recombinant LIF, and 15% FBS on gelatin-coated culture dish. After a 24-h incubation period, the cells were washed twice with phosphate-buffered saline (PBS) and incubated with fresh serum-free media (5% serum replacement instead of 15% FBS) including all supplements and designated agents for the indicated period.
Measurement of glucose production
Glucose production was measured as previously described [22]. Briefly, cells were washed three times with warm PBS to remove medium, followed by treatment with GlcN for 0–24 h in glucose-free medium (Gibco 11966-025). Glucose concentration in medium was determined with a glucose assay kit from Roche Applied Science (Cat. No. 0716251) and was normalized to the cellular protein concentration.
Real-time reverse transcriptase–polymerase chain reaction
Total RNA was extracted from cells, and real-time quantification of RNA targets was then performed in a Rotor-Gene 6500 real-time thermal cycling system (Corbett Research) by using the QuantiTect SYBR Green RT-PCR kit (Qiagen) according to manufacturer's protocol. The resulting data were analyzed using the software provided by the manufacturer. The primers used are described in Table 1.
Western blotting and immunoprecipitation
Cell lysate protein concentration was determined by the Bradford method [23]. Equal amounts of protein (30 μg) were resolved by 8%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were incubated with 5% skim milk, appropriate primary Abs, and goat anti-rabbit IgG or goat anti-mouse IgG conjugated to HRP secondary Abs. The bands were visualized with enhanced chemiluminescence (Amersham Pharmacia Biotech) and quantified with TINA version 2.0 program package (Raytest).
For immunoprecipitation, cell lysates were incubated with both target Abs and protein A/G-sepharose beads with gentle shaking overnight. A 2×SDS-PAGE sample buffer was added into beads and incubated at 100°C for 5 min to release the protein sample and then western blotting was performed with target Abs. Lysate expression levels were used as a loading control. We confirmed specific protein–protein binding using IgG (data not shown).
Detection of lectin-binding N-glycan-specific Concanavalin A using lectin blot
Cell lysates were resolved by SDS-PAGE and transferred to PVDF membrane and then the membrane was incubated by Carbo-Free Blocking Solution (Vector Laboratories; Cat. No. SP-5040), 5 μg/mL biotinylated Con A (Cat. No. B-1005), or Vectastain Elite ABC (peroxidase; Cat. No. AK-5000). Image was acquired using DulLuX Chemiluminescent/Fluorescent Substrate for peroxidase (Cat. No. SK-6604). A sample incubated without lectin and with β-actin is prepared as a loading control.
Cellular reactive oxygen species assay
Detection of intracellular hydrogen peroxide (H2O2) utilized 5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate (CM-H2DCF-DA; Molecular Probes), which acts as an H2O2-sensitive fluorophore. DCF-DA (10 mM) was added to cells, which were then incubated in the dark for 30 min at room temperature. Cells were then viewed using IX71 confocal microscope (Olympus) with a 400× objective for imaging, the fluorescence was excited at 488 nm, and the light emitted was observed at 515–540 nm. To quantify the intracellular H2O2 levels, the cells treated with DCF-DA were rinsed twice with ice-cold PBS and then scraped. A 100 μL of cell suspension was loaded into a 96-well plate and examined using a luminometer (Victor3; Perkin-Elmer) and a fluorescent plate reader at excitation and emission wavelengths of 485 and 535 nm, respectively.
Cell cycle analysis using propidium iodide
Cells were incubated in freshly prepared nuclei staining buffer [250 μg/mL propidium iodide (PI) and 100 μg/mL RNase] for 30 min at 37°C. Cell cycle histograms were generated after analyzing the PI-stained cells by flow cytometry (Beckman Coulter). At least 104 events were recorded for each sample. The samples were analyzed using CXP software (Beckman Coulter), and proliferation index [(S+G2/M)/(G0/G1+S+G2/M)]×100 was calculated.
Cell number counting
Cells were incubated with GlcN for 24 h as described previously and washed twice with PBS. Cells were then detached from the culture dishes utilizing a 0.05% trypsin and 0.5 mM EDTA solution, and the detachment was quenched with Soybean trypsin inhibitor (0.05 mg/mL). Subsequently, 0.4% (w/v) trypan blue solution (500 μL) was added to the cell suspension and the cells were counted on a hemocytometer under optical microscopy, while keeping a separate count of the blue cells. Cells failing to exclude the dye were considered nonviable. The data are expressed as the percentage of viable cells.
Preparation of subcellular fraction
Cell fractions were prepared using a slightly modified methodology of that reported by Mackman et al. [24]. Medium was then removed and cells were washed twice with ice-cold PBS, scraped, harvested by microcentrifugation, and resuspended in a buffer A [137 mM NaCl, 8.1 mM Na2HPO4, 2.7 mM KCl, 1.5 mM KH2PO4, 2.5 mM EDTA, 1 mM dithiothreitol, 0.1 mM PMSF, and 10 μg/mL Leupeptin (pH 7.5)]. Resuspended cells were then lysed mechanically on ice by trituration with a 21.1-G needle and lysates were initially centrifuged at 8,000 rpm for 5 min at 4°C and pellet was the nuclear fraction. The supernatant was centrifuged at 15,000 rpm for 1 h at 4°C to prepare cytosolic and total particulate fractions. The particulate fraction, which contained the membrane fraction, was washed twice and resuspended in buffer A containing 1% (v/v) Triton X-100.
Small interference ribonucleic acid transfection
After cells were grown until 75% of the surface of the culture dish, they were transfected with either a small interference ribonucleic acid (siRNA) specific for FoxO1 CSL (10 nM; Dharmacon), or nontargeting (Nt) siRNA (as a negative control, 10 nM; Dharmacon) for 24 h with hiperfectamine (Qiagen) according to the manufacturer's instructions. Transfected siRNA sequence is indicated in Table 2.
Alkaline phosphatase staining
Cells were washed twice with PBS and fixed for ∼15 min with 4% formaldehyde at room temperature. After washing with PBS, cells were incubated with 2 mL AP substrate solution (200 μg/mL Naphthol AS-MX phosphate, 2% N,N-dimethylformamide, 0.1 M Tris, and 1 mg/mL Fast Red TR salt) for 10 min and, if stained, then cells were washed with PBS and photographed.
Immunofluorescence staining
Cells were fixed with 3.5% paraformaldehyde, 0.1% Triton X-100, and 5% bovine serum albumin, and were then treated with a 1:50 dilution of primary antibody against target protein. Each sample was then incubated for 30 min with a 1:100 dilution of FITC and TRITC-conjugated secondary antibody or incubated with FITC and PI (10 μg/mL). Fluorescence images were visualized with a FluoView fluorescence microscope (Olympus).
Duolink® fluorescence assay (proximity ligation assay)
CSL/cleaved Notch1 or CSL/FoxO1 interactions were detected in situ using Duolink II secondary antibodies and detection kits (Olink Bioscience; Cat. No. 92001, 92005, and 92007) according to the manufacturer's instructions. Briefly, primary antibody against CSL (anti-rabbit) and cleaved Notch1 or FoxO1 (anti-mouse) was applied under standard conditions. Duolink secondary antibody against the particular primary antibody was then added. These secondary antibodies were provided as conjugates to oligonucleotides that were ligated together in a closed circle by Duolink ligation solution if the antibodies were in close proximity (<40 nm). Finally, polymerase was added, which amplified any existing closed circles, and detection was achieved with complementary, fluorescently labeled oligonucleotides (green dot). PI staining (red) was used to verify cell morphology and confocal images were acquired.
Statistical analyses
All results are expressed as mean±standard error (SE). All experiments were analyzed by analysis of variance, followed in some cases by a comparison of the treatment and the control using the Bonnferroni–Dunn test. A P value of <0.05 was considered to be significant.
Results
Effect of GlcN on gluconeogenesis and cell proliferation
To examine the effect of GlcN on gluconeogenesis, the level of glucose production and the level of gluconeogenic enzymes (G6Pase and PEPCK) were examined. Cells were cultured in glucose-free medium during treatment with GlcN to prevent the interference effect of glucose in basal medium and then glucose production in the medium was measured. Compared with nontreated control, GlcN increased the level of glucose in the culture medium (Fig. 1A) and stimulated the mRNA and protein expression of G6Pase and PEPCK enzymes (Fig. 1B, C), suggesting that GlcN-induced increase in glucose production is mediated by gluconeogenic enzymes. To determine the specific role of GlcN in G6Pase and PEPCK expression, we examined the G6Pase and PEPCK expression after removal of GlcN. The expression of G6Pase and PEPCK fell back to its normal level after 12 h of GlcN removal (Supplementary Fig. S3). In the experiments of cell cycle analysis using PI staining, GlcN increased the cell proliferation index, which was blocked by a gluconeogenesis inhibitor (3-mercaptopicolinic acid; 10−3 M) or GLUT-1 neutralizing antibody (1 μg/mL) (control: 60.55%±5.01%; GlcN: 81.2%±7.98%; 3-mercaptopicolinic acid+GlcN: 65.11%±2.95%; and GLUT-1 Ab+GlcN: 64.97%±5.24%) (Fig. 1D). Moreover, the cell count using trypan blue dye and analysis of cell cycle regulatory proteins by western blot confirmed the effect of GlcN on cell proliferation (Fig. 1E–G). Taken together, GlcN induced an increase in glucose production through G6Pase and PEPCK and then stimulated cell proliferation.
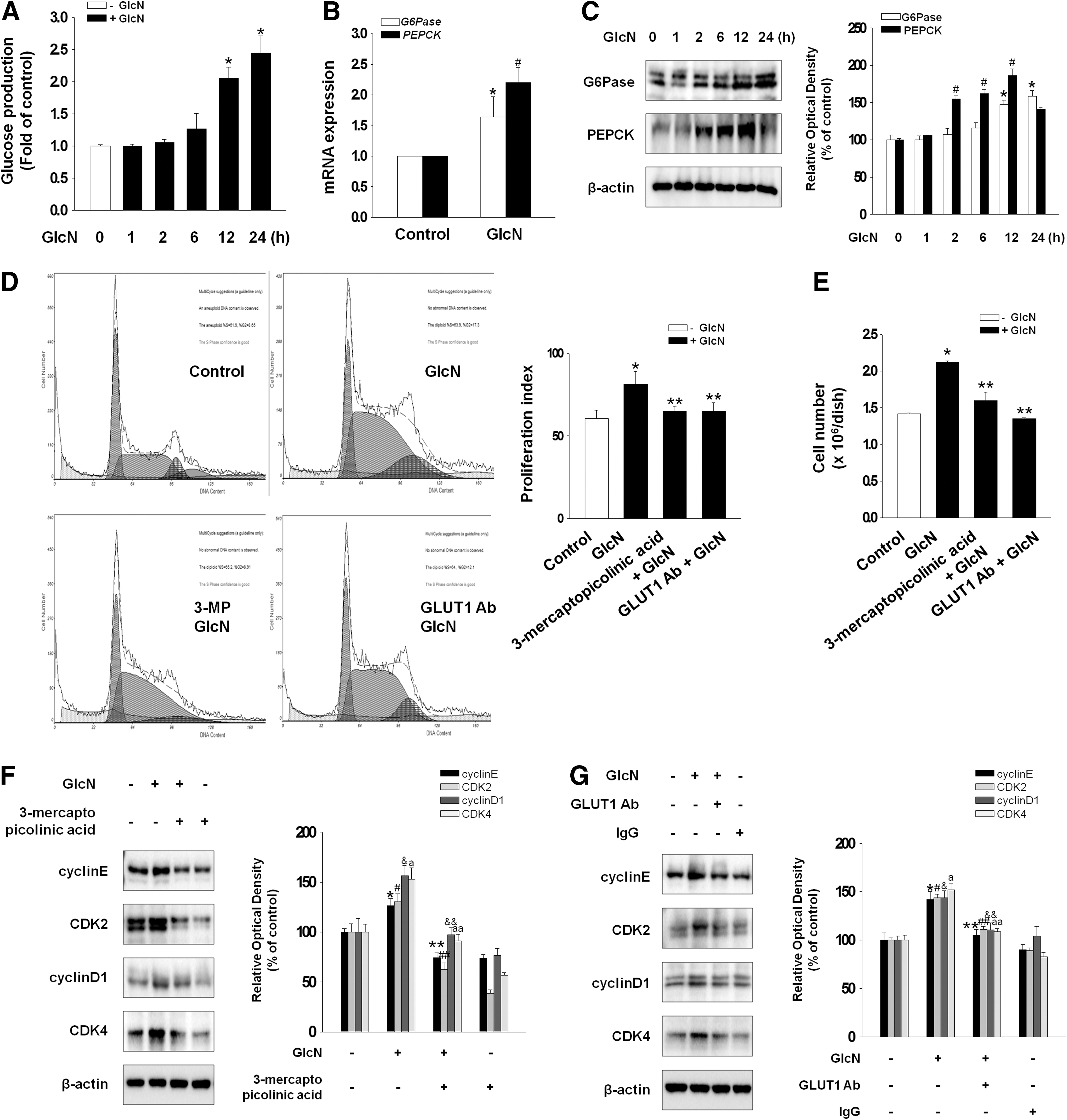
Effect of GlcN on gluconeogenesis and cell proliferation. Mouse embryonic stem cells (mESCs) were treated with GlcN (2 mM) for 0–24 h.
To examine the involvement of ROS in GlcN-induced signaling pathway, the level of DCF-sensitive cellular ROS was quantified using a luminometer and visualized using confocal microscopy. Both experiments showed that GlcN increased the DCF-sensitive fluorescence, which was blocked by an antioxidant (NAC; 10−5 M) (Fig. 2A, B). Using two O-GlcNAc antibodies (CTD110.6 and RL2), which recognize different O-GlcNAc motifs, we confirmed that GlcN alone significantly increases overall O-GlcNAc level. GlcN increased the OGT activity and the expression level of CTD110.6 as well as RL2 (Fig. 2C) that were blocked by NAC (Fig. 2D). To examine whether GlcN affects other glycosylation such as N-linked-glycosylation, the expression of AGL1 was measured. However, GlcN did not change the expression of ALG1 (Fig. 2E). We also detected the level of lectin binding Concanavalin A (Con A) that binds to N-glycan. The level of Con A did not change under influence of GlcN (Fig. 2F). In the experiment to examine the relation between GlcN-mediated OGT expression and gluconeogenesis, inhibition of OGT (by ST045849; 2×10−5 M) blocked GlcN-induced increase in glucose production (Fig. 2G). This result suggests that ROS-mediated OGT expression by GlcN is related to signaling molecules involved in glucose production.
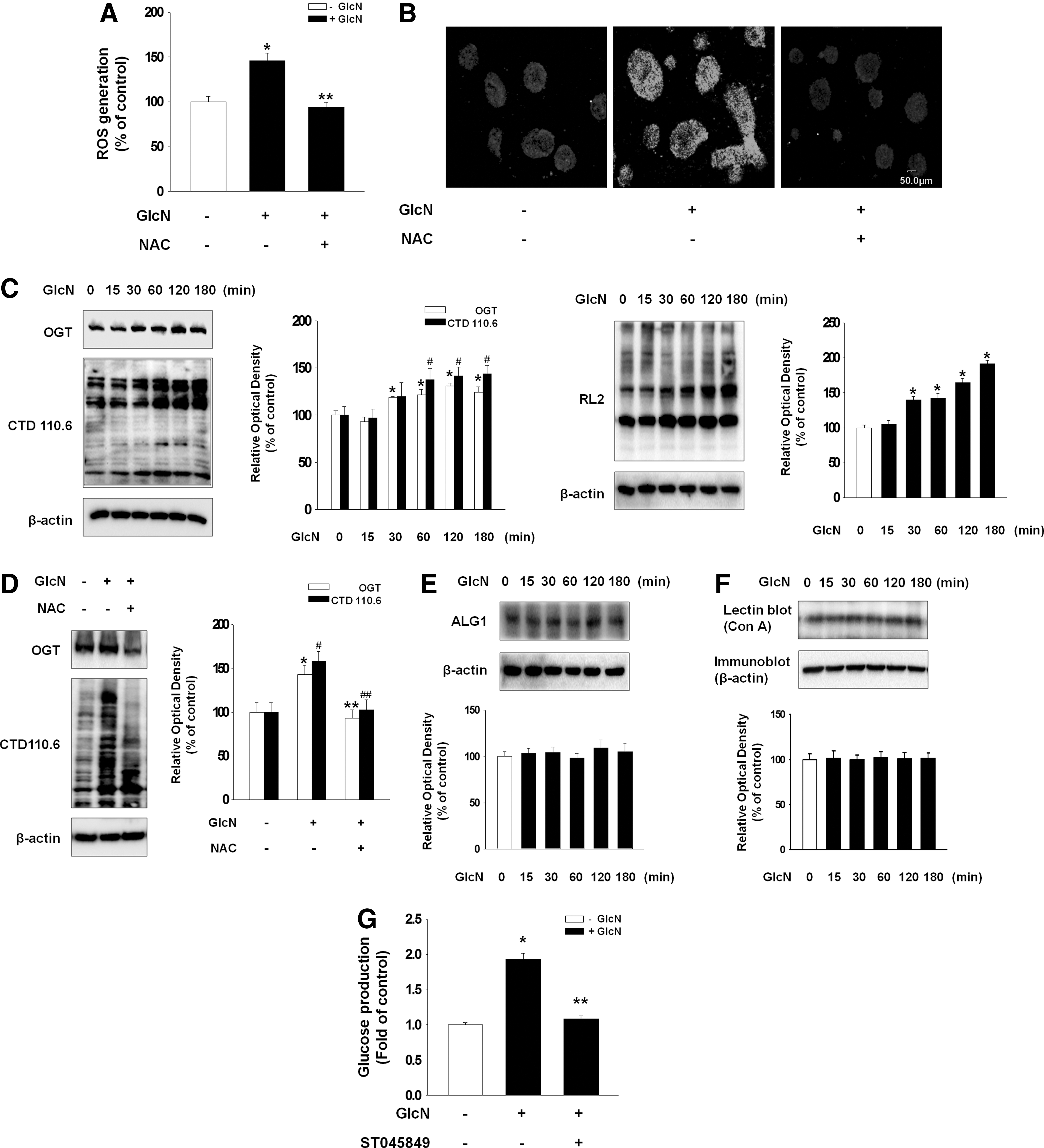
Involvement of ROS-induced OGT on gluconeogenesis.
Effect of O-GlcNAcylated Notch1 and FoxO on G6Pase and PEPCK expression
Experiments to determine the direct relationship between Notch and O-GlcNAc showed that GlcN increased O-GlcNAcylated Notch1 and Notch1 protein expression in the plasma membrane as well as translocation of cleaved Notch1 to the nucleus, which were blocked by ST045849 (Fig. 3A, B). Immunofluorescence staining confirmed these results that inhibition of OGT attenuated the fluorescence intensity indicating Notch glycosylation in plasma membrane (Fig. 3C). Next, we examined the involvement of MAPKs as regulators of γ-secretase for the cleavage event of Notch. GlcN increased the phosphorylation of JNK after 15 min, that of ERK at 15 min until 60 min, and that of p38 after 120 min (Fig. 3D). Moreover, increased phosphorylation of JNK was blocked by NAC while increased phosphorylation of ERK and p38 was not blocked by NAC (Fig. 3E). In addition, GlcN enhanced the translocation of cleaved Notch1 to the nucleus, which was blocked by a JNK inhibitor (SP600125; 10−6 M) and not by an ERK (PD98059; 10−5 M) or a p38 (SB203580; 10−6 M) inhibitor (Fig. 3F). These results indicate that γ-secretase-induced Notch cleavage was dependent on ROS-mediated JNK signaling. To determine the relation between cleaved Notch1 and gluconeogenic enzymes, we examined whether cleaved Notch1 translocated to the nucleus binds to CSL, a transcription factor. GlcN increased the binding of cleaved Notch1 with CSL, which was blocked by L-685,458 (γ-secretase inhibitor; 10−5 M) (Fig. 3G). Proximity ligation assay supported this result that pretreatment of L-685,458 decreased the GlcN-induced dot in nucleus, which indicates the binding of CSL and cleaved Notch1 (Fig. 3H). Inhibition of cleaved Notch1 blocked GlcN-induced increase in G6Pase and PEPCK activity (Fig. 3I).

Effect of Notch1 glycosylation on GlcN-induced gluconeogenesis.
Next, as FoxO1 is known as a regulator of glucose metabolism, we examined whether FoxO1 is involved in GlcN-induced glucose production. GlcN increased O-GlcNAcylated FoxO1 and FoxO1 translocation to the nucleus, which were blocked by ST045849 (Fig. 4A, B). GlcN also increased the total amount of FoxO1 (Fig. 4A). Immunofluorescence staining confirmed these results that inhibition of OGT attenuated the GlcN-induced FoxO1 glycosylation (Fig. 4C). To examine whether translocated FoxO1 to the nucleus binds to CSL, binding of FoxO1 and CSL in the nucleus was examined by immunoblot and proximity ligation assay. GlcN increased the binding of FoxO1 with CSL and this increase was blocked by OGT inhibitor (ST045849) (Fig. 4D, E). Moreover, knockdown of FoxO1 decreased the GlcN-induced increase in G6Pase and PEPCK expression (Fig. 4F). To examine the direct role of CSL in G6Pase and PEPCK expression, cells were transfected with CSL-specific siRNA. Knockdown of CSL resulted in decrease of gluconeogenic enzyme expression (Fig. 4G). The just discussed results showed that both cleaved Notch1 and FoxO1 regulate the activity of gluconeogenic enzymes (G6Pase/PEPCK) through binding with CSL. Therefore, to elucidate whether these signaling pathways are dependent or independent, both Notch1 and FoxO1 were inhibited simultaneously or Notch1 alone or FoxO1 alone was inhibited and then the mRNA expression level of gluconeogenic enzymes and glucose production were examined. Simultaneous blockage of cleaved Notch1 and FoxO1 decreased the expression of G6Pase/PEPCK and glucose production more significantly than that by inhibition of cleaved Notch1 alone or FoxO1 alone (Fig. 4I, J), suggesting that these two signaling pathways act coordinately. The transfection efficiency of FoxO1 siRNA was measured using western blotting (Fig. 4H).
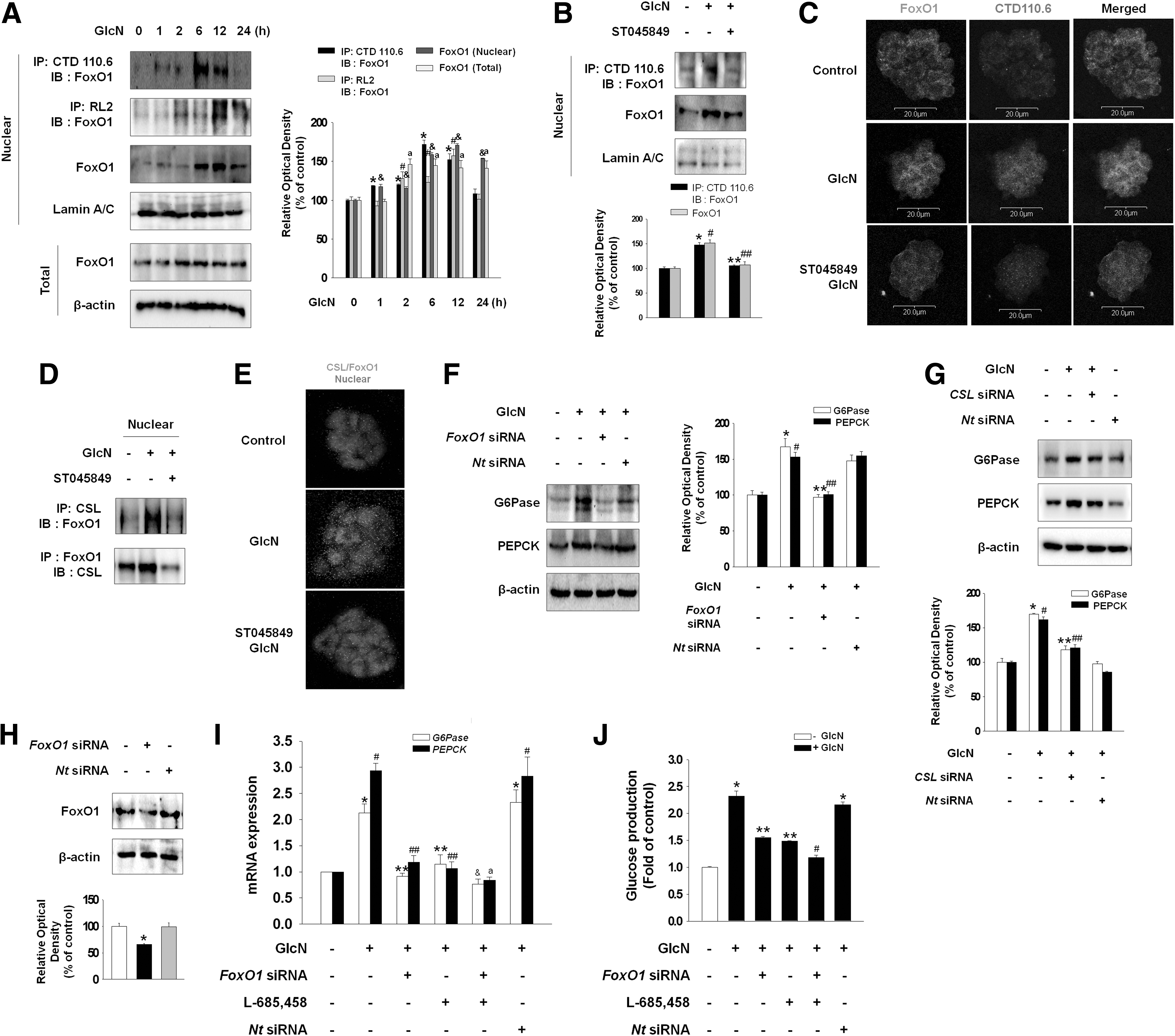
Effect of FoxO1 glycosylation on GlcN-induced gluconeogenesis.
Role of GlcN-induced cleavage of Notch1 and FoxO1 in the maintenance of self-renewal
We found that GlcN increased glucose production through cleaved Notch1 and FoxO1. Therefore, we wondered whether cleaved Notch1 and FoxO1 were involved in the maintenance of undifferentiation status of mESCs. Treatment with GlcN maintained Oct4 expression for 4 days, whereas inhibition of cleaved Notch1 and/or FoxO1 decreased Oct4 expression below the control level after 2 or 3 days (Fig. 5A). Moreover, alkaline phosphatase (AP) staining and immunofluorescence staining against SSEA-1 and Oct4 showed that the expression of undifferentiation markers was decreased at 3 days after depletion of cleaved Notch1 and FoxO1 (Fig. 5B). Western blot confirmed the relation between Notch1, FoxO1, and undifferentiation status of mESCs. GlcN ameliorated the L-685,458- and/or FoxO1 siRNA-induced decrease in Oct4 protein expression (Fig. 5C). On measuring the differentiation markers, depletion of cleaved Notch1 and FoxO1 increased the mRNA expression of differentiation markers [GATA4 (endoderm), Tbx5 (mesoderm), Cdx2 (trophectoderm), and Fgf5 (ectoderm)] (Fig. 5D). These results suggest that mESCs were maintained in an undifferentiated state with GlcN treatment, and Notch1 and FoxO1 were required for the maintenance of self-renewal in mESCs.
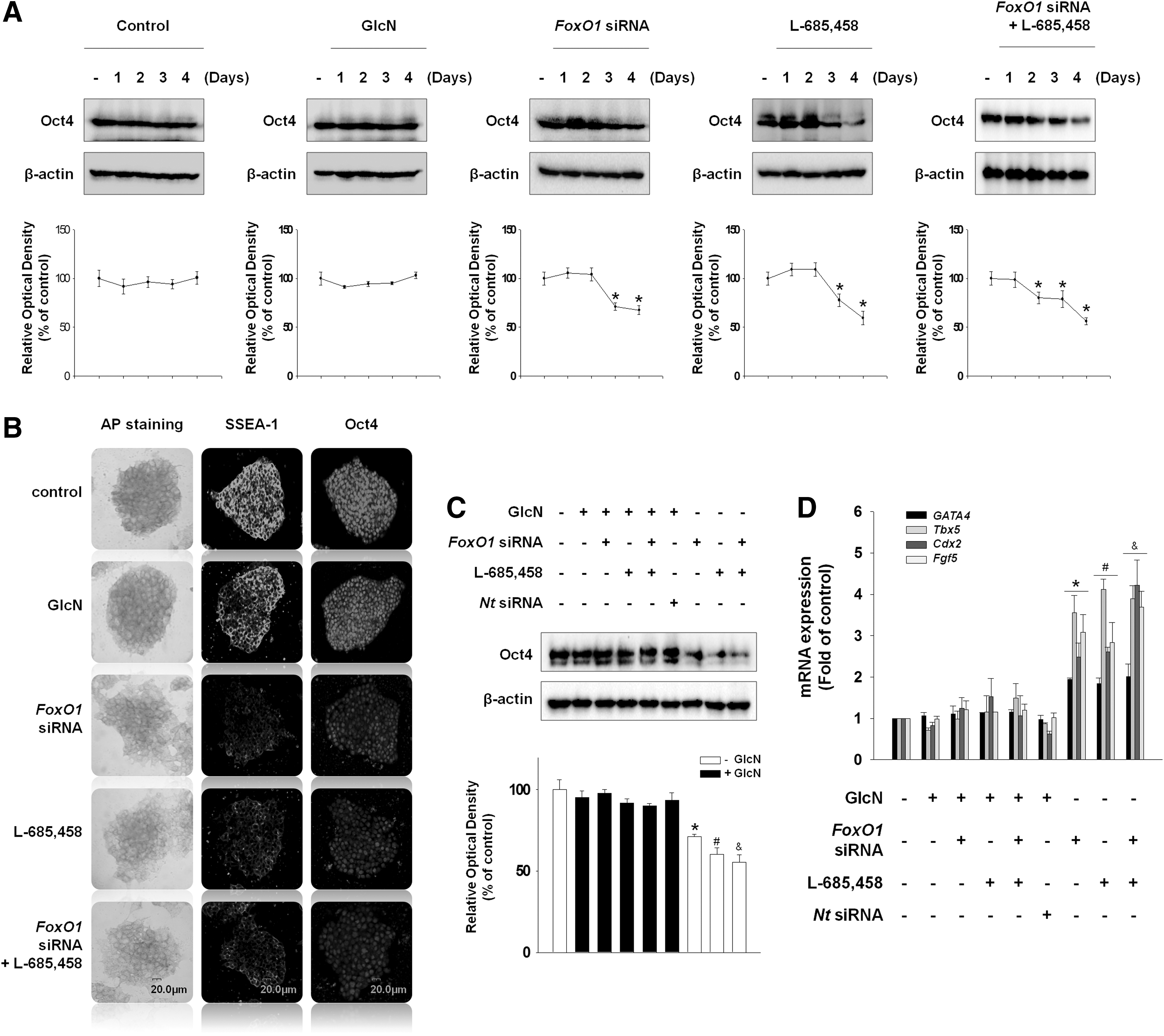
Effect of FoxO1 and Notch1 on maintenance of mESC self-renewal.
Discussion
The present study demonstrated that GlcN-induced OGT activation mediated glucose production through cleaved Notch1 and FoxO1, which contributes to the regulation of maintenance of self-renewal in mESCs. As the present study showed that GlcN increased the activities of G6Pase and PEPCK along with increased glucose production, we hypothesized that GlcN might regulate some of the transcription factors, which affect gluconeogenic enzyme gene expression. Among the various functions of GlcN, evidence has accumulated for a role as a cellular function transducer whereby multiple signals such as O-GlcNAc modification are initiated [25]. Disruption of O-GlcNAc cycling through pharmacological manipulation or overexpression of OGT or O-GlcNAcase (OGA) results in major cell cycle defects [26]. Also, it is well known that OGA induced by GlcN is essential for embryonic development and maintenance of genomic stability. [27]. Glycosylation is the most complicated posttranslational modification with an extremely high level of structural diversity. The heterogeneity and the large structural diversity have made oligosaccharide analysis significantly more difficult than other biopolymers. Although recent reports show the protein modification of O-GlcNAc using mass spectrometry [28,29], we have not solved the problems of complete structural identification. Therefore, we confirmed the modification of O-GlcNAc using both CTD110.6 and RL2 antibodies (O-GlcNAc-specific antibody recognizing other motif) as well as immunofluorescence staining consistent with previous study [30 –33]. These results provide evidence that O-GlcNAc regulates the activation of major signaling pathways and then manipulates the stem cell functions. It has been reported that reactive oxygen species (ROS) lead to the activation of HBP and increased O-GlcNAc synthesis [34,35]. Although we know relatively little regarding the regulation of OGT and OGA, we proved the possibility that ROS could directly modulate their activities.
The extracellular portion of Notch undergoes extensive N- and O-linked glycosylation during Notch synthesis and secretion, which is critical for proper folding of the receptor and its subsequent interactions with ligands [36]. Following export to the cell surface, Notch signal generates the forces needed to expose an otherwise inaccessible cleavage site in the extracellular portion of the Notch C-terminal fragment, which is subsequently cleaved by the intramembrane γ-secretase aspartyl protease complex, leading to the release of the Notch intracellular signal transducing fragment of the Notch intracellular domain (cleaved Notch) [37]. According to the previous studies, O-GlcNAcylated Notch1 seems to be indirectly cleaved Notch1 through γ-secretase. Interestingly, γ-secretase is regulated by MAPKs with type-specific different aspect. C-Jun N-terminal kinase (JNK) activates γ-secretase [38] whereas ERK1/2 is an endogenous negative regulator of the γ-secretase activity [39]. In the experiments to elucidate the regulatory mechanism of γ-secretase activation, we found that GlcN-induced JNK activation mediates translocation of cleaved Notch1 to the nucleus. The demonstration that FoxO1 directly interacts with CBF1/RBP-Jκ/Suppressor of Hairless/LAG-1 (CSL) provides a molecular mechanism by which FoxO1 could modulate Notch1 signaling. Moreover, we found that the simultaneous blockage of cleaved Notch1 and FoxO1 decreased the expression of G6Pase and PEPCK more significantly than that by inhibition of cleaved Notch1 alone or FoxO1 alone. Kitamura et al. [21] reported that Notch/FoxO cooperation integrates environmental cues through Notch with metabolic cues through FoxO1 to regulate progenitor cell maintenance. These data indicate that cleaved Notch1 and FoxO1 signaling is functionally connected in the maintenance of embryonic stem cells. These functions are probably elicited by cell-type-specific upstream regulators and cofactors as well as by crosstalk with other pathways.
Developmental pathways regulating self-renewal interact with nuclear transcription factors to keep the cells in an “undifferentiated” state as well as to determine cell fate [40]. Oct4 is necessary to maintain pluripotency and self-renewal of stem cells through positive regulation of its own gene and genes encoding the components of key signaling pathways [41]. It controls the expression of many genes (SOX2, FGF4, Opn, STAT3, and Utf1) that are essential for making stem cells capable of continuous self-renewal and maintaining pluripotency [42,43]. Accordingly, a recent study reported that FoxO1 activates Oct4- and SOX2-dependent promoters by upregulating their expression in embryonic stem cells, thus acting as an essential regulator of pluripotency [34]. Although we did not examine the direct relation between O-linked glycosylation and Oct4, we found that inhibition of FoxO1 disrupts the pluripotency, suggesting that GlcN-induced O-GlcNAcylated FoxO1 might be critical for Oct4 transcriptional activity. FoxOs, especially FoxO1, influence stem cell fate, and we cannot rule out the possibility of crosstalk with Notch pathway because of the association of Oct4 with Notch signaling pathway [44]. Consistent with previous reports, we also found that cleaved Notch1 and FoxO1 play an important role in maintenance of self-renewal in mESCs. The data suggest that FoxO1 binding to the CSL domain stabilizes the Notch/CSL complex and promotes corepressor clearance. These findings also suggest a mechanism by which the two major pathways cleaved Notch1 and FoxO1 act in a synergistic manner to control stem cell self-renewal. Although it remains to be seen whether other FoxO and Notch isoforms also interact and how they contribute to this process, we proved the hypothesis that GlcN regulates cleaved Notch1 and FoxO1 and affects PEPCK and G6Pase gene expression. Taken together, GlcN increased OGT-dependent glucose production, and its related genes Notch1 and FoxO1 played an essential role in maintenance of mESC self-renewal (Fig. 6). Therefore, it is conceivable that the coordination between Notch1 and FoxO1 would lead to maximum self-renewal capacity and maintenance of the stem cell pool. In conclusion, GlcN-induced OGT activation mediated glucose production through cleaved Notch1 and FoxO1, which contributed to the regulation of maintenance of self-renewal in mESCs.
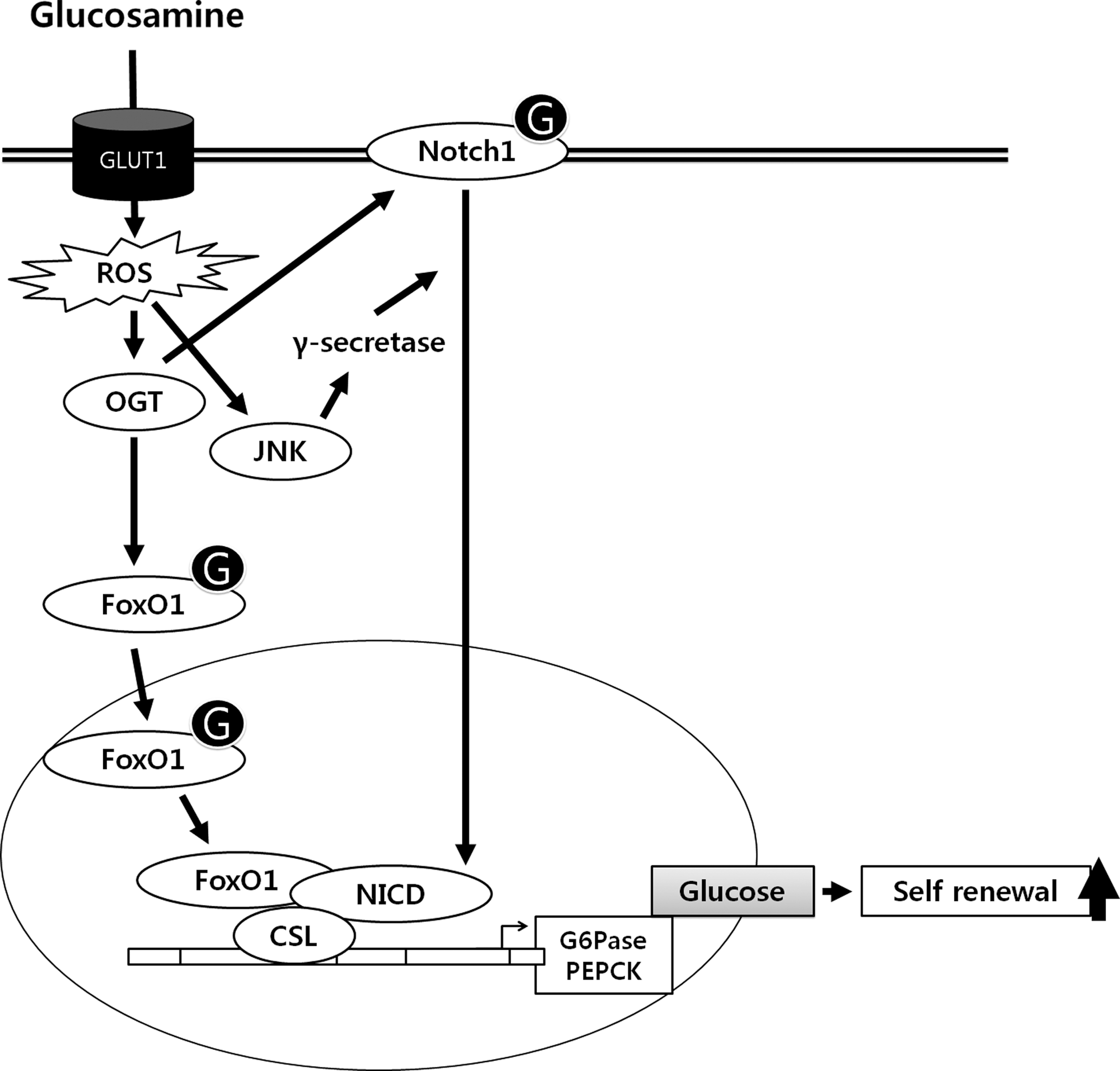
Hypothesized model for signal pathways underlying GlcN-mediated glucose production through cleaved Notch1 and FoxO1 coordinately contribute to regulate maintenance of self-renewal in mouse embryonic stem cells. GlcN entered into the cell through GLUT1 enhances ROS and then OGT activation. Subsequently, GlcN increases Notch1 glycosylation and JNK-dependent translocation of cleaved Notch1 into nucleus. Moreover, GlcN increases FoxO1 glycosylation and translocation into nucleus. Translocated cleaved Notch1 and FoxO1 bind to CSL transcription factor and then stimulates glucose production and self-renewal. In conclusion, GlcN-induced OGT activation mediated glucose production through cleaved Notch1 and FoxO1, which contribute to regulate maintenance of self-renewal in mESCs.
Footnotes
Acknowledgment
This research was supported by a grant of the Korean Health Technology R&D Project (A120216) and National Research Foundation (NRF-2013M3A9B4076520), Korean government.
Author Disclosure Statement
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.