
Abstract
While hydroxyurea (HU) is well known to deplete dNTP pools and lead to replication fork arrest in the cell, the mechanisms by which it exerts a cell response are poorly understood. Here, our results suggest that mouse embryonic stem cells (mESCs), unlike terminally differentiated cells such as mouse embryonic fibroblasts (MEFs), rapidly respond to low concentrations of HU by p53 acetylation, leading to activation of the caspase-dependent apoptotic pathway. We show that HU treatment induces the production of nitric oxide (NO), which plays a central role in the rapid induction of apoptosis in mESCs. By contrast, reactive oxygen species, which are expressed at significantly higher levels in mESCs compared with MEFs, are not related to the HU response. Furthermore, on exposure to HU, the p38 signaling pathway becomes activated in a dose-dependent manner, and chemical inhibition of the p38 pathway attenuates HU-dependent apoptosis in mESCs. Our data reveal that acetylation of p53 as a result of HU-dependent NO production plays a key role in the induction of the apoptotic response in mESCs. Finally, p38 signaling appears to be the main pathway underlying the activation of apoptosis in mESCs in response to HU exposure.
Introduction
E
A possible explanation for the low mutational rate of ESCs may be that self-renewing cells with damaged DNA are eliminated from the stem cell population. The unique cell cycle profile of ESCs makes their response to DNA-damaging agents different from that of somatic cell types. For example, while most somatic cells are arrested in the G1 phase of the cell cycle after DNA damage, mESCs lack a G1 checkpoint and, if damaged, are eliminated from the self-renewing population through differentiation or apoptosis [4,5]. Support for this mechanism was derived from independent observations showing that ESCs are hypersensitive to DNA-damaging agents and rapidly undergo caspase-3-dependent apoptosis after DNA damage [6 –11].
Accurate DNA replication is also essential for ESCs to prevent the accumulation of mutations and to maintain genome integrity. During DNA replication, numerous obstacles such as DNA lesions on the DNA template cause replication fork stalling or fork collapse, which can lead to replication-dependent DNA double-strand breaks (DSBs). In mammalian cells, DNA replication stress rapidly activates signaling cascades that halt S-phase progression, facilitate repair, and protect cells from apoptosis [12]. For example, when dNTPs are depleted by hydroxyurea (HU), replication forks become progressively stalled and collapse into DNA DSBs, thereby activating two different Rad51-mediated pathways to restart replication and repair the DSBs [13]. By contrast, ESCs exposed to inhibitors of DNA replication commit to apoptosis instead of activating critical S-phase checkpoints to induce homologous recombination-dependent repair of stalled or collapsed replication forks [14]. However, little is known of the underlying mechanisms for the rapid induction of apoptosis in ESCs exposed to DNA replication stress caused by replication inhibitors such as HU.
p53 has been extensively studied as a transcription factor that activates target genes involved in cell-cycle arrest, apoptosis, and DNA repair [15]. p53-mediated apoptosis can occur by a transcription-dependent mechanism in the nucleus or by a transcription-independent pathway through direct action on mitochondria [16,17]. In the latter context, p53 is translocated to mitochondria in stressed cells, where it induces mitochondrial outer membrane permeabilization through direct physical interactions with BCL-2 family members [18 –21].
Nitric oxide (NO) is a diffusible short-lived signaling molecule that acts in a diverse range of physiological processes, including cell growth, differentiation, and apoptosis depending on the subcellular environment [22]. Previous studies by other investigators have shown that NO induces apoptosis in mESCs by activating the p38 MAP kinase and ERK signaling pathways [23].
Here, we investigated the response of mESCs to the replication inhibitor HU. The data show that HU rapidly induces acetylation of p53, which initiates caspase-dependent apoptosis of mESCs. Moreover, by using chemical inhibitors, we identified NO as a central player in the hypersensitive response of mESCs, and the p38-mediated pathway as the main signaling mechanism inducing apoptosis on HU exposure.
Materials and Methods
Reagents
HU (Cat No. H8627), EX527 (Cat No. E7034), N-acetyl-
Cell culture
mESCs, J1, were cultured on 0.1% gelatin-coated Petri dishes in mESC culture media composed of Dulbecco's modified Eagle's medium (DMEM) plus GlutaMAX™-1 (Cat No. 10569; Gibco Invitrogen, Carlsbad, CA,
RNA interference
Nonspecific siRNAs used as a negative control were purchased from Bioneer (Cat No. SN-1001; Daejeon, Korea,
Sirt1 overexpression
pAd-Track Flag-SIRT1 was purchased (Plasmid #8438; Addgene, Cambridge, MA) to overexpress sirt1 and transfected into mESCs with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Extraction of RNA and real-time PCR
Total cellular RNA was extracted from mESCs using the TRIZOL Reagent (Ambion, Foster City, CA) following the manufacturer's instructions. Total RNA (2 μg) was used for single-stranded cDNA synthesis using an oligo(dT)18 primer and Omniscript Reverse Transcriptase (Qiagen, Valencia, CA). Gene-specific primers were designed using Primer3 (v. 0.4.0), a web-based primer design tool (Supplementary Table S1; Supplementary Data are available online at
Isolation of protein and western blotting
Total protein was isolated from cultured cells that were washed twice with phosphate-buffered saline and then lysed with cell lysis buffer (Cat No. 9803; Cell Signaling Technology, Danver, MA,
Flow cytometry
For cell-cycle analysis, cells were fixed in 70% ethanol and stained with propidium iodide (PI) (Cat No. p4170; Sigma-Aldrich) for 30 min at room temperature under dark conditions. The distribution of cell-cycle phases was analyzed by flow cytometry. An annexin V assay was performed using the ApoScan Annexin V-FITC apoptosis detection kit (Cat No. LS-02-100; BIOBud, SungNam, Korea,
Statistical analysis
The data are presented as the mean±SE, and statistical significance was assessed using one-way ANOVA followed by the Student's t-test for unpaired samples. A probability (P-value) of <0.05 was considered significant.
Results
Apoptosis is induced by HU in mESCs
To examine the response of mESCs to replication stress, we first treated mESCs with various concentrations of HU and examined changes to the cell cycle. As shown in Figure 1A, the cell-cycle distribution of mESCs rapidly changed after administration of 50 μM of HU, with a higher sub-G1 population observed in HU-treated cells than in -untreated cells. The population of sub-G1 phase cells gradually increased from 17.64% to 85.29% in an HU dose-dependent manner. Contrary to mESCs, MEFs were resistant to HU even at a concentration of 100 μM. However, the population of MEFs in the S phase increased at 200 μM HU, indicating that HU induces growth retardation in MEFs rather than growth arrest or apoptosis (Fig. 1A). Annexin V staining and flow cytometry analysis revealed that HU induces apoptosis in a dose-dependent manner in mESCs (Fig. 1B). Consistent with the cell-cycle analysis and annexin V staining, protein procaspase-3 and procaspase-9 levels decreased on HU treatment in mESCs, but not in MEFs (Fig. 1C). These results suggest that mESCs, unlike MEFs, rapidly respond to low concentrations of HU and activate caspase-mediated apoptosis.
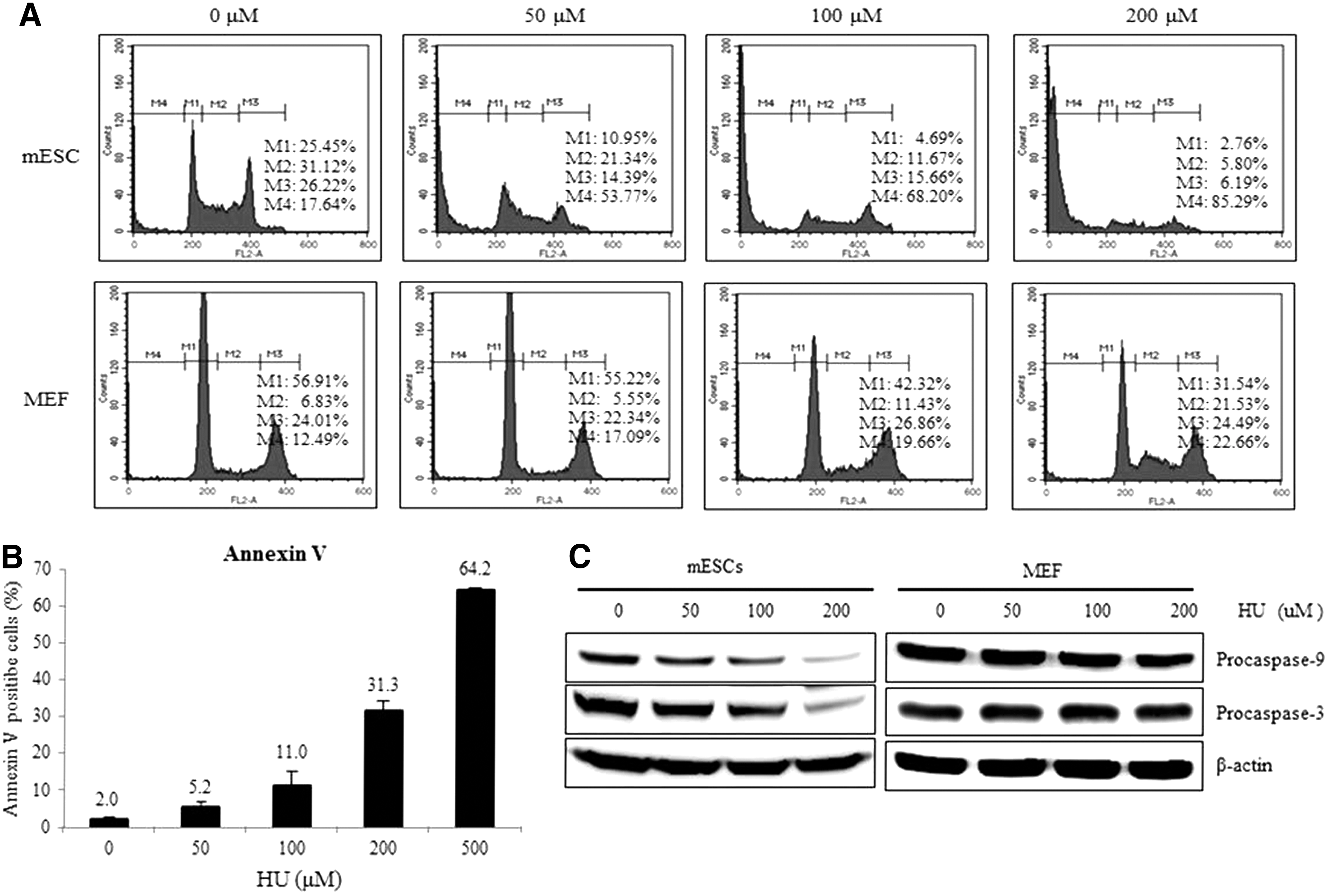
Apoptosis is observed after hydroxyurea (HU) exposure in mouse embryonic stem cells (mESCs).
HU induces apoptosis of mESCs through a p53-dependent pathway
To determine whether HU-mediated apoptosis in mESCs is p53 dependent, we examined the suppression effect of p53 on apoptosis. As shown in Figure 2A, HU-induced apoptosis reduced by about 50% when p53 expression was silenced by siRNA. In addition, HU-mediated caspase activation during apoptosis of mESCs was compromised by the suppression of p53 (Fig. 2B). Furthermore, expression of the p53 target genes, p21, mouse double minute 2 homolog (MDM2), and Noxa, was induced by HU treatment (Fig. 2C). These results indicate that HU-induced apoptosis was regulated by p53-mediated caspase activation in mESCs.
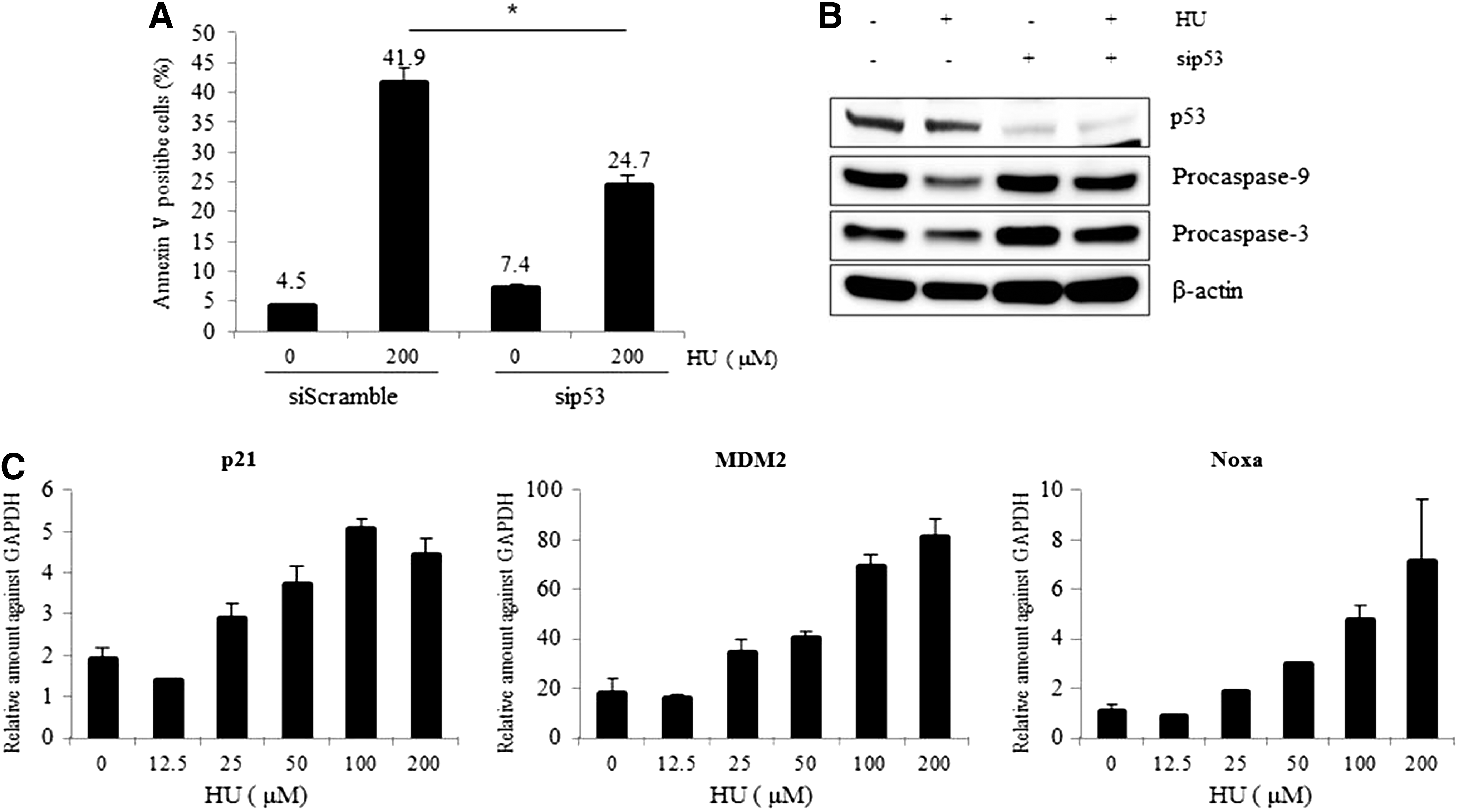
HU-induced apoptosis is mediated by p53.
Acetylation of p53 mediates apoptosis of mESCs through a p53-dependent pathway
Previous studies have provided evidence that DNA damage induces acetylation of p53, leading to p53 activation and either growth arrest or apoptosis of damaged cells [24,25]. To understand the underlying mechanism of the sensitive response of mESCs to HU, we first examined acetylation of p53 on HU treatment. As expected, acetylation of p53 occurred after HU exposure in a dose-dependent manner (Fig. 3A). We next examined the underlying mechanism of HU-mediated acetylation of p53. Notably, expression of sirt1, which is a class III histone deacetylase and functions as a p53 deacetylase, gradually and dose-dependently decreased on HU treatment (Fig. 3B, upper panel). Consistently, sirt1 mRNA levels also decreased on HU treatment (Fig. 3B, lower panel), indicating that the accumulation of acetyl p53 after HU exposure was caused by sirt1 depletion. To understand the importance of p53 acetylation in the HU-dependent induction of apoptosis in mESCs, we co-treated cells with EX527, a chemical inhibitor of sirt1, and HU. As expected, acetylation of p53 was increased dose dependently with the addition of EX527 (Supplementary Fig. S1A). In addition, procaspase-3 levels decreased in EX527-treated cells, indicating that HU and EX527 synergistically activate the caspase pathway (Supplementary Fig. S1B). Consistently, treatment of EX527 significantly augmented HU-induced apoptosis in mESCs, indicating that acetylation of p53 is directly related to HU-mediated apoptosis (Fig. 3C). In line with the chemical inhibition effect of sirt1, siRNA-mediated suppression of sirt1 expression resulted in a dramatic increase in apoptosis on HU treatment, even at 50 μM (Fig. 3D). In addition, acetylation of p53 and activation of caspase-3 on HU treatment were mitigated when Sirt1 was ectopically overexpressed (Fig. 3E). Taken together, we conclude that HU-mediated apoptosis in mESCs is p53 dependent and that acetylation of p53 may be the main cause of the rapid induction of apoptosis after HU exposure in mESCs.
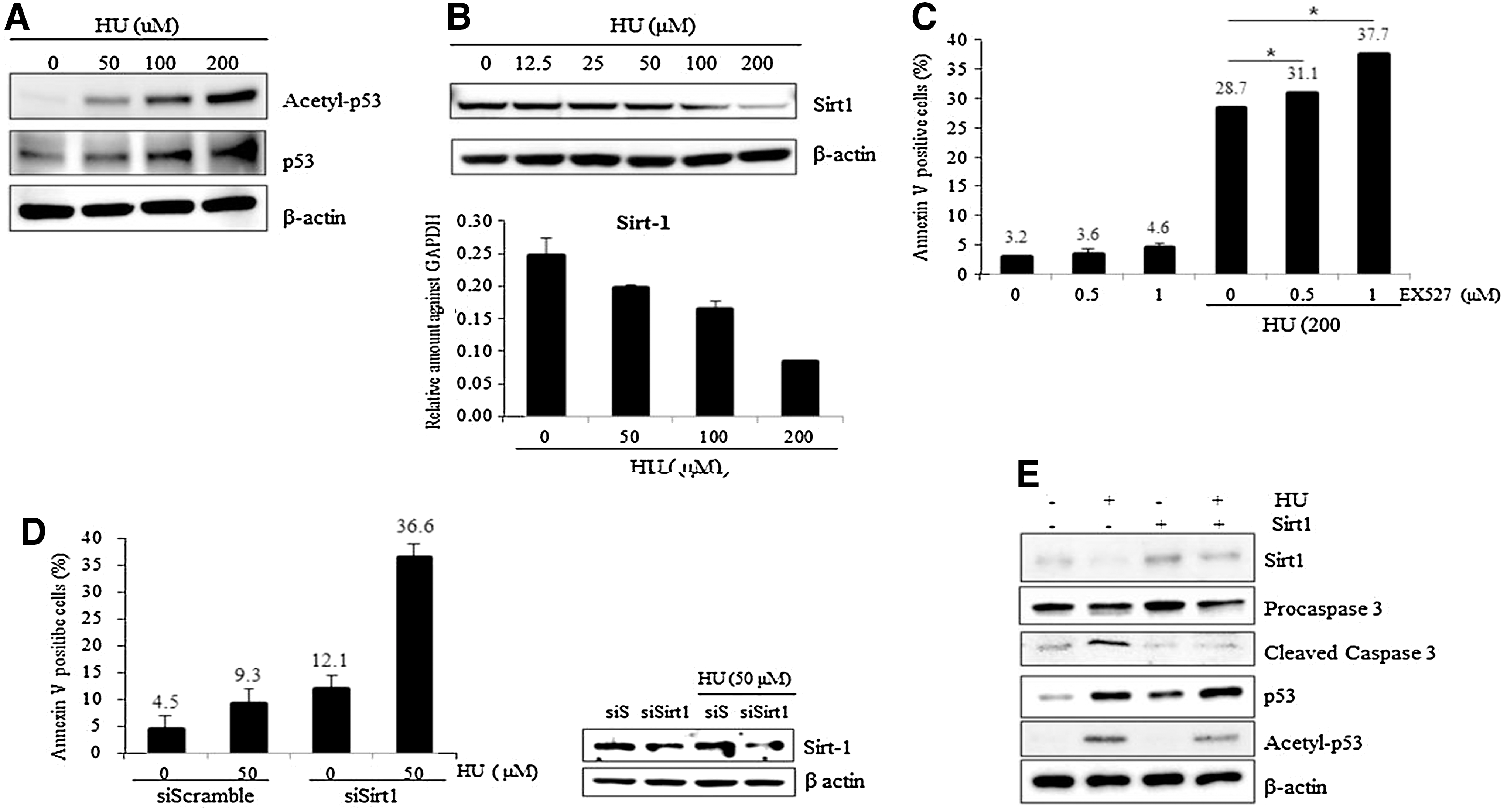
HU induces acetylated-p53 dependent apoptosis of mESCs.
NO produced by HU triggers apoptosis of mESCs
HU-mediated replication stress induces DNA DSBs, resulting in the phosphorylation of the histone H2A variant H2AX to form γH2AX foci [26]. To evaluate whether rapid induction of apoptosis in mESCs on low concentration treatment of HU is caused by damage to DNA, we examined γH2AX foci formation after exposure to HU. As expected, γH2AX levels increased after treatment with 50 μM of HU even though the expression of Rad51, an essential protein in the initiation of homologous recombination to repair DSB, was unchanged even at 200 μM HU (Fig. 4A, left panel). Contrary to the results seen in mESCs, γH2AX was not detected in MEFs exposed to HU at as high a concentration as 1 mM (data not shown). These results suggest that accumulation of DSBs induced by low concentrations of HU is a unique phenomenon to mESCs. The increase in γH2AX levels was not detected at 6 h after HU treatment, but was clearly detected at 24 h after HU exposure (Fig. 4A, middle panel). The signal intensity of γH2AX was quantified by a densitometer (Fig. 4A, right panel).
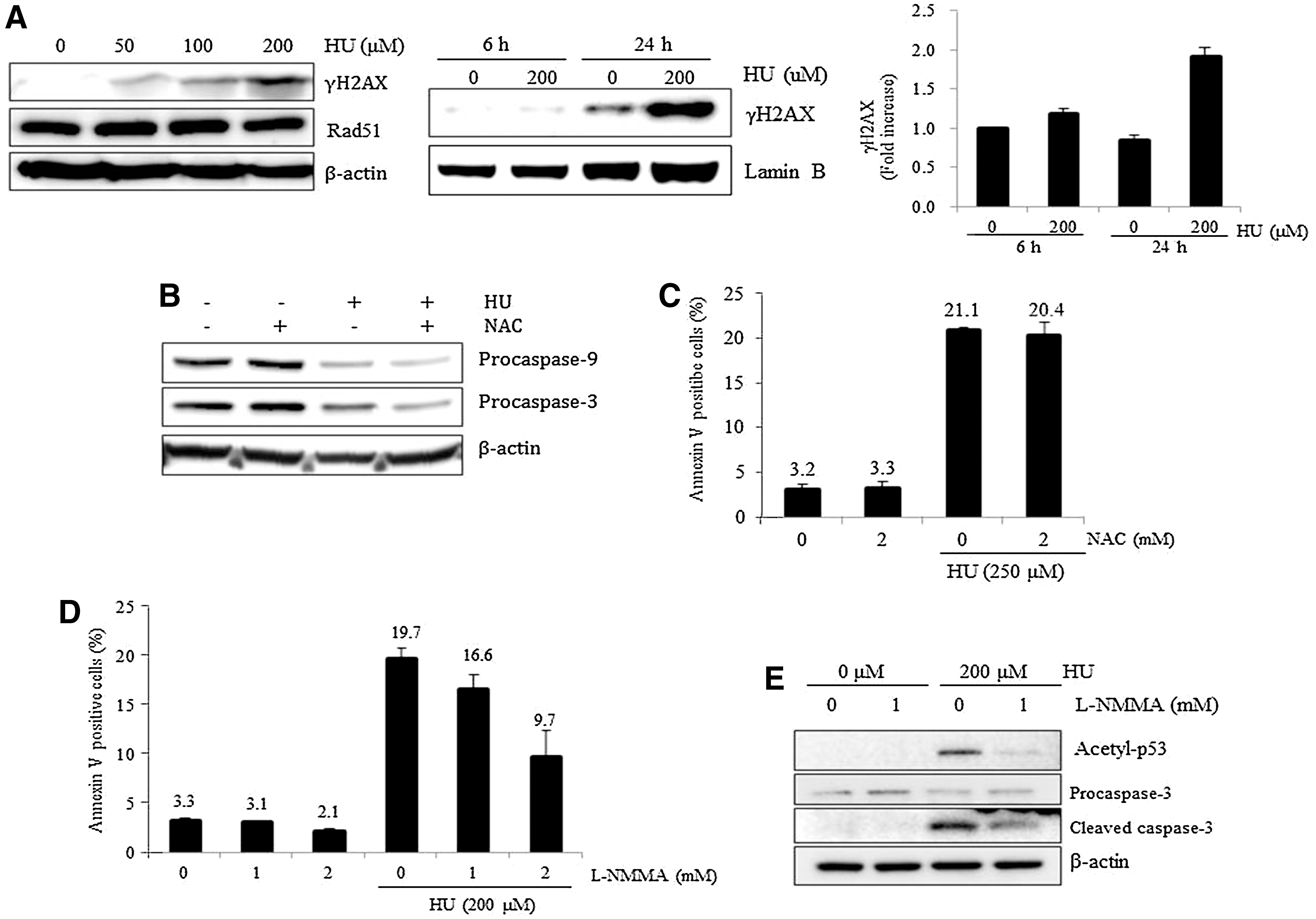
DNA damage, reactive oxygen species (ROS), and nitric oxide (NO) in HU-induced apoptosis.
We next investigated the mechanisms underlying DSBs in genomic DNA after low concentrations of HU in mESCs. Replication stress can trigger apoptosis accompanied by the production of reactive oxygen species (ROS) in mammalian cells and budding yeast [27,28]. In addition, ESCs have high levels of ROS arising from high levels of metabolic activity [29]. Therefore, we assessed whether high levels of endogenous ROS contribute to the sensitive response of mESCs to HU by examining the effect of ROS depletion on HU-mediated apoptosis of mESCs. We first confirmed that 2 mM of NAC treatment, a scavenger of ROS, decreases endogenous ROS levels of mESCs to 20% that of the control within 1 h (data not shown). However, pretreatment with NAC did not prevent the activation of the caspase pathway after HU exposure (Fig. 4B). Consistently, pretreatment with NAC did not compromise the apoptotic effects in mESCs treated with HU (Fig. 4C). These results suggest that the rapid and sensitive apoptotic response to HU is not related to high levels of endogenous ROS in mESCs.
Since NO generated after HU treatment activates the caspase pathway in endothelial cells and triggers apoptotic cell death in mESCs [23,30], we next examined whether NO is the main cause of the rapid induction of apoptosis in mESCs after HU exposure. Notably, when cells were pre-treated with L-NMMA, an NO synthase inhibitor, for 1 h, HU-mediated apoptosis of mESCs was significantly inhibited (Fig. 4D). In addition, pretreatment with L-NMMA led to a decrease in acetylation of p53 as well as to decreased activation of the caspase pathway in response to HU treatment (Fig. 4E). Based on these data, we concluded that NO production is the main cause of the sensitive apoptotic response of mESCs to low concentrations of HU.
p38 pathway plays an important role in HU-induced apoptosis of mESCs
We next studied the underlying signaling pathways that trigger rapid apoptosis induced by HU in mESCs. p38 MAP kinase is involved in NO-mediated cell death of neuronal cells, and NO donors stimulate the phosphorylation of p38 MAP kinase in cultured rat astrocytes [31,32]. Consistent with these reports, phosphorylation of p38 gradually increased in an HU dose-dependent manner (Fig. 5A). To confirm that activation of the p38 pathway is directly associated with HU-induced apoptosis, cells were pre-treated with chemical inhibitors of p38 signaling (SB202190) for 1 h before HU exposure. Notably, inhibition of p38 phosphorylation by SB202190 significantly suppressed HU-induced apoptosis (Fig. 5B). Consistently, pretreatment of mESCs with SB202190 decreased acetylation of p53 and attenuated cleavage of procaspase-9 on HU exposure (Fig. 5C). These data indicate that the p38 pathway is involved in the rapid induction of apoptosis of mESCs on exposure to low concentrations of HU.

p38 mediates HU-induced apoptosis.
Discussion
Appropriate DNA replication is essential to prevent the accumulation of mutations in the genome. HU is a DNA replication inhibitor that causes cell-cycle arrest in the S phase followed by deoxyribonucleotide depletion [33]. HU has been used at a concentration of more than 1 mM to induce cell-cycle arrest in several mammalian cell types [13,34,35]. Compared with somatic cells, ESCs have robust mechanisms to maintain genome stability against replication stress and prevent the transmission of mutations from precursors to adult cell lineages. Consistent with this notion, Figure 1A clearly shows a significant difference between mESCs and MEFs in response to HU. MEFs initiated growth arrest at 100 μM HU, whereas mESCs initiated apoptosis at 50 μM HU. Recently, a study showed that human ESCs under replication stress fail to activate the CHK1 pathway, which leads to S-phase arrest and replication restart through a homologous recombination repair pathway, and instead committed to apoptosis [14]. Our study revealed that p53 becomes rapidly acetylated in response to HU treatment, and acetylated p53 initiates caspase-dependent apoptosis in mESCs.
Although p53 protein is abundant in ESCs, there are contradictory reports about its role in response to DNA damage. One study reported that mESCs do not activate the p53-dependent checkpoint pathway to repair DNA damage and undergo p53-independent apoptosis in response to DNA damage or nucleotide depletion [6]. By contrast, other studies have reported that p53 accumulates in the nucleus of mESCs to activate its target genes p21, mdm2, puma, and noxa in response to UV light or after treatment with doxorubicin [36,37]. The present study suggests that mESCs respond to HU via the activation of a p53-dependent caspase pathway. The transcriptional suppression of sirt1 in mESCs on HU exposure suggests that decreased sirt1 expression is directly related to the increased acetylation of p53. However, we cannot exclude the possibility that other mechanisms, such as activation of acetyltransferase p300/CREB-binding protein by HU, lead to the observed increase in acetyl-p53 levels.
Interestingly, genomic DNA damage represented by γH2AX foci occurred in mESCs after treatment with 200 μM HU, at which concentration MEFs were not affected. This suggests that the apoptotic response of mESCs to low concentrations of HU may be due to the vulnerability of genomic DNA against external toxic reagents such as HU. Although the level of ROS in mESCs is relatively high compared with somatic cells, our data suggest that ROS concentration is not related to the response of mESCs to HU treatment. Instead, our study revealed that NO expression, which results from HU exposure, initiates the apoptotic response in mESCs. Consistent with our data, one of the therapeutic mechanisms of HU in the treatment of sickle cell anemia is the production of NO [30]. Moreover, NO is reported to induce activation of caspase-3, -8, and -9 through the p38 pathway in mESCs [23], which is in line with our results.
Currently, the underlying mechanism for the activation of p38 signaling in response to HU is the main focus of our research. It will be interesting to study whether the p38 signaling pathway functions as a sensor for damaged DNA, or whether it plays an active role in inducing the sensitive response by mESCs after HU exposure.
Footnotes
Acknowledgments
The authors thank Hee-Jin Ahn, Dae-Kwan Kim, and Seong Kyu Yang for their technical assistance. This work was supported by the Korea Science and Engineering Foundation (KOSEF) of the Korean government (MOST) (NRF-2012R1A1A3003070 and 2012M3A9C6050367). This work was also supported by the Priority Research Centers Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (20120006679). This work was also carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (PJ010033)” Rural Development Administration, Republic of Korea.
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.