
Abstract
In this study we have generated canine mesenchymal stromal cells (MSCs), also known as mesenchymal stem cells, from canine induced pluripotent stem cells (ciPSCs) by small-molecule inhibition of the transforming growth factor beta (TGFβ)/activin signaling pathway. These ciPSC-derived MSCs (ciPSC-MSCs) express the MSC markers CD73, CD90, CD105, STRO1, cPDGFRβ and cKDR, in addition to the pluripotency factors OCT4, NANOG and REX1. ciPSC-MSCs lack immunostaining for H3K27me3, suggesting that they possess two active X chromosomes. ciPSC-MSCs are highly proliferative and undergo robust differentiation along the osteo-, chondro- and adipogenic pathways, but do not form teratoma-like tissues in vitro. Of further significance for the translational potential of ciPSC-MSCs, we show that these cells can be encapsulated and maintained within injectable hydrogel matrices that, when functionalized with bound pentosan polysulfate, dramatically enhance chondrogenesis and inhibit osteogenesis. The ability to efficiently derive large numbers of highly proliferative canine MSCs from ciPSCs that can be incorporated into injectable, functionalized hydrogels that enhance their differentiation along a desired lineage constitutes an important milestone towards developing an effective MSC-based therapy for osteoarthritis in dogs, but equally provides a model system for assessing the efficacy and safety of analogous approaches for treating human degenerative joint diseases.
Introduction
M
While it has been demonstrated that human placental- and fetal-derived MSCs have superior proliferative and differentiative abilities as compared with MSCs sourced from adult tissues [9 –11], their collection has associated practical and, in the case of fetal MSCs, ethical concerns. Further, it has been demonstrated that only around 30% of umbilical cord blood samples yield MSCs [10,12]. Thus, the focus has been primarily on the harvesting of MSCs from adult bone marrow and adipose tissue. However, these adult sources also have significant drawbacks. Firstly, MSCs represent a very small fraction of the cells isolated from both bone marrow and adipose tissue—0.001%–0.01% and 0.05%, respectively [10,12]; secondly, the quality and quantity of MSCs that can be collected decline with increasing age of the donor [13,14], which is of particular concern given that the majority of patients requiring stem cell-based therapies are aged; and finally, the harvesting of MSCs from bone marrow and adipose tissue is invasive and so has associated risks.
To circumvent both the ethical concerns associated with fetal-derived MSCs, and the decreased proliferative and differentiative abilities of adult MSCs, Chen et al. [15] recently described a methodology whereby human induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) could be induced to differentiate into MSCs via inhibition of the transforming growth factor beta (TGFβ)/activin signaling pathway and maintenance in culture conditions that support an epithelial-to-mesenchymal transition. By exposing the cells to the TGFβ/activin type I receptor inhibitor SB431542 for just 10 days, and then transitioning the cells into typical MSC culture conditions, Chen et al. [15] generated iPSC- and ESC-derived MSCs that resembled primary MSCs in terms of their immunophenotype and in their ability to differentiate into mesodermal derivatives. Significantly, from a translational perspective, this system is rapid and highly efficient and yields large numbers of cells with a superior proliferative ability [15].
MSCs have been the focus of the vast majority of stem cell-based therapies targeting human musculoskeletal defects and diseases. A similar surge in interest in the potential of MSC-based therapies has occurred in the veterinary field, in particular for the treatment of musculoskeletal conditions in dogs. Apart from the more immediate benefit to canine patients, dogs are a valuable experimental model for understanding the etiology and underlying pathology of many of the significant musculoskeletal conditions in humans, including osteoarthritis and intervertebral disc disease, both of which are highly prevalent in dogs and humans [1,16 –18]. Thus, research into MSC-based regenerative therapies in dogs has the potential to inform the development of similar treatments in human patients. Several studies have described the isolation and characterization of MSCs from a range of adult canine tissues, including adipose tissue [19 –23], bone marrow [21 –23], muscle [21], periosteum [21] and synovium [24]. Adult canine MSCs from adipose tissue are also available commercially worldwide. However, the same limitations that apply to adult human MSCs (hMSCs) also apply to adult canine MSCs: namely, low yields and a marked decline in the ability of the cells to proliferate and differentiate with increasing passage of cells in vitro and age of the donor [21,24,25].
In order to generate canine MSCs that are unencumbered by the diminished proliferative and differentiative abilities synonymous with harvested MSCs, we have applied the SB431542 induction method, initially described for human pluripotent stem cells [15], to canine iPSCs (ciPSCs) [26]. Using this methodology we have produced ciPSC-derived MSCs (ciPSC-MSCs) that have an immunophenotype similar to tissue-derived canine MSCs, a high proliferative rate that does not diminish with progressive passage, and an ability to give rise to the three mesoderm derivatives of cartilage, bone and adipose tissue, but which do not form teratoma-like tissues in vitro. Finally, as a prelude to trialing these cells in a translational context, we examined the behavior of the ciPSC-MSCs when incorporated into injectable hydrogel scaffolds designed to optimize chondrogenesis within the milieu of a joint [27].
It is anticipated that the combination of an efficient and scalable means of producing high-quality canine MSCs and a means by which to deliver them, and direct their differentiation, at a specific site within the body will enable us to progress towards more effective MSC-based therapies for musculoskeletal disease in dogs and, ultimately, humans.
Materials and Methods
All use of animals, and tissues obtained from animals, was approved by the University of Queensland Animal Ethics Committee.
Generation and maintenance of ciPSC-MSCs
Two fully characterized clones of ciPSCs, generated from female adult dermal fibroblasts by lentiviral delivery of human reprogramming factors, were cultured as previously described [26]. Both ciPSC clones, at passage 53, were induced to undergo an epithelial-to-mesenchymal transition using the protocol described by Chen et al. [15]. Colonies of ciPSCs were enzymatically passaged with TrypLE (Life Technologies) to yield large clumps of cells that were then plated onto T25 tissue culture flasks (Costar) coated with Matrigel (BD Biosciences). Cells were cultured for 10 days with 10 μM of the TGFβ/activin type I receptor inhibitor SB431542 (Stemgent) in the medium previously used to support the ciPSCs [KnockOut Dulbecco's modified Eagle's medium (DMEM; Life Technologies), 20% (v/v) KnockOut Serum Replacement (KSR; Life Technologies), 0.1 mM nonessential amino acids (NEAAs; Life Technologies) and 2 mM
Commercially available adult canine adipose-derived MSCs were kindly supplied by Regeneus Pty Ltd. Vials were thawed and plated onto tissue culture flasks and maintained in MSC medium as described for the ciPSC-MSCs. Adult MSCs were extracted from the bone marrow of an adult female dog according to the methodology outlined in Soleimani and Nadri [28], and maintained in tissue culture flasks with MSC medium as described above.
Karyotyping
Metaphase spreads were prepared for the adipose-derived MSCs, bone marrow-derived MSCs, and each of the ciPSC-MSC lines according to standard protocols, and a minimum of 15 metaphase spreads were examined for normal chromosome number and morphology. Karyograms were commercially prepared by Sullivan Nicolaides Pathology (Taringa, QLD, Australia).
Growth kinetics
After the third passage in MSC medium, ciPSC-MSCs derived from both iPSC clones, adult adipose-derived MSCs, and adult bone marrow-derived MSCs were plated in triplicate at a density of 1×104 cells per cm2 in T75 tissue culture flasks. When cells reached 80% confluency, they were dissociated with TrypLE, stained with trypan blue, and counted in an automated cell counter (Bio-Rad Laboratories). Successive subcultures were plated at the same density, and passaged and counted as above for a period of 30 days. The cumulative cell number and cumulative population doublings were calculated as described in Barlow et al. [29].
RNA isolation, cDNA synthesis and polymerase chain reaction
Total RNA was isolated from ciPSCs, ciPSC-MSCs and adult MSCs using the Qiagen RNeasy Mini Kit (Qiagen), as per the manufacturer's protocol. Complementary DNA was synthesized using the Bio-Rad iScript Reverse Transcriptase Kit (Bio-Rad Laboratories) according to the manufacturer's instructions. The polymerase chain reaction (PCR) primers used, and their product sizes, are listed in Supplementary Table S1 (Supplementary Data are available online at
Fluorescence immunocytochemistry
Fluorescence immunocytochemistry was performed for markers of pluripotency (OCT4, NANOG and REX1) and for markers that are typically associated with MSCs (CD73, CD90, CD105 and STRO1). Samples were fixed with 4% paraformaldehyde at room temperature (RT) for 10 min. For the detection of nuclear proteins, cells were permeabilized with 0.1% Triton in phosphate-buffered saline (PBS) for 20 min before blocking with 5% goat serum (Life Technologies) in PBS (Life Technologies). Primary antibodies were diluted in 3% goat serum in PBS. Primary antibodies, and their dilutions, are as follows: anti-OCT4 (1:50, MAB4401; Millipore), anti-NANOG (1:150, ab80892; Abcam), anti-REX1 (1:50, ab50828-50; Abcam), anti-CD73 (1:50, sc-25603; Santa Cruz Biotechnology), anti-CD90 (1:50, SM420P; Acris Antibodies), anti-CD105 (1:50, ab156756; Abcam) and anti-STRO1 (1:50, MAB1038; R&D Systems). Negative controls were incubated with 3% goat serum in PBS in place of primary antibody. All secondary antibodies were diluted to 1:1,000 in PBS: Alexa Fluor 488 goat anti-mouse IgGH&L (Invitrogen), Alexa Fluor 488 goat anti-mouse IgMμ (Invitrogen), Alexa Fluor 568 goat anti-rabbit IgGH&L (Invitrogen), and Alexa Fluor 488 goat anti-rat IgGH&L (Invitrogen). Nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI).
X chromosome inactivation was examined by fluorescence immunocytochemistry using an antibody to the trimethylated histone H3K27 (anti-H3K27me3, ab6002; Abcam), which is an XIST-dependent chromatin mark that directly identifies the inactive X chromosome [30]. Samples were fixed, permeabilized and blocked as described above before incubation with the primary antibody at a dilution of 1:100. The secondary antibody (Alexa Fluor 488 goat anti-mouse IgGH&L) was applied as described above.
Flow cytometry
Cells were dissociated into a single-cell suspension with Cell Dissociation Buffer (Life Technologies). A total of 2×106 cells were used for each sample. Cells were fixed in 2% paraformaldehyde at RT for 20 min before blocking in 5% goat serum in PBS–1% bovine serum albumin (PBA) for 30 min at RT. Primary antibodies against CD73, CD90, CD105 and STRO1, as described above, were diluted 1:50 in 5% goat serum in PBA and applied to the cells for incubation at 4°C overnight. The following day, cells were washed in PBA before addition of the secondary antibody (as above), diluted 1:1,000 in PBA, for 1 h at RT. Negative controls were incubated with 5% goat serum in PBA in place of primary antibody. Immunostained cells were identified by flow cytometry using a BD CSampler Accuri C6 fluorescent activated cell sorting analyzer.
In vitro osteogenic, chondrogenic and adipogenic differentiation assays
The ability of the ciPSC-MSCs to differentiate into mesodermal derivatives was assessed using the Stem-Pro Osteogenesis, Chondrogenesis and Adipogenesis Differentiation Kits (Life Technologies), according to the manufacturer's instructions. To assess for osteogenesis and chondrogenesis, cultures were stained with Alizarin red S and Alcian blue (pH 1.0), respectively, as per standard protocols. Adipogenesis was determined by staining with HCS LipidTOX Red (Life Technologies) according to the manufacturer's instructions. Nuclei were visualized with DAPI.
In vitro terminal differentiation assays
Due to the increasing pressure to utilize alternative methods to live animal experimentation, and because of the poor survival of our ciPSCs in NOD/SCID mice [26], we have devised and fully characterized a methodology for generating teratoma-like, terminally differentiated tissues in vitro (Fortuna et al., unpublished data). While originally this protocol was applied to human ESCs and iPSCs, we have also used it successfully with horse [31] and platypus iPSCs (Whitworth, unpublished data). ciPSCs, ciPSC-MSCs and adult MSCs were passaged with TrypLE. Approximately 4×105 cells were sandwiched between two layers of 10% (w/v) low-molecular-weight methylcellulose (Sigma-Aldrich) dissolved in KnockOut DMEM. Cells were maintained in KnockOut DMEM supplemented with 20% (v/v) KSR, 0.1 mM NEAAs, 2 mM
Encapsulation and culture of ciPSC-MSCs in hydrogels
Hydrogels were prepared as described in Frith et al. [27]. Hydrogels of three different compositions were used: (a) poly(ethylene glycol) (PEG) (JenKem); (b) PEG and hyaluronic acid (HA) (Lifecore), and (c) PEG, HA and HA–pentosan polysulfate (PPS) (HA conjugated to PPS as prepared in Frith et al. [32]). ciPSC-MSCs were encapsulated in the hydrogels at a density of 1×107 cells/mL according to the methodology described by Frith et al. [27]. Fifty microliters of cell/hydrogel composite was spotted onto glass coverslips placed into 48-well low-binding tissue culture plates (Costar). Triplicates of each of the three types of hydrogel were cultured in each of three types of medium: basal medium [DMEM supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin and 10% FBS (all Life Technologies)]; osteogenic medium [DMEM supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 10% FBS, 100 ng/mL dexamethasone (Sigma-Aldrich) 50 μM ascorbate-2-phosphate (Sigma-Aldrich), and 10 mM β-glycerophosphate (Sigma-Aldrich)], and chondrogenic medium [DMEM high-glucose supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 10 mg/mL insulin (Life Technologies), 5.5 mg/mL transferrin (Life Technologies), 5 ng/mL sodium selenite (Life Technologies), 3 mM linoleic acid (Life Technologies), 3 mM oleic acid (Life Technologies), 1 mg/mL bovine serum albumin (Life Technologies), 10 μg/mL sodium pyruvate (Life Technologies), 4 mg/mL
Results
The TGFβ/activin type I receptor inhibitor (SB431542) induces ciPSCs to differentiate into cells with an MSC-like morphology
ciPSC colonies are slightly domed and consist of uniformly round cells with a typical pluripotent stem cell morphology of a large nuclear-to-cytoplasmic ratio (Fig. 1A) [26]. During 10 days of culture with SB431542, ciPSCs became organized into a monolayer and assumed a cuboidal to stellate appearance, with cells becoming more stellate rather than cuboidal at the periphery of the colony (Fig. 1B). After passage into MSC medium, and plating onto uncoated plastic, stellate-shaped cells were uniformly distributed as single cells (Fig. 1C, D), rather than an epithelial-like sheet, and closely resembled both bone marrow- and adipose-derived primary adult MSCs (Fig. 1E, F).
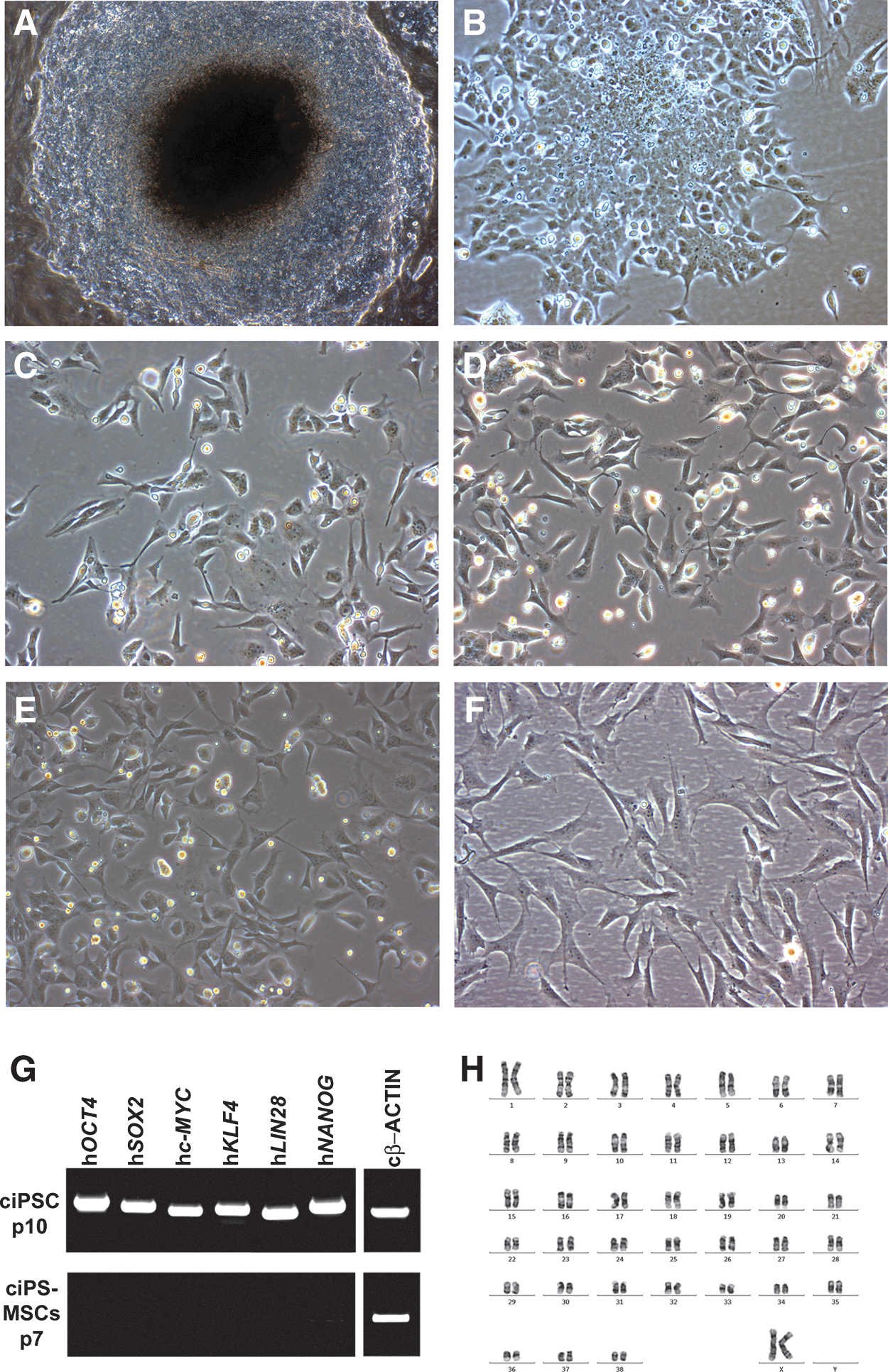
Transforming growth factor beta (TGFβ)/activin type I receptor inhibitor (SB431542) induces canine induced pluripotent stem cells (ciPSCs) to differentiate into cells with a mesenchymal stromal cell (MSC)–like morphology.
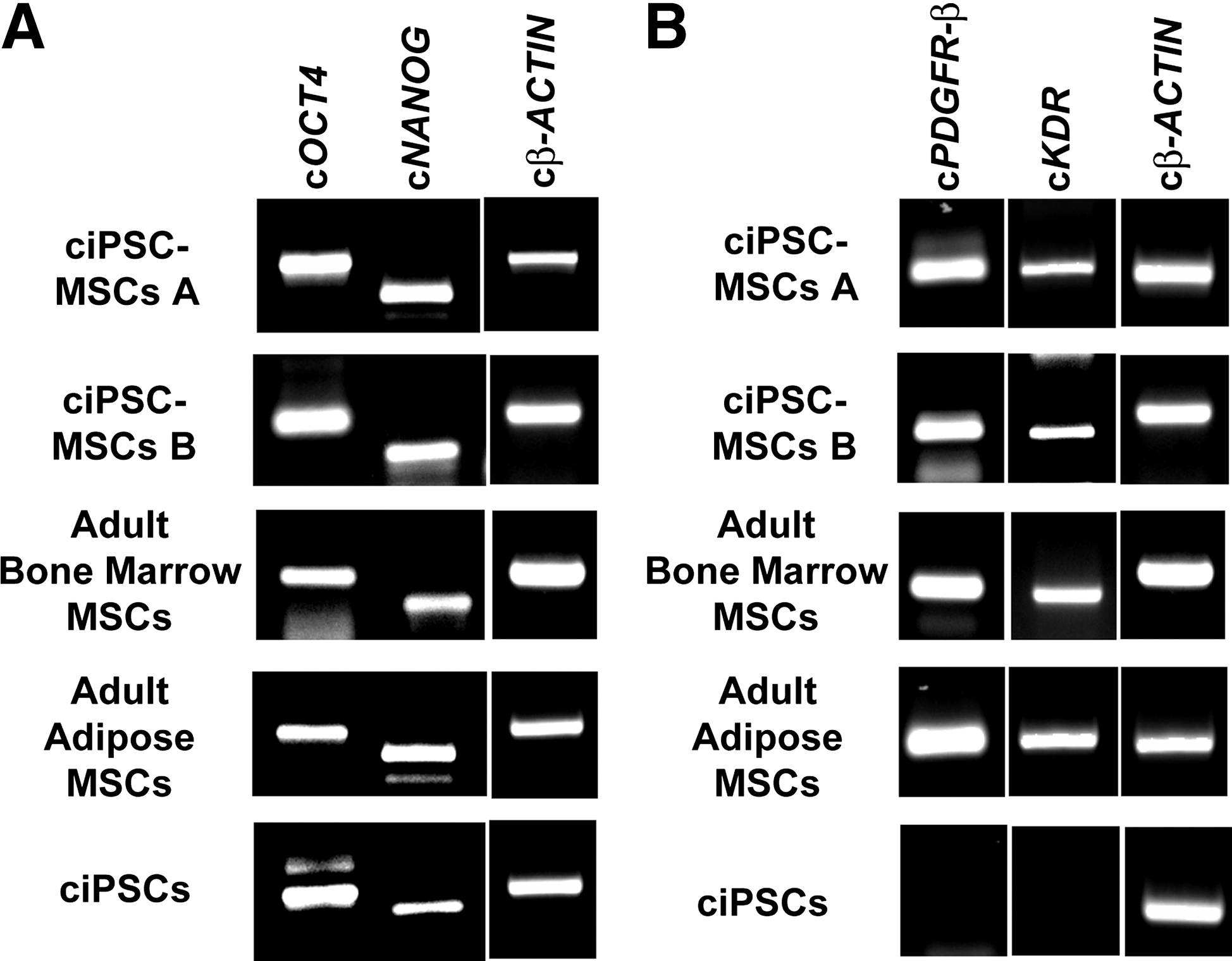
Because the ciPSCs were generated using lentivirally delivered transgenes, it was important to determine whether the transgenes had been reactivated during the differentiation into MSC-like cells. We have previously shown that all six transgenes, while expressed in ciPSCs at passage 10 (Fig. 1G) [26], were transcriptionally silenced at passage 26 [26]. Similarly, ciPSC-MSCs, which were generated from ciPSCs at passage 57, maintained a lack of transgene expression (Fig. 1G).
Metaphase spreads of ciPSC-MSCs derived from both iPSC clones, and adult adipose- and bone marrow-derived MSCs demonstrated a normal chromosome number of 78 in all of the metaphase spreads examined (Fig. 1H, data not shown). All cell lines were confirmed as being female with two X chromosomes. It should be noted that the commercial preparation of adipose-derived MSCs (Regeneus Pty Ltd.) is a pooled sample containing MSCs harvested from several individuals, all of them female.
ciPSC-MSCs express both MSC and pluripotency markers
ciPSC-MSCs from both iPSC clones express the typical MSC surface markers CD73, CD90 and CD105 (Fig. 2A, B, D, E, G, H), as do the adult bone marrow-derived MSCs (Fig. 2C, F, I), adipose-derived MSCs (Supplementary Fig. S1A–C) and the ciPSCs (Supplementary Fig. S2A–C). In contrast, expression of the MSC marker STRO1 has been acquired by the ciPSC-MSCs (Fig. 2J, K) since it is not expressed by the ciPSCs (Supplementary Fig. S2D). Negative controls for non-specific immunostaining by the secondary antibodies are shown in Supplementary Fig. S3.
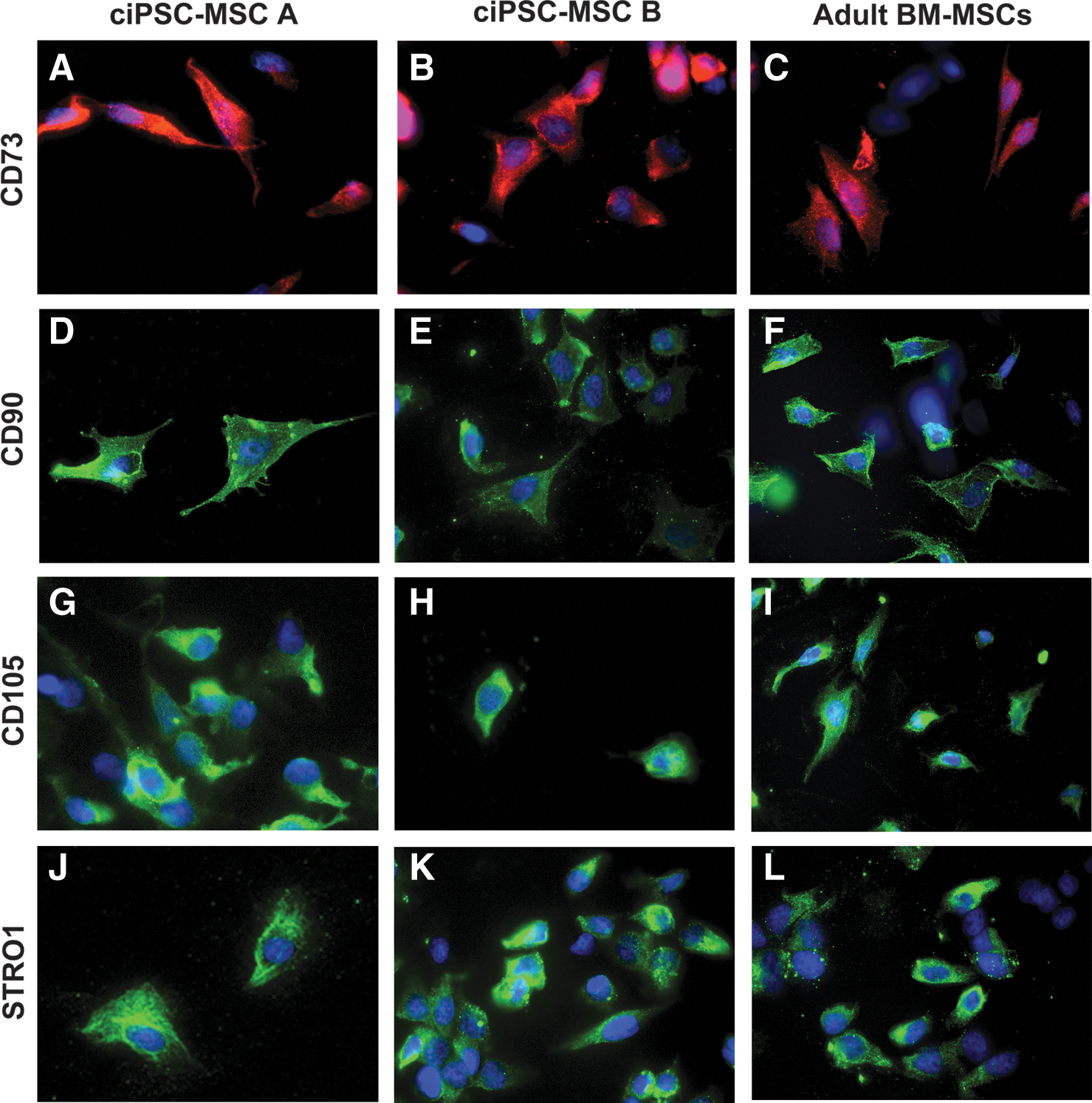
ciPSC-MSCs express MSC surface markers. ciPSC-MSCs from both iPSC clones (ciPSC-MSC A and B), and adult bone marrow-derived MSCs (BM MSCs), express MSC surface markers CD73
Flow cytometry confirmed that the majority of the ciPSC-MSCs from both iPSC clones, and the bone marrow-derived MSCs, are positive for the cell surface markers CD73, CD90, CD105 and STRO1 (Fig. 3A, B).

The majority of ciPSC-MSCs express MSC surface markers.
ciPSC-MSCs, and adult bone marrow- and adipose-derived MSCs, also express the pluripotency factors OCT4, NANOG and REX1 (Fig. 4 and Supplementary Fig. S1D–F). Expression of cOCT4 and cNANOG was confirmed at the transcriptional level for both the ciPSC-MSCs and adult MSCs, with the ciPSCs serving as a positive control (Fig. 5A).
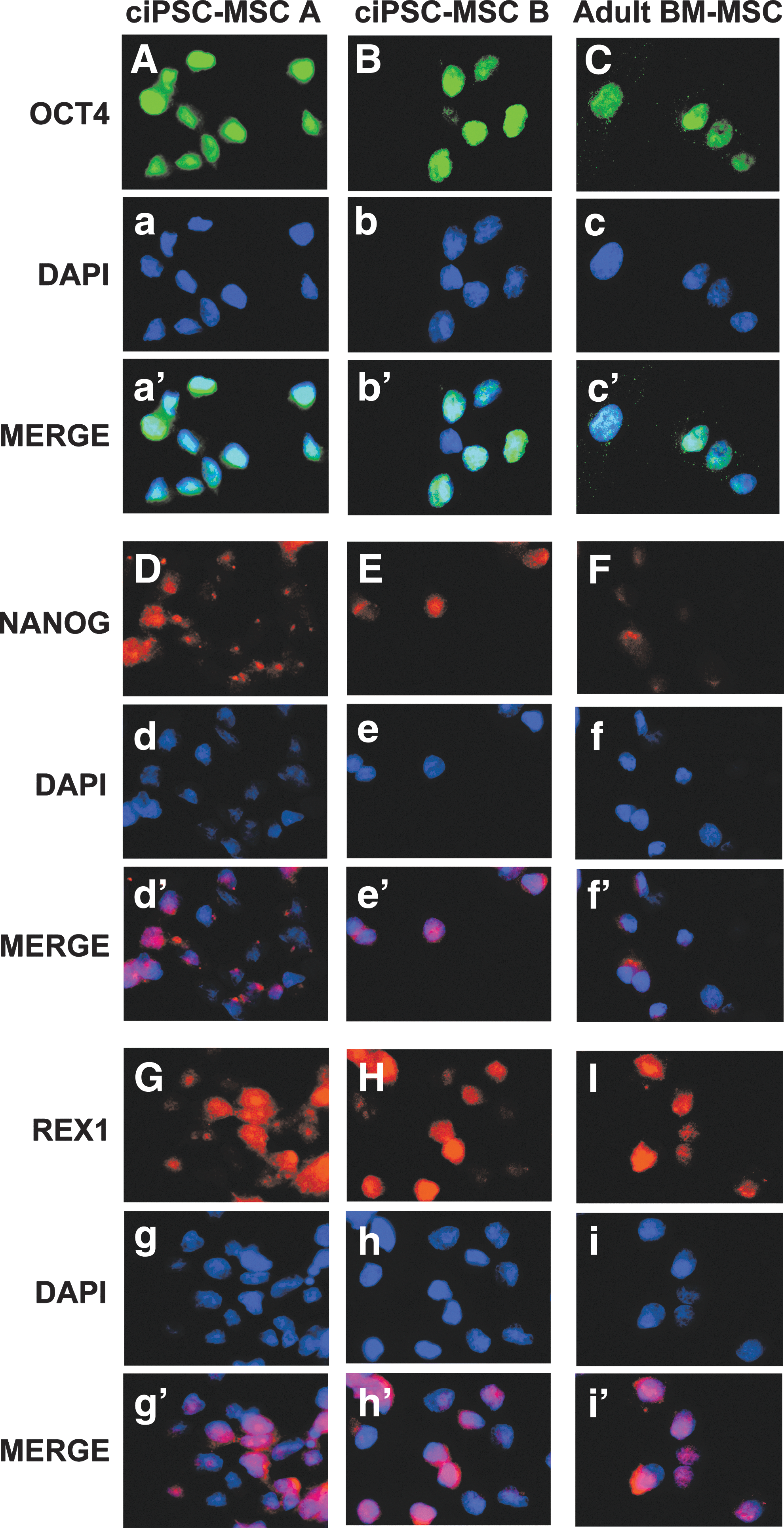
ciPSC-MSCs express pluripotency factors. ciPSC-MSCs and adult bone marrow-derived MSCs show robust expression of the pluripotency factors OCT4
ciPSC-MSCs express both pluripotency and MSC-specific factors.
Although the ciPSC-MSCs differ from the ciPSCs in their expression of STRO1, we sought to identify other MSC-linked markers that might be unique to the ciPSC-MSCs (and adult MSCs), thus supporting the argument that these cells are distinct from the ciPSCs from which they were derived. To this end, cPDGFRB and cKDR were identified as being expressed by both the ciPSC-MSCs and adult MSCs, but not by the ciPSCs (Fig. 5B).
ciPSC-derived and adult MSCs possess two active X chromosomes
As described previously [26], the ciPSCs were generated from adult female dermal fibroblasts that, as with all female somatic cells, have undergone X chromosome inactivation (Fig. 6A). However, during reprogramming into ciPSCs, the inactive X chromosome had undergone reactivation so that all ciPSCs possess two active X chromosomes [26]. We confirmed that at passage 65, beyond the passage from which the ciPSC-MSCs were generated, ciPSCs still possessed two active X chromosomes (Fig. 6B). Perhaps not surprisingly, given their expression of OCT4, NANOG and REX1, ciPSC-MSCs from both ciPSC clones similarly retain two active X chromosomes (Fig. 6C, D). Adult MSCs derived from adipose tissue and bone marrow similarly express OCT4, NANOG, and REX1 and also have two active X chromosomes (Fig. 6E, data not shown).
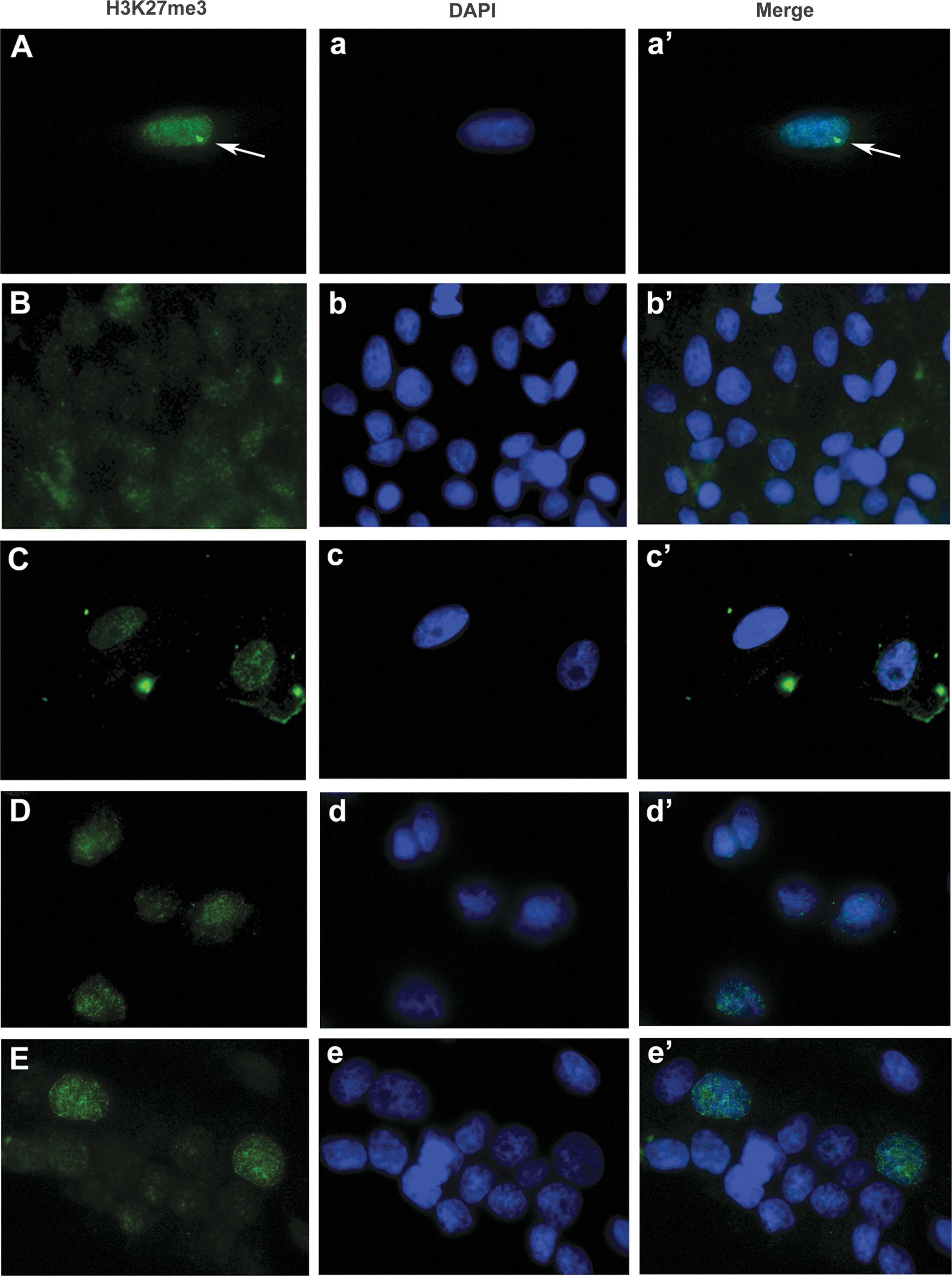
ciPSC-MSCs and adult MSCs possess two active X chromosomes.
ciPSC-MSCs display rapid growth kinetics
After 30 days, ciPSC-MSCs from iPSC clone A achieved a cumulative cell number of 1.74×1013 and reached a cumulative population doubling of 23 (Fig. 7A, B). Similarly, ciPSC-MSCs from iPSC clone B had a cumulative cell number of 2.37×1013 and also had a cumulative population doubling of 23 (Fig. 7A, B). The live cell count was consistently between 92% and 98% for ciPSC-MSCs from both clones. In contrast, the adult bone marrow-derived MSCs had a cumulative cell number of 2.07×109 and a cumulative population doubling of 10 (Fig. 7A, B), while the adipose-derived MSCs failed to proliferate and retained similar, or reduced, cell numbers (Fig. 7A, B) with a live cell count ranging from 80% to 97%, for the entirety of the 30-day culture period. The ciPSC-MSCs have been maintained to passage 15 without any evident loss of cell viability or proliferative ability (data not shown).
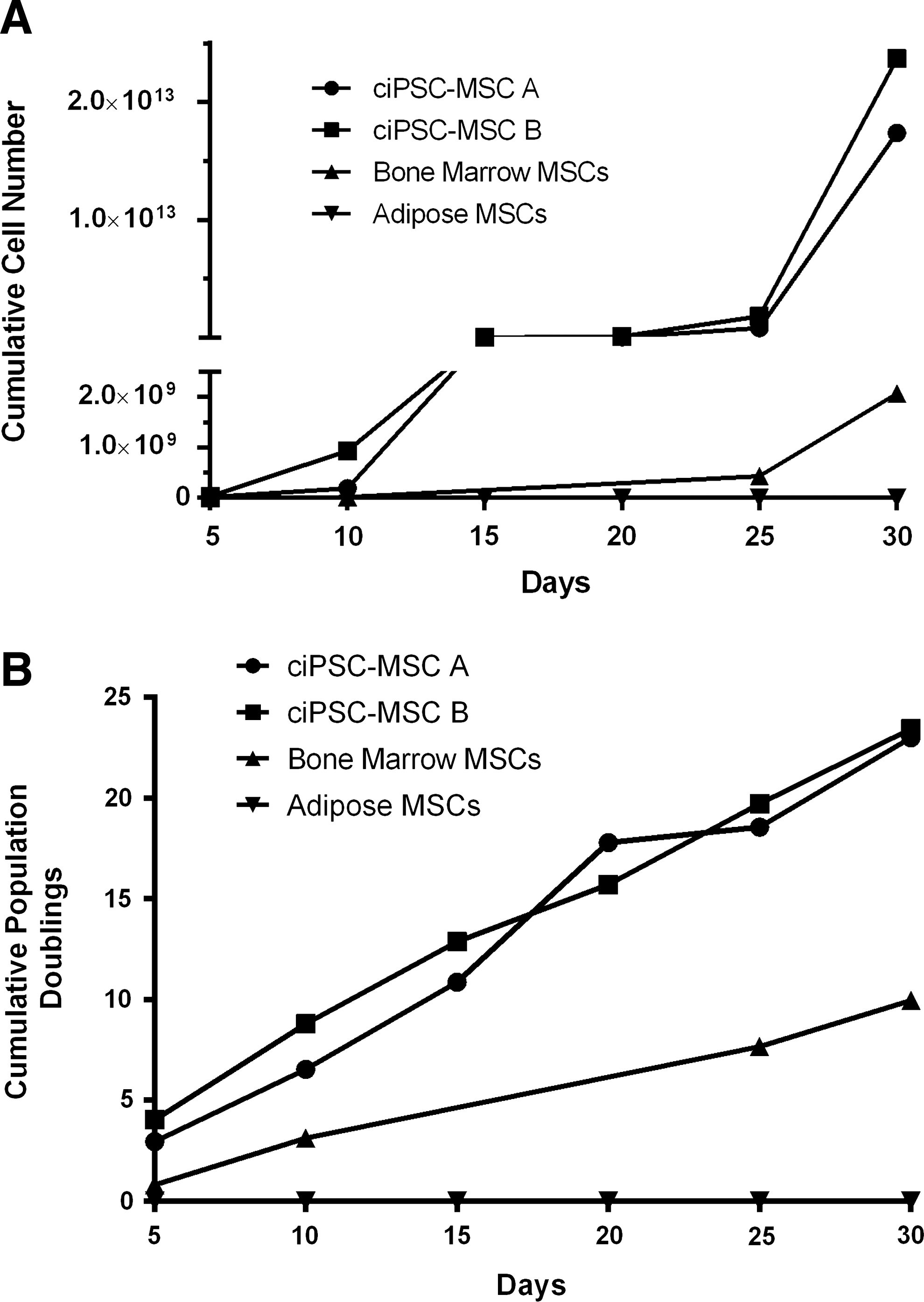
ciPSC-MSCs display robust growth kinetics.
ciPSC-MSCs undergo mesodermal differentiation but do not form teratoma-like tissues
ciPSC-MSCs undergo robust osteogenic, chondrogenic and adipogenic differentiation in vitro when maintained in the appropriate inductive medium (Fig. 8A–C).
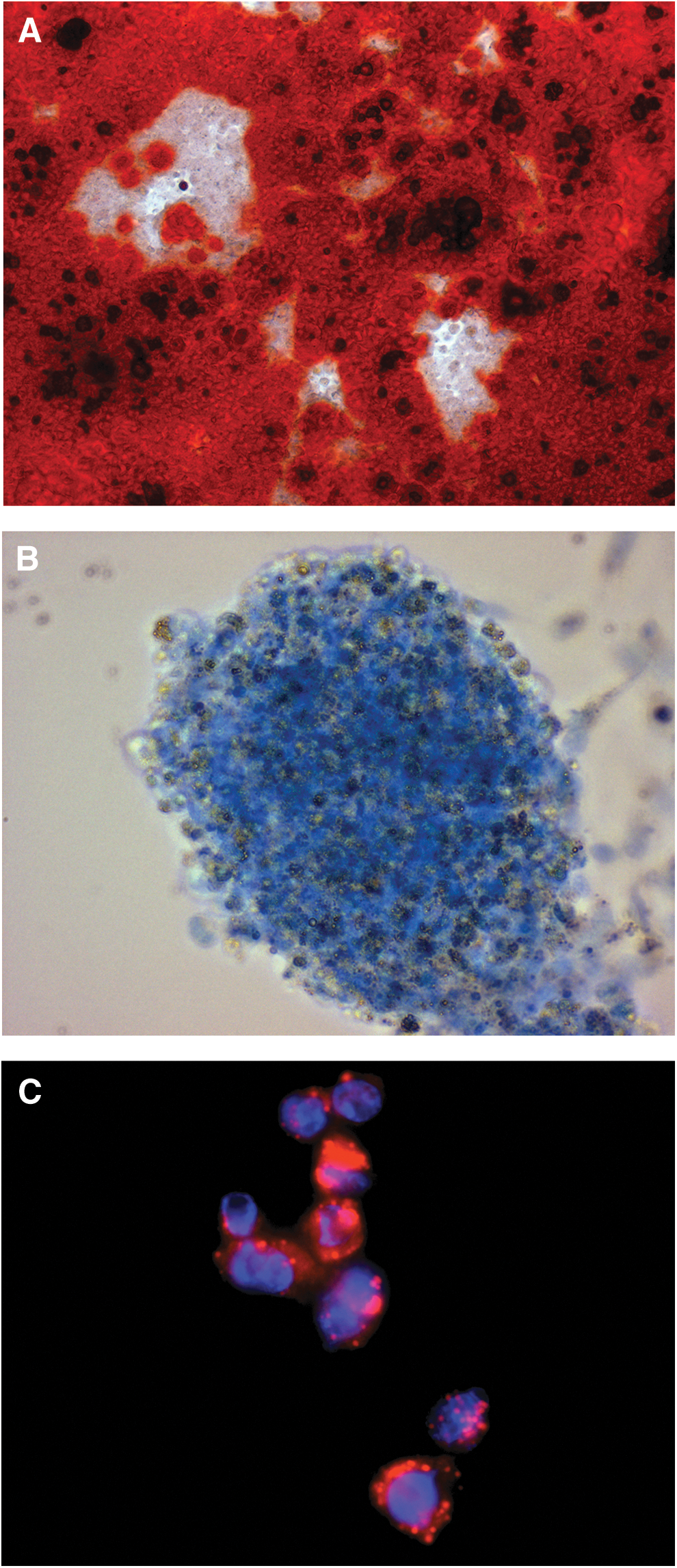
ciPSC-MSCs undergo osteogenesis, chondrogenesis and adipogenesis.
Because it is proposed to use the ciPSCs-MSCs in a clinical context, it was important to verify whether or not they are capable of forming teratomas—an undesirable trait in MSCs destined for in vivo transplantation. ciPSCs form teratoma-like tissues in vitro that contain derivatives of all three germ layers, including adipose tissue, erythrocytes, and bone from mesoderm (Fig. 9A–C); ciliated columnar epithelium from endoderm (Fig. 9D); and keratin from ectoderm-derived cells (Fig. 9E). In contrast, both the ciPSC-MSCs and adult adipose-derived MSCs formed only derivatives from the mesoderm (Fig. 9F, G). The similarity in the histology between the ciPSC-MSCs and adult adipose-derived MSCs is striking: both cell types formed isolated populations of adipose cells, and small, localized regions of bone-like tissue, among undifferentiated cells (Fig. 9F, G).
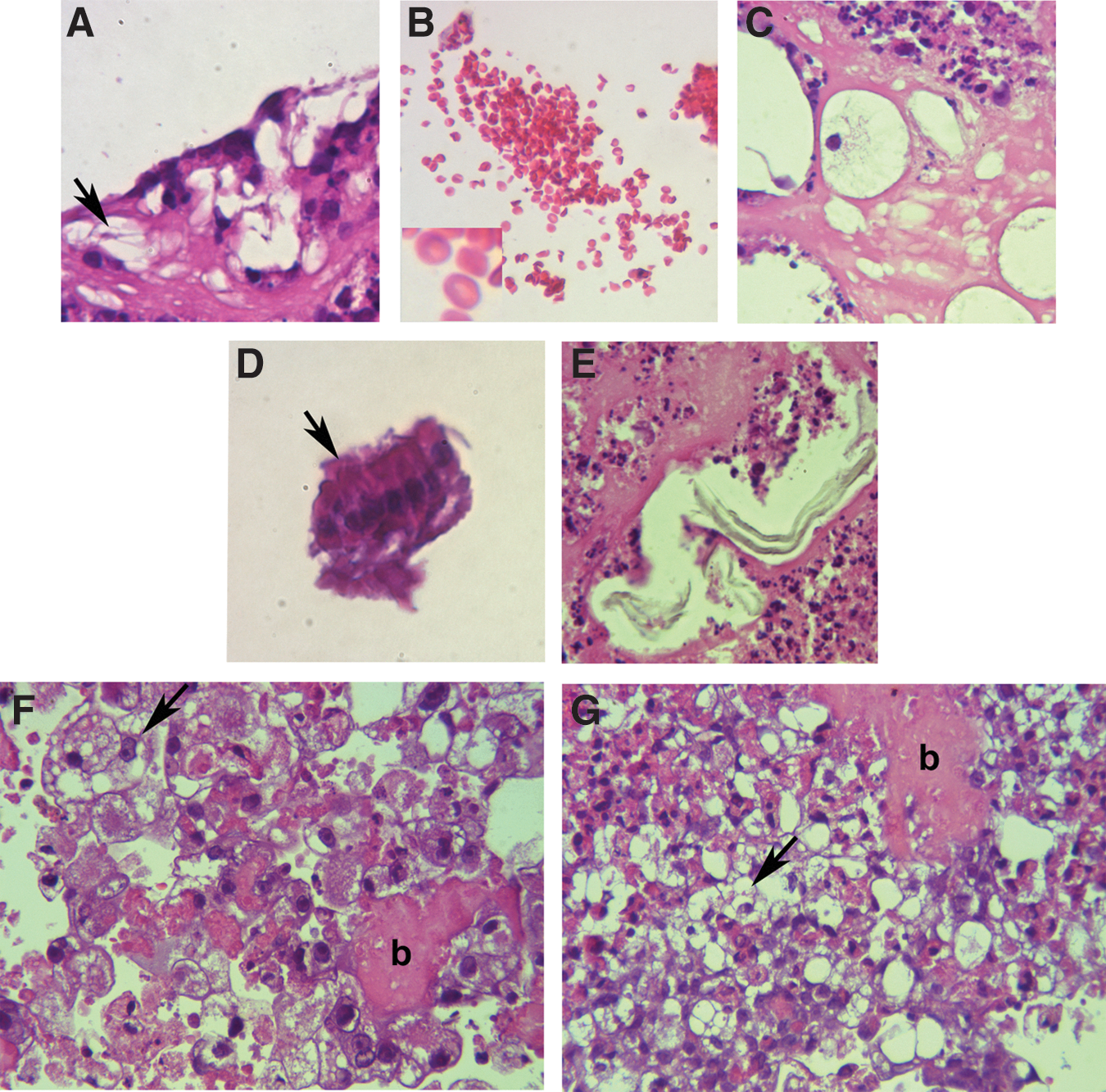
ciPSCs, but not ciPSC-MSCs, form teratoma-like tissues in vitro. ciPSCs form teratoma-like structures in vitro that contain tissue derivatives of the three germ layers, including
Hydrogels support chondrogenesis and osteogenesis in vitro
To demonstrate the potential utility of these cells with biomaterials destined for clinical applications, the morphology and differentiation of ciPSC-MSCs was investigated after encapsulation within injectable hydrogel materials. PEG hydrogels are of significant interest for stem cell encapsulation and delivery due to their bioinert and low fouling properties that reduce the likelihood of an immune response [33]. Indeed, recent work has demonstrated the potential of hydrogels formed from both PEG and HA for MSC encapsulation [27]. For this reason, both PEG and HA/PEG hydrogels were examined. As well as its widespread use in the veterinary field for applications in joint repair, there is further evidence for the utility of PPS for chondrogenic differentiation of MSCs [34] and so recently developed gels containing a form of PPS covalently bound to the HA backbone of the hydrogel matrix (HA-PPS) were also used [31]. ciPSC-MSCs encapsulated in each of these three hydrogel types (PEG, PEG/HA and PEG/HA+HA-PPS) were cultured in basal medium, as well as medium supplemented with osteo- or chondrogenic factors.
After encapsulation in the hydrogel matrix, ciPSC-MSC/hydrogel composites were spotted onto glass coverslips in tissue culture plates (Fig. 10A). Under these three-dimensional conditions, the cells adopted a rounded morphology that was maintained for the 21-day culture period (Fig. 10B). The structure of the cell/hydrogel composites was assessed by hematoxylin and eosin staining after 21 days and showed a relatively even distribution of cells throughout all of the hydrogel matrices (Fig. 10C), with the rounded cells residing within lacuna-like structures. Interestingly, the size of these lacunae was much larger in the gels containing PEG alone than in those containing both PEG and HA. There were no significant differences in cell morphology between any of the hydrogel compositions or medium formulations.
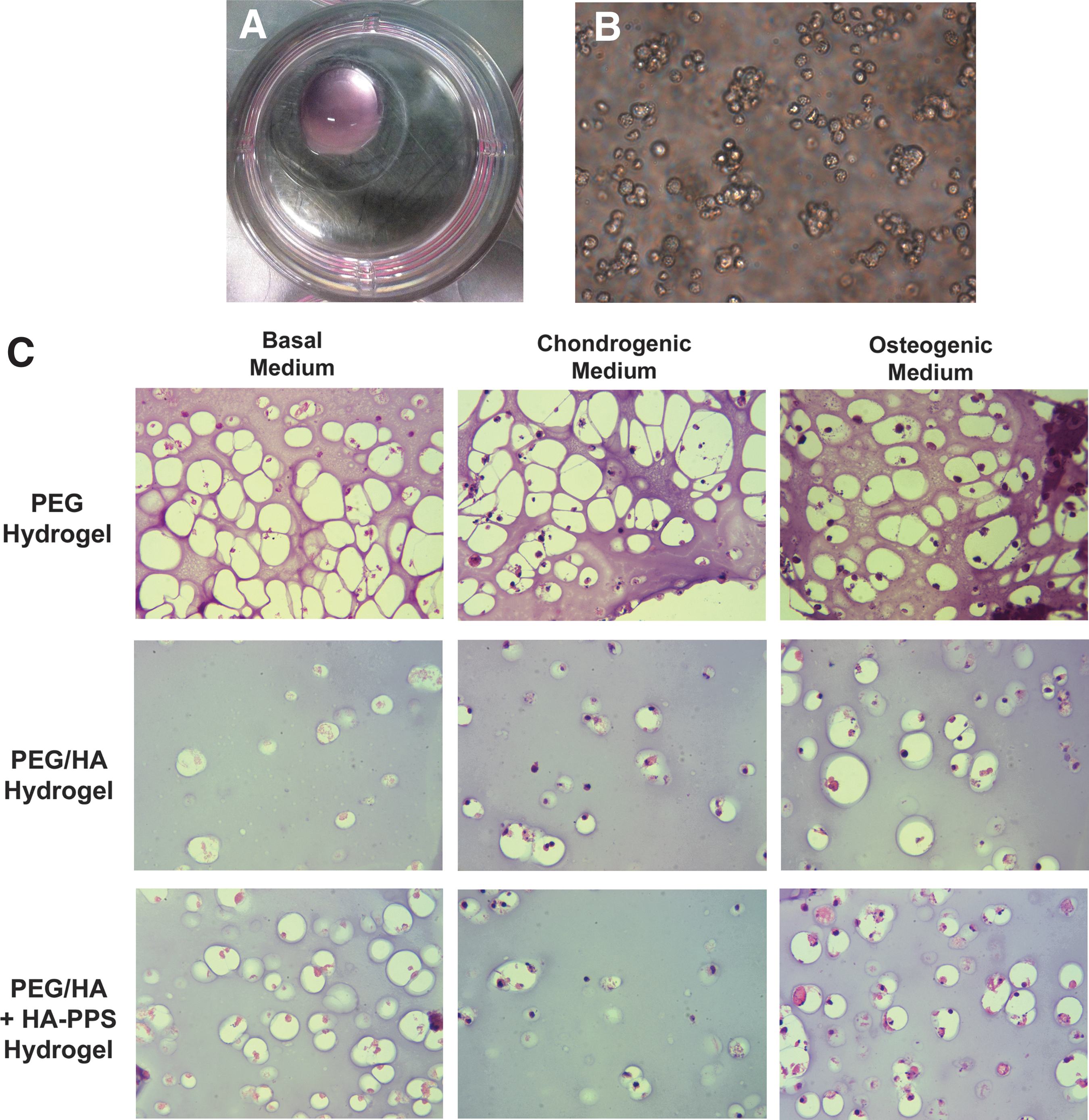
ciPSC-MSCs survive when encapsulated in hydrogels.
Alcian blue staining for the deposition of glycosaminoglycans (GAGs) was used as a marker of chondrogenic differentiation. Due to the background staining of the hydrogels (and particularly those containing HA, which is itself a GAG), positive staining was determined to be any area more intense than the overall background matrix. Little GAG deposition was observed around cells encapsulated in either the PEG or PEG/HA gels, even in the presence of chondrogenic supplements (Fig. 11A, B, D, E). However, there were many areas of intense Alcian blue staining both surrounding and between ciPSC-MSCs in PEG/HA+HA-PPS gels in the presence of chondrogenic supplements (Fig. 11H), suggestive of chondrogenic differentiation. Conversely, while there was an abundance of Alizarin red-positive deposits in the PEG hydrogels cultured in osteogenic medium (Fig. 11C), osteogenic differentiation was significantly reduced in the PEG/HA hydrogels (Fig. 11F) and completely abolished by HA-PPS (Fig. 11I).
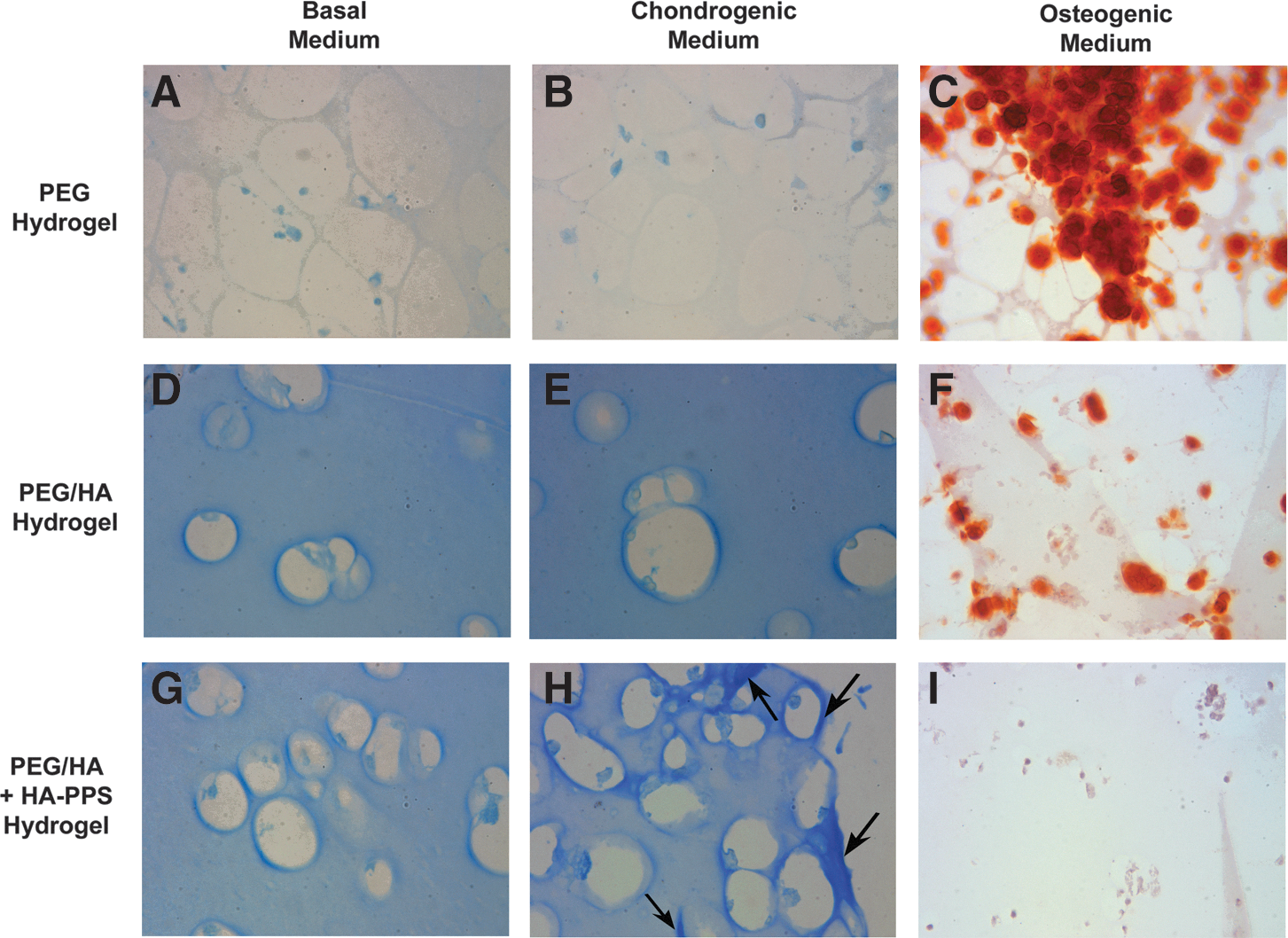
Hydrogels support chondrogenesis and osteogenesis in vitro. Alcian blue staining for the deposition of GAG was used as a marker of chondrogenic differentiation.
Discussion
In this study, we have generated ciPSC-MSCs that resemble tissue-harvested canine MSCs in terms of their morphology, adherence to plastic, immunophenotype, and multipotential differentiative ability [19 –24,35]. Perhaps surprisingly, given the substantive number of publications that describe the isolation of MSCs from various human tissues, there are no definitive criteria for the identification of MSCs. In 2006, the International Society for Cellular Therapy defined a number of minimal criteria that must be met in order for a human cell type to be described as an MSC [36]. Specifically, they must be adherent to plastic; express the cell surface markers CD73, CD90, and CD105; lack expression of the hematopoietic cell surface markers CD34 and CD45; and have a demonstrated ability to differentiate into osteogenic, chondrogenic, and adipogenic tissues. Our ciPSC-MSCs fulfill all of these positive-selection criteria. The only criterion that we were unable to address, due to a lack of positive control material in the dog, was the absence of CD34 and CD45 expression. Several studies have described the isolation and characterization of MSCs from canine tissues, including their immunophenotype [21 –24,35]. All of these studies report their cells as being positive for CD90 expression, and all but one [22] describe their cells as being negative for CD34 and CD45 [21,23,24,35]. However, none of these groups describe the use of a positive control cell type in their assessment of CD34 and CD45 immunoreactivity, and so a lack of immunostaining might be attributable to either an absence, or lack of recognition, of the immunogen; in the latter instance, this is of particular significance when primary antibodies are raised against epitopes from alternative species such as mouse and human. Thus, without a suitable positive control, we are unable to comment upon the presence or absence of CD34 and CD45 expression in our cells. They do, however, robustly express all three of the positive-selection cell surface markers for MSCs: CD73, CD90 and CD105.
Because our ciPSC-MSCs were generated through induced transdifferentiation of pluripotent ciPSCs, it was important to identify markers that were specifically indicative of the cells having assumed a mesenchymal identity, rather than being simply aberrant iPSCs. To this end, we have identified the additional MSC markers STRO1, cPDGFRβ and cKDR as being expressed only by the ciPSC-MSCs, and adult MSCs, and not the parent ciPSCs, further confirming that we have, indeed, produced MSCs with a gene expression signature significantly different from the parental ciPSCs.
Several studies of adult canine MSCs, derived from a variety of tissue sources, describe the expression of pluripotency markers normally associated with ESCs, such as OCT4, SOX2, NANOG and REX1 [19,21,23,24]. Our ciPSC-MSCs similarly express OCT4, NANOG and REX1, as do the adult bone marrow- and adipose-derived MSCs that we examined. Although the ciPSCs from which the ciPSC-MSCs were generated were reprogrammed through the expression of transgenes encoding human OCT4 and NANOG [26], the production of these factors by our ciPSC-MSCs cannot be attributable to persistent transgene expression since we have shown that the transgenes are transcriptionally silent within both the ciPSCs (this study and Whitworth et al. [26]) and ciPSC-MSCs, and that canine-specific OCT4 and NANOG transcripts are detectable within our cells, and also within the adult adipose- and bone marrow-derived MSCs. Thus, in addition to the previously described cell surface markers, a further defining characteristic of adult canine MSCs is the expression of pluripotency factors OCT4, SOX2, NANOG and REX1. Adult hMSCs, isolated from various tissues, have also been shown to express varying combinations of OCT4, NANOG, SOX2 and REX1 [37]. Interestingly, Chen et al. [15] describe a concomitant decline in the expression of pluripotency factors with the transition from iPSCs/ESCs to MSCs during their culture with SB431542. The significance of the expression of various pluripotency factors in adult human, and canine, MSCs is yet to be fully elucidated; however, it appears that in human umbilical cord MSCs, and those derived from adult adipose tissue, but not bone marrow, REX1 positively regulates cell proliferation [38]. Nanog, also, has been implicated as a positive regulator of proliferation in murine MSCs [39].
We originally described the production of ciPSCs that had been reprogrammed to a naive state of pluripotency, as evidenced by the presence of two active X chromosomes (XaXa) [26]. Hence, we were intrigued to see whether the ciPSC-MSCs were similarly XaXa, or whether they had initiated inactivation of one of the X chromosomes (XaXi). Perhaps not surprisingly, given the expression of pluripotency factors in the ciPSCs-MSCs, both X chromosomes appear to be active. Further, both populations of adult MSCs, from adipose tissue and bone marrow, appear to also have two active X chromosomes. To our knowledge, this is the first account of the X chromosome status in MSCs of any species. In mouse ESCs, REX1 plays an important role in inhibiting X chromosome inactivation by repressing transcription of Xist, and by activating Tsix [40]. Given that our ciPSC-MSCs and the adult MSCs express REX1 and appear to be XaXa, it is tempting to speculate that this functional relationship may also exist in canine MSCs.
An important aspect of MSC biology is an ability to differentiate into mesodermal derivatives: minimally, cartilage, bone and adipose tissue. Our ciPSCs-MSCs showed robust differentiative ability in assays for all three tissue types. Of particular consideration, given both their derivation from iPSCs, and our eventual intent to transplant these cells in vivo, is the ability of the ciPSC-MSCs to form teratoma-like tissues. Using an in vitro terminal differentiation assay, the ciPSCs gave rise to tissues that were derivatives of all three germ layers, although the mesodermal lineage was more extensively represented than the endoderm or ectoderm; we have observed a similar bias toward mesoderm derivatives in iPSCs from other domestic species assayed with this system [31]. However, of key importance was the observation that neither the ciPSC-MSCs nor the adult adipose-derived MSCs formed teratoma-like tissues. At the histological level, the similarities between the ciPSC-MSCs and the adult MSCs are striking. Both showed isolated populations of adipose cells and small, localized regions of calcified bone-like matrix among undifferentiated MSCs. The tendency to form adipose tissue and bone is not surprising given that the inherent ability to form both of these tissues is among the defining features of MSCs. However, in a translational context, the spontaneous formation of bone-like tissue is potentially undesirable, especially if the cells have been delivered within a joint. To this end, it should be noted that the commercial preparations of adult MSCs used in this study have been used in clinical trials for osteoarthritis since 2012 with no adverse events recorded (regeneus.com.au; Regeneus, pers. comm.). Of clinical significance is the observation that the differentiation of MSCs along a particular pathway is very much influenced by both the mechanical and biochemical properties of their environment [41 –45]. Thus, we sought to determine whether we could direct the differentiation of our ciPSC-MSCs along the chondrogenic, rather than osteogenic or adipogenic, pathway by a combination of mechanical and biochemical means; specifically, by encasing them in hydrogel scaffolds that have previously been shown to support chondrogenesis [27].
Injectable hydrogels are receiving increasing interest for cellular delivery due to their highly hydrated and porous nature, which provides an ideal environment for the encapsulated cells. Further, these materials can be injected into the body, removing the need for extensive surgery, and can mould to the precise shape of any defect. Importantly, when looking toward future clinical application of these cells, we showed that ciPSC-MSCs could be encapsulated within a range of hydrogel compositions and were capable of differentiation along both the osteo- and chondrogenic lineages.
Our work also demonstrated the requirement to match the biomaterial system used to the specific application, as the ciPSC-MSCs showed differentiation along the chondrogenic lineage only in PEG/HA+HA-PPS gels while their responsiveness to osteogenic factors was limited to the PEG hydrogels. The potent inhibitory effect of the HA-PPS upon osteogenesis is similar to that noted for unconjugated PPS, which has been shown to decrease osteogenic differentiation of hMSCs [34]. The exact mechanism by which this occurs is presently unknown, but is an active focus of our laboratory. Intriguingly, we have previously demonstrated chondrogenesis of hMSCs when encapsulated in PEG/HA+HA-PPS hydrogels, even in the absence of chondrogenic medium [32], which we suggest may be due to endogenous factors being captured and presented to cells (in both an autocrine and paracrine manner) by the HA-PPS in these systems. The requirement here for the addition of chondrogenic medium highlights differences between the two species, most likely due to differences in the type, or amounts, of soluble factors secreted into the surrounding extracellular space and thus the relative levels of stimulation provided by these functional hydrogels.
Conclusions
In this study we have demonstrated that ciPSCs can be used to efficiently generate MSCs that are highly proliferative, express MSC and pluripotency markers, undergo robust osteo-, chondro-, and adipogenesis but do not form teratoma-like tissues. We have also shown that when incorporated into hydrogels containing PPS in the form of HA-PPS, these iPSC-derived MSCs are stimulated to differentiate along the chondrogenic pathway, providing the first step in the process towards developing an effective MSC-based therapy for osteoarthritis in dogs, and serving as a model system for human degenerative joint disease.
Footnotes
Acknowledgments
The authors are grateful to Clay Winterford of QIMR Berghofer Medical Research Institute for his expertise in performing the histological processing of the hydrogels. This work was supported by grants 2011000286 from the Australian Companion Animal Health Foundation; 2011000284, 2011002440, and 2012002150 from the John and Mary Kibble Trust to D.J.W.; and 2012002076 from the University of Queensland Collaboration and Industry Engagement Fund to D.J.W., E.J.W., and J.J.C.-W. J.E.F. is supported by an ARC DECRA Fellowship (DE13010098).
Author Disclosure Statement
The authors each declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.