
Abstract
Glutaminase is the enzyme that converts glutamine into glutamate, which serves as a key excitatory neurotransmitter and one of the energy providers for cellular metabolism. Previous studies have revealed that mice lacking glutaminase 1 (GLS1), the dominant isoform in the brain and kidney, died shortly after birth due to disrupted glutamatergic transmission, suggesting the critical role of GLS1 in the physiological functions of synaptic network. However, whether GLS1 regulates neurogenesis, a process by which neurons are generated from neural progenitor cells (NPCs), is unknown. Using a human NPC model, we found that both GLS1 isotypes, kidney-type glutaminase and glutaminase C, were upregulated during neuronal differentiation, which were correlated with the expression of neuronal marker microtubule-associated protein 2 (MAP-2). To study the functional impact of GLS1 on neurogenesis, we used small interference RNA targeting GLS1 and determined the expressions of neuronal genes by western blot, real-time polymerase chain reaction, and immunocytochemistry. siRNA silencing of GLS1 significantly reduced the expression of MAP-2, indicating that GLS1 is essential for neurogenesis. To unravel the specific process(es) of neurogenesis being affected, we further studied the proliferation and survival of NPCs in vitro. siRNA silencing of GLS1 significantly reduced the Ki67+ and increased the TUNEL+ cells, suggesting critical roles of GLS1 for the proliferation and survival of NPCs. Together, these data suggest that GLS1 is critical for proper functions of NPCs, including neuronal differentiation, proliferation, and survival.
Introduction
N
Glutaminase 1 (GLS1) is an enzyme that converts glutamine into glutamate [15]. In the central nervous system (CNS), GLS1 is expressed at high levels, particularly in neurons [16]. Based on the in situ hybridization data provided by “Allen Brain Atlas: Developing Mouse Brain” at
GLS1 is critical for synaptic integration in the CNS. Gls1 knockout mice died at postnatal day 1 due to impairment in respiratory function that is controlled by glutamatergic synaptic network [26]. Although GLS1 is critical for CNS synaptic transmission and glutamate has been extensively studied for embryonic and adult CNS neurogenesis, the specific role of GLS1 in neurogenesis has not been identified. In this study, we report that GLS1 is an essential enzyme for the neuronal differentiation, proliferation, and survival of human NPCs in vitro.
Materials and Methods
Isolation and culture of human NPC
Human fetal brain tissue (12–16 weeks postconception) was obtained from elective abortions carried out at the University of Washington in full compliance with the National Institute of Health (NIH), University of Nebraska Medical Center (UNMC), and University of Washington ethical guidelines. Human cortical NPCs were isolated from human fetal brain tissue, as previously described [27]. Briefly, NPCs were cultured in substrate-free tissue culture flasks and grown as neurospheres in a neurosphere initiation medium (NPIM), which consisted of X-Vivo 15 (BioWhittaker, Walkersville, ME) with N2 supplement (Gibco BRL, Carlsbad, CA), neural cell survival factor-1 (NSF-1; BioWhittaker), basic fibroblast growth factor (20 ng/mL; Sigma-Aldrich, St. Louis, MO), epidermal growth factor (20 ng/mL; Sigma-Aldrich), leukemia inhibitory factor (10 ng/mL; EMD Millipore Corporation, Billerica, MA), and 60 ng/mL N-acetylcysteine (Sigma-Aldrich). Cells were passaged at 2-week intervals, as previously described [27].
Human NPC differentiation
Differentiation of NPCs to neurons was performed as previously described [27]. Briefly, trypsin- and mechanically dissociated NPCs were plated on poly-
siRNA knockdown of glutaminase
siRNA knockdown in NPCs was performed, as previously described [28]. Briefly, siRNA targeting GLS1 (ID# s5840) was purchased from Applied Biosystems, Inc. (Foster City, CA). Before they were changed into the serum-free Neurobasal medium for neuronal differentiation, NPCs were transfected with 100 nM siRNA duplexes for 24 h in the presence of siIMPORTER (Upstate Cell Signaling Solutions, Charlottesville, VA) according to the manufacturer's instructions. Nonspecific control siRNA from Applied Biosystems, Inc. (ID# AM4635) was transfected with the same concentration as controls to GLS1 siRNA.
Protein extraction and western blot
Cells were rinsed twice with 1× phosphate-buffered saline (PBS) and lysed by M-PER Protein Extraction Buffer (Pierce, Rockford, IL) containing 1× protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Protein concentrations were determined using a BCA Protein Assay Kit (Pierce). Proteins (20–30 μg) from cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. After electrophoretic transfer to polyvinyldifluoridene membranes (Millipore Corporation and Bio-Rad, Hercules, CA), proteins were treated with purified primary antibodies for microtubule-associated protein 2 (MAP-2) (mouse, cat#MAB3418, 1:1,000; Millipore Corporation), KGA (rabbit, 1:1,000; Dr. N. Curthoys, Colorado State University, Fort Collins, CO), GAC (rabbit, 1:500; Dr. N. Curthoys, Colorado State University), GAD67 (mouse, cat#MAB5406, 1:500; Millipore Corporation), poly ADP ribose polymerase (PARP) (rabbit, cat# 9542s, 1:1,000; Cell Signaling Technologies, Beverly, MA), or β-actin (Sigma-Aldrich) overnight at 4°C followed by a horseradish peroxidase-linked secondary anti-rabbit or anti-mouse antibody (1:10,000; Cell Signaling Technologies). Antigen–antibody complexes were visualized by the Pierce ECL Western Blotting Substrate (Pierce). For data quantification, films were scanned with a CanonScan 9950F scanner; the acquired images were then analyzed on a Macintosh computer using the public domain NIH ImageJ program (at
RNA extraction and TaqMan real-time RT-PCR
RNA was isolated with the TRIzol Reagent (Invitrogen Corp, Carlsbad, CA) and RNeasy Kit according to the manufacturer's protocol (Qiagen, Inc., Valencia, CA). Primers used for real-time reverse-transcription–polymerase chain reaction (RT-PCR) were GAC (ID# 528445: forward sequence 5-TATGGAAAAAAGTGTCACCTGAGTCA-3, reverse sequence 5-GCTTTTCTCTCCCAGACTTTCCATT-3, probe sequence 5-AATGAGGACATCTCTACAACTGTA-3); KGA (ID# 489954: forward sequence 5-CGAAGATTTGCTTTGTCAGCTATGG-3, reverse sequence 5-CTCTGCAGCAGCTACATGGA-3, probe sequence 5-CAGCGGGACTATGATTC-3); MAP-2 (ID# hs00258900) and GAPDH (ID# 4310884E) were commercial products from Applied Biosystems, Inc. Real-time RT-PCR was carried out using the one-step quantitative TaqMan assay in a StepOne™ Real-Time PCR system (Applied Biosystems, Inc.). Relative KGA and GAC mRNA levels were determined and standardized with a GAPDH internal control using the comparative ΔΔCT method. All primers used in the study were tested for amplification efficiencies and the results were similar.
Immunocytochemistry
Cells were fixed in 4% PFA and washed in PBS, as previously described [27]. Cells were then incubated overnight at 4°C with primary antibodies, followed by goat anti-mouse IgG Alexa Fluor 488 secondary antibodies (1:1,000; Molecular Probes, Eugene, OR) for 1 h at 25°C. Primary antibodies included mouse MAP-2 (1:500; Millipore Corporation), rabbit GFAP (1:2,000; Dako, Carpinteria CA), and mouse anti-Ki67 (1:500; BD Biosciences, San Diego, CA). All antibodies were diluted in 0.1% Triton X-100 and 2% bovine serum albumin in PBS. Cells were counterstained with DAPI (1:1,000; Sigma-Aldrich) to identify nuclei. Morphological changes were visualized and captured with a Nikon Eclipse E800 microscope equipped with a digital imaging system or a Zeiss META 510 confocal microscope (Carl Zeiss MicroImaging, LLC, Thornwood, NY). Images were imported into Image-Pro Plus, version 7.0 (Media Cybernetics, Silver Spring, MD), for quantification. A total of 500–1,000 immunostained cells from 10 random fields per culture were manually counted using magnifications of 20× objective lens.
Analyses of glutamate by Amplex Red Glutamic acid/Glutamate oxidase Assay Kit
Intracellular glutamate levels in the cells were determined by Amplex Red Glutamic acid/Gutamate oxidase Assay Kit (Invitrogen Corp) based on the manufacturer's instruction. Cell lysates were diluted to the same protein concentration before entering the assay.
In situ TUNEL assays
Primary human NPCs were transfected with siRNA for control siRNA or GLS1 siRNA. Three days after transfection, cells were stained with an in situ TUNEL assay (Roche Diagnostics, Indianapolis, IN). TUNEL+ NPCs and total cells were counted after acquiring random images (10) from immunostained fields using a Nikon Eclipse TE2000E microscope. A minimum of 10 fields was counted for each treatment condition.
Statistical analysis
Data were analyzed as mean±SEM. The data were evaluated statistically by the analysis of variance followed by the Tukey test for paired observations. The two-tailed Student's t-test was used to compare two groups. Significance was considered when P<0.05. All experiments were performed with at least three donors to account for any donor-specific differences. Assays were performed at least three times in triplicate or quadruplicate.
Results
GLS1 splice variants KGA and GAC were upregulated during neuronal differentiation
Human NPCs were differentiated to neurons in adherent cultures for 2 weeks. Total RNA was isolated at various time points, including day 0, 4, 7, and 13 after differentiation. The gene expression of neuronal marker MAP-2 continued to increase throughout this period of time (Fig. 1A), suggesting successful and continuous neuronal differentiation of the culture. Interestingly, both KGA and GAC mRNA were upregulated during neuronal differentiation (Fig. 1B, C). To confirm the upregulation of these genes after neuronal differentiation, we collected parallel protein samples and determined protein levels of MAP-2, KGA, and GAC through western blot (Fig. 1D–G). The neuronal differentiation resulted in a sharp increase of MAP-2 (Fig. 1D, E). Similarly, the KGA protein had 3- and 4.5-fold upregulation at 7 and 13 days after differentiation, respectively (Fig. 1D, F). The GAC protein had 2- and 3.5-fold upregulation at 4 and 7 days after differentiation, respectively (Fig. 1D, G). Both mRNA and protein levels of KGA were positively correlated with the MAP-2 levels (Fig. 1H and Supplementary Fig. S1A; Supplementary Data are available online at
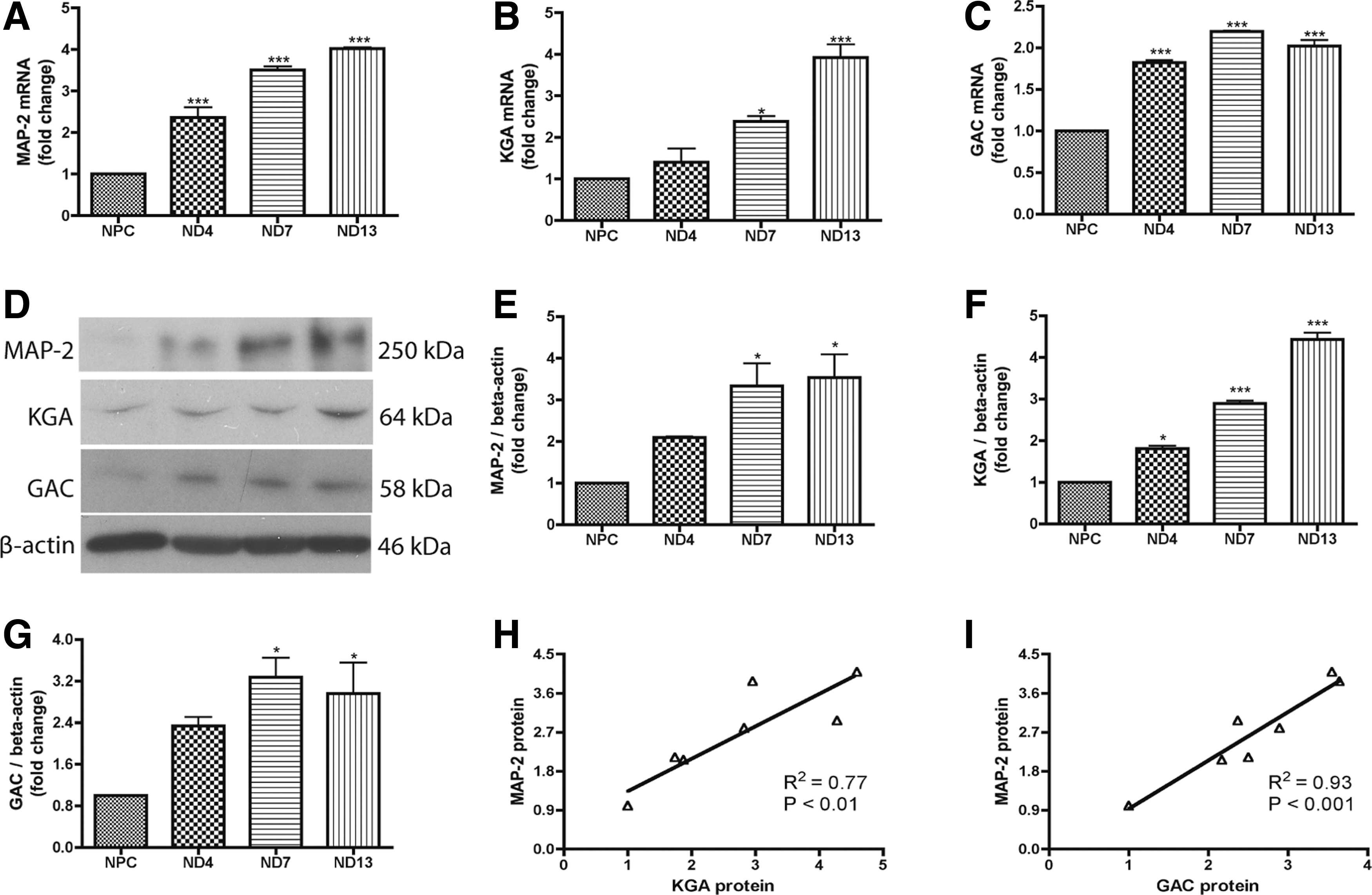
Glutaminase 1 (GLS1) isotypes, kidney-type glutaminase (KGA), and glutaminase C (GAC) were upregulated during neural progenitor cell (NPC) differentiation to neurons.
siRNA knockdown of GLS1 impaired neuronal differentiation
To test whether KGA and GAC were required for neuronal differentiation, we used siRNA to specifically silence GLS1 expression in NPCs. The GLS1 siRNA-transfected NPCs were induced to differentiate in neuronal differentiation media for 6 days. The knockdown of GLS1 resulted in significant decreases of both KGA (Fig. 2A, D, E) and GAC (Fig. 2B, D, F) mRNA and protein levels compared with the control siRNA group. The knockdown also resulted in a decrease of intracellular glutamate (Fig. 2G), indicating that the knockdown had reduced the enzymatic function of KGA and GAC. After siRNA knockdown of GLS1, MAP-2 protein and mRNA were reduced by more than 50% of that in control siRNA-transfected cells (Fig. 2C, D, H). The reduction of the mature neuronal marker MAP-2 in GLS1-deficient cells indicates that GLS1 is required for neuronal differentiation.
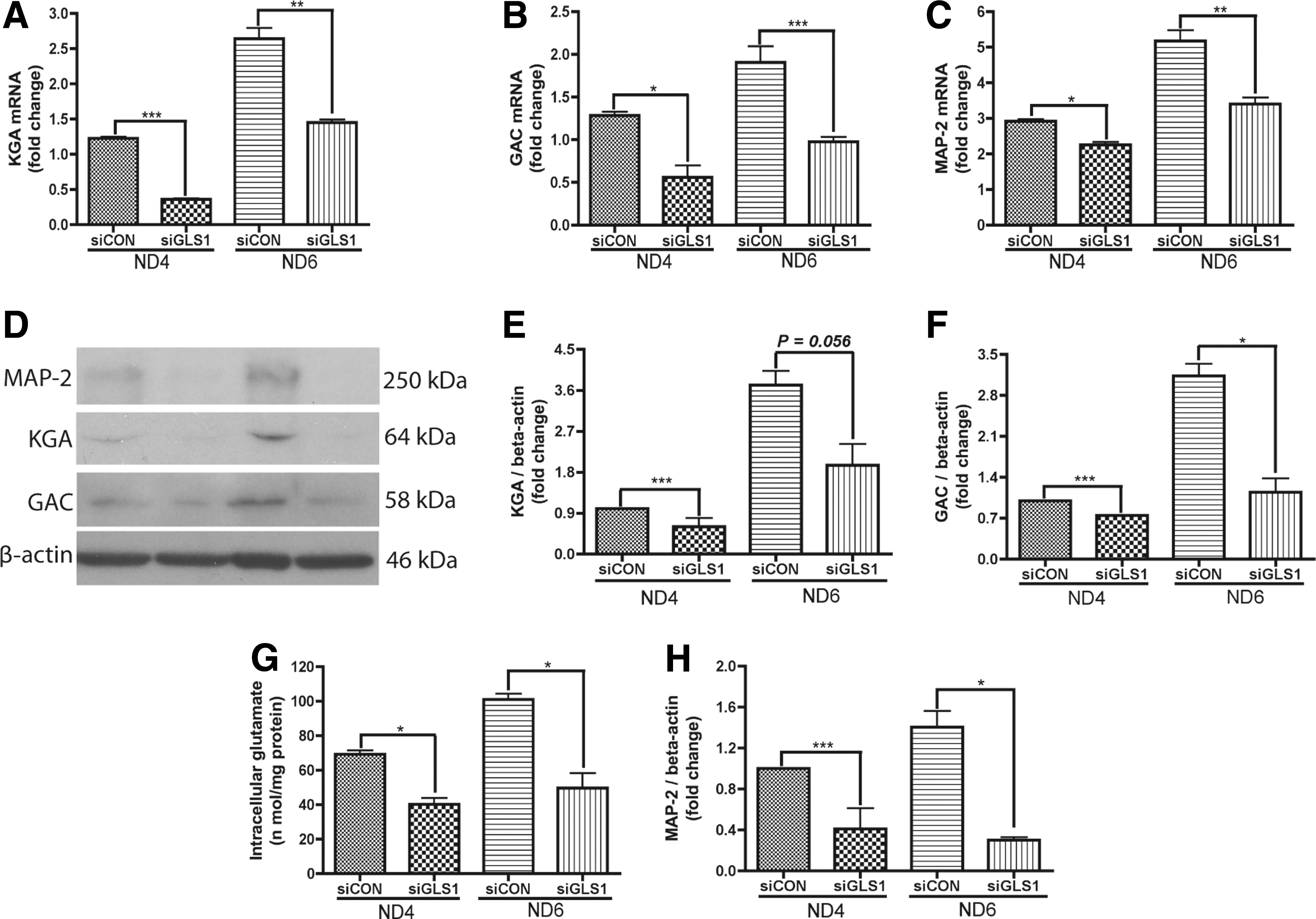
Lack of GLS1 impaired the expression of MAP-2 during neuronal differentiation.
To further characterize the morphological changes of neurons after GLS1 knockdown, we labeled MAP-2 in the differentiated cultures through immunocytochemistry (Fig. 3A). Human NPCs transfected with control siRNA had ∼70% of the cells positive for MAP-2 (Fig. 3B) at 6 days after differentiation. In contrast, NPCs transfected with GLS1 siRNA had a significantly lower percentage (40%) of cells positive for MAP-2 (Fig. 3B). The impairment was specific to neuronal differentiation since the astrocyte differentiation, determined by GFAP immunostaining, did not change after GLS1 knockdown when compared with the control siRNA group (Fig. 3C). The number of the total counted cells is provided in Supplementary Table S1. These data suggest that GLS1 is required for the formation of mature neurons.
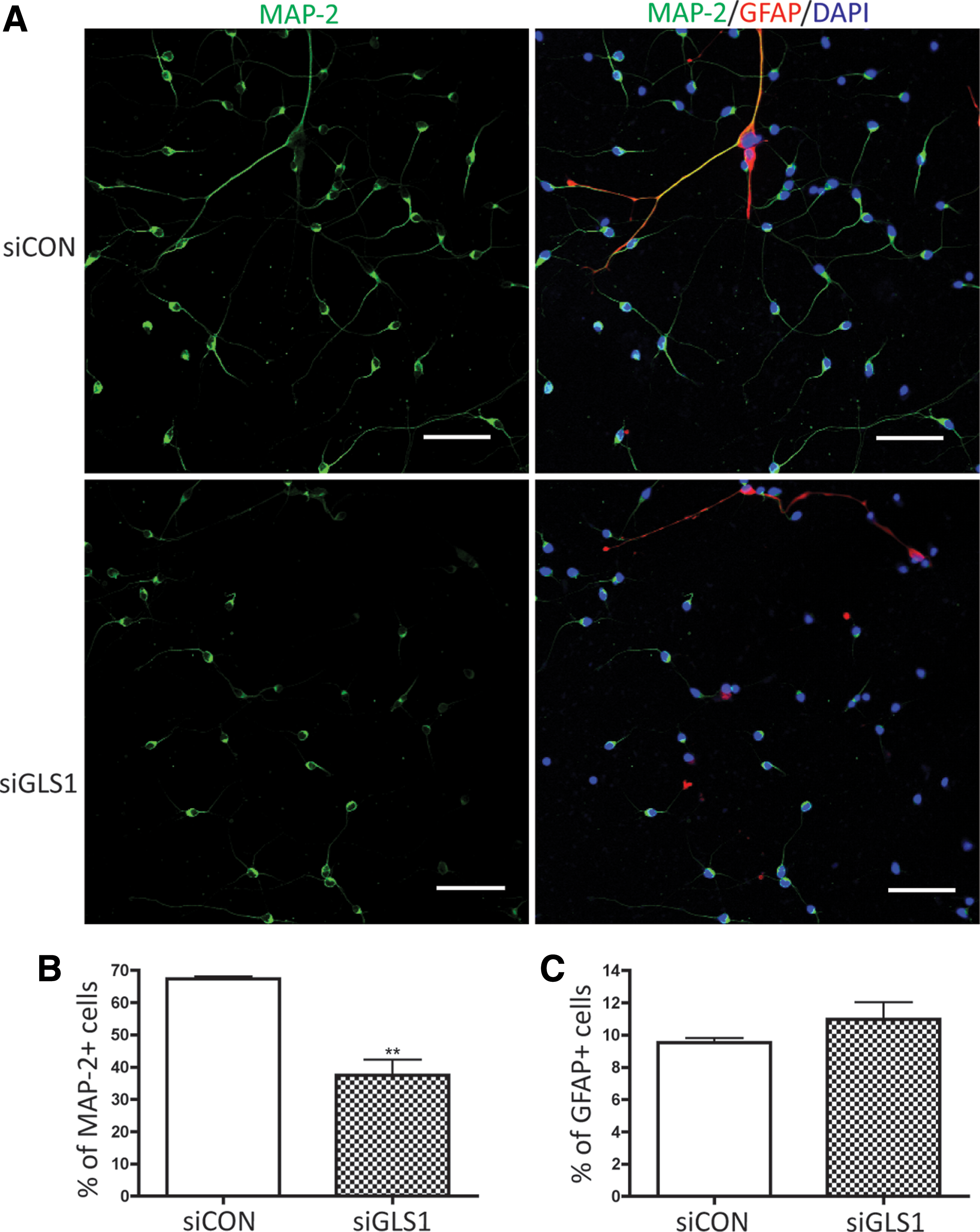
Lack of GLS1 specifically impaired neuronal differentiation. Human NPCs were transfected by control siRNA or GLS1 siRNA and then exposed to the neuron differentiation medium for 6 days. The cells were fixed and stained with MAP-2 to determine the levels of neuronal differentiation.
Knockdown of GLS1 impaired NPC proliferation and survival
Neuronal differentiation is a key step of neurogenesis. However, proper NPC functions also include proliferation and survival. To determine whether lack of GLS1 affected NPC proliferation, we transfected NPCs with siRNA targeting GLS1 and maintained the culture under growth media for 3 days. Through immunocytochemistry that labeled the Ki67+ cells, we were able to identify the levels of NPC proliferation in the cultures. The percentage of Ki67+ cells was significantly decreased when GLS1 was silenced (Fig. 4A, B), suggesting a critical role of GLS1 in the NPC pool expansion. The number of the total counted cells is provided in Supplementary Table S1. The decrease of KGA and GAC mRNA levels by GLS1 siRNA transfection in NPCs was confirmed by real-time RT-PCR (Fig. 4C, D).

GLS1 silencing reduced NPC proliferation. Human NPCs were transfected by control siRNA or GLS1 siRNA, then maintained in growth media for 3 days. NPC proliferation was determined by immunolabeling the Ki67 of proliferating cells in culture.
Next, we determined NPC survival after GLS1 was silenced by siRNA. The levels of apoptosis were identified by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. TUNEL detects DNA fragmentation by labeling the terminal end of nucleic acids. We applied TUNEL assay to undifferentiated NPCs kept in growth media. siRNA targeting GLS1 significantly increased the percentage of TUNEL+ cells in the human NPC culture compared with the siRNA control group (Fig. 5). The number of the total counted cells is provided in Supplementary Table S1. Therefore, the TUNEL assay revealed the occurrence of more cellular apoptosis when GLS1 was deficient. To further confirm the apoptosis, we determined PARP cleavage in the NPCs that lack GLS1. PARP is deactivated and cleaved by active caspase 3 (cleaved caspase 3) in the apoptosis cascade; thus, cleaved PARP represents ongoing apoptosis. Western blot analysis revealed the presence of cleaved PARP in both control and GLS1 siRNA groups. However, cleaved PARP was at significantly higher levels in the GSL1 siRNA group compared with the control siRNA group (Fig. 6). These data demonstrate the occurrence of cellular apoptosis after GLS1 was knocked down.

GLS1 silencing increased NPC apoptosis. Human NPCs were transfected by control siRNA or GLS1 siRNA, then maintained in growth media for 3 days. NPC apoptosis was determined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
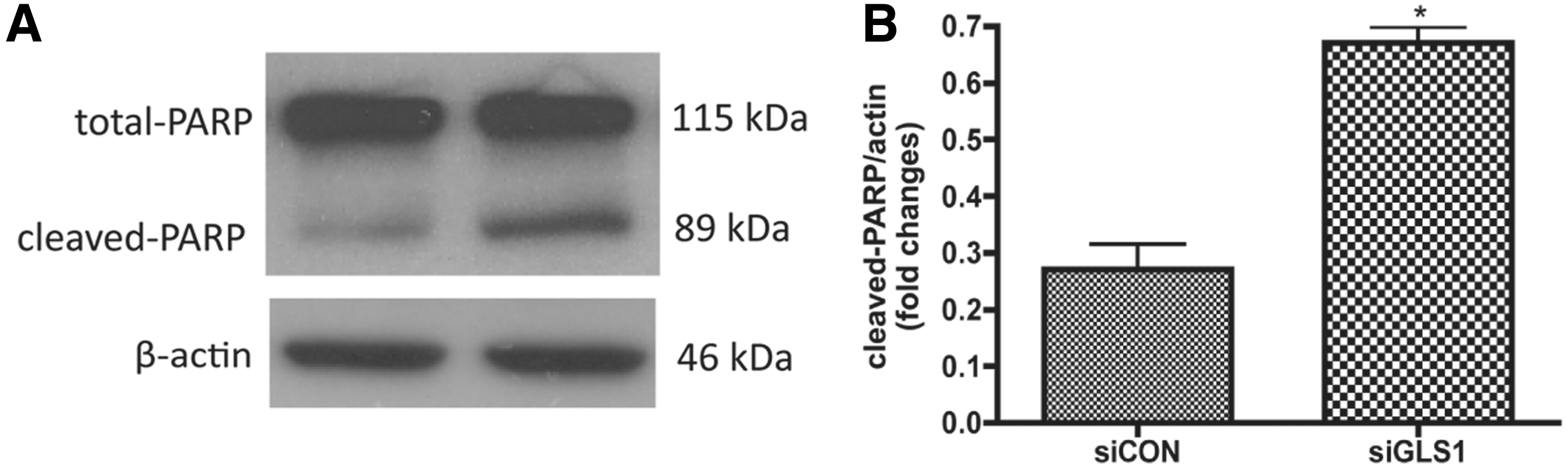
Lack of GLS1 increased poly ADP ribose polymerase (PARP) cleavage in NPCs. Human NPCs were transfected by control siRNA or GLS1 siRNA, then maintained in growth media for 3 days.
Discussion
Neurogenesis is a highly orchestrated process that includes maintenance and expansion of NPCs, NPC migration, and differentiation to neurons, neuronal survival, and integration of new neurons to the existing synaptic networks [6]. As a result, impairment in neurogenesis often causes brain malformations, neurotransmission disruptions, and cognitive difficulties and deterioration, as reviewed by Jessel and Sanes [2,6 –8]. Despite the importance of neurogenesis to the health and diseases, factors involved in this process remain to be fully elucidated. In the current study, we show that GLS1 isoforms were upregulated and correlated with MAP-2 during neuronal differentiation (Fig. 1). siRNA silencing of GLS1 in NPCs impaired their subsequent neuronal differentiation, suggesting that GLS1 is critical for neuronal differentiation (Figs. 2 and 3). Furthermore, we showed reduced proliferation and increased apoptosis in GLS1-deficient cells, suggesting that GLS1 is required for NPC proliferation and survival (Figs. 4 –6). Collectively, we characterized GLS1 regulation in NPCs and identified GLS1 as one of the essential factors for neurogenesis.
GLS1 is a critical mitochondrial enzyme for cellular metabolism. Its expression increases during mammalian neural development and remains high in postnatal CNS, indicating a functional relevance of GLS1 to brain development. We report for the first time that lack of GLS1 impairs NPC differentiation, proliferation, and survival. This finding is consistent with the report of a functional role of GLS2 on neuronal differentiation [29]. Since GLS1 expression is at a much higher level in mammalian CNS compared with GLS2, tight regulation of GLS1 is of critical importance to brain homeostasis.
The identification of GLS1 functions in neurogenesis has an important clinical implication. GLS1, particularly the GAC isotype, has been shown to regulate oncogenic transformation [30] and mediate excitotoxicity in HIV-1-associated neurocognitive disorders [31]. Therefore, inhibiting GLS1 may serve for therapeutic purposes in cancer treatment or neurodegenerative diseases. Indeed, inhibition of GLS1 by either a potent chemical inhibitor or siRNA silencing slowed the growth of glioma cells [32] or blocked oncogenic transformation of human breast cancer cells and B lymphoma cells [33]. Also, knocking down GAC in HIV-infected microglia or macrophages alleviated the damage to neurons in the in vitro cocultured settings [31]. Although targeting GLS1 clearly provides benefits in the treatment of cancer or neurodegenerative diseases, our studies with neurogenesis suggest that it may have unintended consequences on the NPCs, as the properties and functions of NPCs were impaired with deficient GLS1 expression. Therefore, genetic, immunological, or pharmacological inhibition of GLS1 needs to be cautious on the potential complications. Furthermore, Gls1 KO mice died at postnatal day 1 due to glutamatergic synaptic transmission disruptions [26], and Gls1+/− mice displayed deteriorated hippocampal activity and developed schizophrenia-like symptoms [34]. These studies on GLS1 knockout (Gls1 KO) mice are consistent with our evaluation of the critical role of GLS1 on neurogenesis, particularly on the formation of neuronal network.
Neuronal differentiation, proliferation, and survival are three components of neurogenesis examined in the current study. How GLS1 functions to change each of those components is currently unclear. However, there are several potential pathways that GLS1 may take to assert its function. First, the natural product of GLS1 is glutamate, and glutamate is known to regulate neurogenesis. Interestingly, glutamate may have dichotomous effects on neurogenesis, depending on its concentrations. Low concentrations of exogenous glutamate (10 μM) introduced to cell or slice culture led to increased NPC proliferation and neurogenic potentials [19 –21], whereas high concentrations of exogenous glutamate (300 μM) introduction resulted in impaired DNA synthesis and reduced cellular proliferation [25]. Second, glutamate has a direct neurotoxic effect on mature neurons [35 –38]. Excess glutamate production by activated microglia and brain macrophages has been documented in several neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, stroke, and HIV-associated neurocognitive dementia [31,39]. Third, glutamate may serve as an alternative energy substrate for the citric acid cycle. Therefore, GLS1 deficiency may lead NPCs into a starvation-like state where normal activities like cellular proliferation and differentiation are attenuated. Furthermore, cells lacking energy are known to switch to the apoptotic signaling cascade. It is likely that more than one pathway play out in the NPCs that lack GLS1. More studies on glutamate signaling pathways leading to the impairment of NPC functions are needed.
In summary, our study demonstrated that GLS1 is required for neurogenesis in vitro because GLS1 deficiency impaired NPC differentiation, proliferation, and survival. These data shed light on a novel pathway to regulate neurogenesis and further suggest that great caution should be taken when targeting GLS1 in cancer or neurodegenerative disease therapy.
Footnotes
Acknowledgments
The authors thank Dr. Norman Curthoys for providing the KGA and GAC antibodies. They thank Dr. Hui Peng, Dr. Santhi Gorantla, and Ms. Li Wu for the technical support of this work. Julie Ditter, Lenal Bottoms, Myhanh Che, Johna Belling, and Robin Taylor provided outstanding administrative and secretarial support. This work was supported, in part, by research grants by the National Institutes of Health: R01 NS 41858-01, R01 NS 061642-01, P01 NS043985, and P20 RR15635-01 (J.Z.).
Author Disclosure Statement
No competing financial interests exist with the submitted manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.