
Abstract
Reduction of estradiol production and high serum concentrations of follicular stimulating hormone (FSH) are endocrine disorders associated with premature ovarian failure. Here, we report that transplantation of ovarian-like cells differentiated from stem cells restored endogenous serum estradiol levels. Stem cells were isolated from postnatal mouse skin and differentiated into ovarian-cell-like cells that are consistent with female germ, and ovarian follicle somatic cells. The ovarian-cell-like cells were transplanted into ovariectomized mice (Cell Trans), whereas control mice were subjected to bilateral ovariectomies without cell transplantation (OVX). Using vaginal cytology analysis, it was revealed that in 13 out of 19 Cell Trans mice, estrus cycles were restored around 8 weeks after cell transplantation and were maintained until 16 weeks post-transplantation, whereas in the OVX group, all mice were arrested at metestrus/diestrus of the estrus cycle. The uterine weight in the Cell Trans group was similar to sham operation mice (Sham OP), while severe uterine atrophy and a decreased uterine weight were observed in the OVX group. Histologically, ectopic follicle-like structures and blood vessels were found within and around the transplants. At 12–14 weeks after cell transplantation, mean serum estradiol level in Cell Trans mice (178.0±35 pg/mL) was comparable to that of the Sham OP group (188.9±29 pg/mL), whereas it was lower in the OVX group (59.0±4 pg/mL). Serum FSH concentration increased in the OVX group (1.62±0.32 ng/mL) compared with the Sham OP group (0.39±0.34 ng/mL). Cell Trans mice had a similar FSH level (0.94±0.23 ng/mL; P<0.05) to Sham OP mice. Our results suggest that ovarian somatic cells differentiated from stem cells are functional in vivo. In addition to providing insights into the function of ovarian somatic cells derived from stem cells, our study may offer potential therapeutic means for patients with hypo-estradiol levels like those encountered in premature ovarian failure.
Introduction
I
It is known that gonadotropins from the host pituitary gland can support morphogenesis of Graafian follicles and estradiol production in xenografts. For example, grafting cortical slices of either cat or sheep ovaries under the kidney capsules of ovariectomized mice has resulted in serum estradiol detection in response to follicle-stimulating hormone (FSH), which is accompanied by the restoration of vaginal patency. The grafts also became vascularized and large follicles were present in the transplant nine months later [16]. These results suggest that ovarian xenografts can respond to the host pituitary gland and have endocrine function. To mimic a condition similar to ovarian hypofunction in premature ovarian failure patients, ovariectomized animals were used in the current study to test the hypothesis that transplantation of ovarian-cell-like cells differentiated from SSCs may re-establish pituitary-ovarian communication to restore estradiol production in vivo.
Materials and Methods
Isolation and culture of SSCs
All animal-related experiments in the study were conducted according to the Care and Use of Experimental Animals of the Canadian Council on Animal Care Guidelines, and have been approved by the University of Guelph Animal Care and Use Committee and the Animal Center for Medical Experimentation at Gyeonsang National University. Postnatal female mouse SSCs were isolated and cultured as previously described [17,18] at 37°C and 5% CO2 air atmosphere. These stem cells have been characterized previously [18,19]. Cells were passaged twice prior to use. Stem cells from four to six fetuses were combined and used in each of the experiments. The porcine follicular fluid (PFF) was harvested from both small (1–3 mm) and large (4–6 mm) follicles of prepubertal gilts, adhering to a collection ratio of three small follicles for each large follicle. It was then centrifuged at 1500 g for 30 min; the supernatant was passed through a 0.22-μm filter, and stored at −80°C.
In vitro differentiation into PGC-like cells from SSCs
SSCs at the fourth day after passage 2 were isolated with the same method as previously described [17,18]. The SSCs (3×105 cells/well) were differentiated into mouse PGC-like cells (PLCs) using specific culture media consisting of high-glucose DMEM (Life Technologies, Carlsbad, CA) supplemented with 5% heat-inactivated fetal bovine serum (FBS; Life Technologies; Lot No. 914847), 5% filtered PFF, 0.1 mM nonessential amino acids (Life Technologies), and 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), similar to the previously described media for porcine oocyte-like cell (OLC) differentiation from SSCs [8,20]. The cells were cultured in a 24-well plate adherent dish (Sarstedt, Montreal, Canada) at 37°C and 5% CO2 in air atmosphere for 12 days, with half the medium changed every 3 days. At day 12 (D12) of differentiation, PLCs and plate-adherent fibroblast-like supporting cells were harvested with 0.1% trypsin for 3 min at 37°C. A total of 1×106 cells were plated with 150 μL of Matrigel™ matrix (BD Biosciences, Bedford, MA) in a well of a 24-well plate containing 200 μL of fresh and 200 μL of spent medium. Cells were cultured with Matrigel scaffold for an additional 6 days, to D18 of differentiation, while changing half of the medium every 3 days. The spent medium was collected at each time point, and the isolated supernatant after centrifugation (500 g for 5 min) was stored at −80°C for estradiol analysis by ELISA. Differentiated PLCs and OLCs at D18 were collected for analysis and in vivo transplantation. To compare the effect of Matrigel matrix and culture duration, SSCs were differentiated into PLCs for D18 and D24 in the same manner as described above without Matrigel.
Real-time PCR for differentiated cells
Differentiated cells at D18 and D24 with or without Matrigel were harvested and total RNA was isolated using the Total RNA Kit (Norgen Biotek Corporation, Thorold, Canada) according to manufacturer's protocol. Reverse transcription was performed as previously described [19]. Samples were DNase treated by adding 1 μL of 10× DNase buffer and 1 μL of amplification grade DNase (Life Technologies) and then incubated for 15 min at RT. One microliter of EDTA (25 mM) was then added and the samples were incubated for 10 min at 65°C. RT was then performed by adding 0.5 μL H2O, 5 μL 5× first strand buffer, 1.25 μL of random hexamer primers, 6.25 μL of 2 mM dNTPs, and 1 μL MMLV reverse transcriptase to the sample. The samples were then incubated at 25°C for 10 min, 37°C for 50 min, and 70°C for 15 min. Real-time PCR was carried out on an Mx3005P™ System (Stratagene, La Jolla, CA) by using the Quantitect SYBR green PCR kit (Takara Bio, Otsu, Japan). A total of 500 ng of DNase-treated cDNA was added to 6.25 μL of SYBR green mix, 0.25 μL ROX, and 0.25 μL each forward and reverse primers at 10 μM (final reaction volume of 12.5 μL). Product sizes were confirmed on 1.2% agarose gel. The RNA polymerase II (RPII) reference gene was amplified for each sample to verify the presence of cDNA and as an internal control to calculate the relative level of target gene expression using the 2−▵▵Ct method [21]. Primers, expected product size, and accession numbers are presented in Supplementary Table S1 (Supplementary Data are available online at
Immunocytochemistry
Immunocytochemistry for DAZL and MVH in OLCs and for AROMATASE in PLCs
SSCs were cultured with 5% PFF and FBS media for 12 days, and then the cells were cocultured with Matrigel (Matri+) or without Matrigel (Matri−) to D18. Differentiated PLCs and OLCs (≥40 μm diameter) at D18 were collected with 0.25% trypsin, and immunocytochemistry (ICC) analyses were performed. Cells were washed twice with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde (PFA; Fisher Scientific, Nepean, Canada) in PBS for 20 min. Cells were then washed three times in PBS with 0.1% Tween 20 (Fisher Scientific) and incubated for 10 min, and then for 20 min in PBS with 0.1% Triton-X-100 (Fisher Scientific). Next, cells were blocked for 2 h in PBS with 2% BSA and 0.05% Triton-X-100 (blocking solution), followed by an incubation with primary antibody, 1:400 rabbit polyclonal anti-DAZL (Abcam, Cambridge, MA) or 1:400 rabbit polyclonal anti-DDX/MVH (Abcam), overnight at 4°C. Cells were then washed in blocking solution and incubated with 1:300 diluted goat anti-rabbit IgG-R-phycoerythrin (PE; Sigma-Aldrich) for 2 h at RT. This was followed with a blocking solution wash and incubation with Hoechst333258 (Sigma-Aldrich) for 5 min, followed by three washes with PBS containing 2% BSA. Cells were mounted using fluorescent mount medium (DakoCytomation, Carpinteria, CA) and viewed using an Olympus BX-UCB microscope (Olympus, Cetner Valley, PA) and MetaMorph analysis software (Universal Imaging Corporation, Downingtown, PA).
To detect aromatase activity in Matri+ and Matri− PLCs at D18, cells from each group were incubated with 1:400 diluted rabbit anti-aromatase (Acris GmbH, Herford, Germany) and 1:300 anti-rabbit IgG-PE (Sigma-Aldrich). After nuclear staining with Hoechst, aromatase-positive cells were counted. At least three independent experiments were performed for statistical analysis.
Double ICC for STELLA and DAZL or MVH in PLCs
Differentiated PLCs with or without Matrigel at D18 were harvested and double ICC analyses were performed for codetection of STELLA and DAZL or DDX/MVH similar to the previous procedures. Cells were co-incubated in primary antibodies overnight at 4°C and in secondary antibodies for 2 h at RT. Primary antibodies: mouse monoclonal anti-STELLA (1:400; Chemicon, Temecula, CA) and rabbit polyclonal anti-DAZL (1:400; Abcam) or rabbit polyclonal anti-DDX/MVH (1:400; Abcam). Secondary antibody co-incubations were performed with anti-mouse FITC (1:300; Sigma-Aldrich) and anti-rabbit IgG-R-PE (1:300; Sigma-Aldrich).
Analysis of estradiol concentration in spent media by ELISA
Spent media was collected during culture when replacing half of the media and stored at −80°C until analysis. Estradiol levels were determined using the Estradiol EIA kit (Oxford Biomedical Research, Oxford, MI) according to the manufacturer's protocol.
In vivo transplantation of differentiated PLCs in ovariectomized mice
A total of 32 female CD1 mice (10–12 weeks old) were used for in vivo experiments. Three animals were used for the sham surgery group (Sham OP), 10 for the ovariectomized group (OVX), and 19 for the ovariectomy and cell transplantation group (Cell Trans).
Animals were anesthetized using an intraperitoneal injection of Avertin (0.75 mg/g) and OVX mice had both ovaries removed through a dorsal skin incision. In the Sham OP group, ovaries were exposed using the same approach as the OVX mice but then repositioned. For in vivo transplantation of PLCs (Cell Trans group), SSCs were differentiated into mouse PLCs in the 5% PFF and FBS containing media. At D12 of differentiation, PLCs and supporting fibroblast-like cells were collected and labeled with the fluorescent lipophilic carbocyanine dye PKH26 (Sigma-Aldrich) according to the manufacturer's instruction. A total of 1×106 PKH26-labeled cells were resuspended in 150 μL of Matrigel scaffold in a well of the 24-well plate for culture until D18 with half media changes every 3 days. At D18 of culture, the differentiated PLCs and Matrigel were collected. After bilateral ovariectomies were done in Cell Trans mice, the right kidney was exposed and ∼50 μL of the cultured cells and Matrigel scaffold was injected into the caudal fat tissues and under the capsule of exposed kidney using a 24 G syringe. Approximately 100 μL of the cultured cells and Matrigel was injected into the subcutaneous fat tissue. Some mice received one treatment while others received injections at both sites.
Blood samples (200 μL) were taken from experimental animals' saphenous vein at 4, 8, and 12 weeks after surgery. These blood samples were kept at RT for 30 min to allow clot formation, followed by centrifugation at 2200 g for 10 min. Blood serum was stored at −80°C, and serum estradiol and FSH concentrations were analyzed by ELISA. For each measurement, 50 μL of serum was used in ELISA. Estradiol EIA kits (Oxford Biomedical Research) and FSH ELISA kits (Endocrine Technologies, Inc., Newark, CA) were used for analysis of serum estradiol and FSH levels.
For analysis of the estrus cycle, vaginal smears were performed on experimental animals as previously described [22,23]. The smear was done for five consecutive days between 11:00 a.m. and 12:00 noon every day starting beginning 8 weeks after surgery.
Twelve weeks postsurgery, some animals were euthanized and the uterus and kidneys were harvested. The harvested uterus was weighed. RNA was isolated from the harvested uterus of experimental animals and real-time PCR was done for analysis of estrogen receptor (ER) mRNA levels. Cell-transplanted kidneys and subcutaneous fat tissues of the Cell Trans group mice were excised and fixed with 10% neutral buffered formalin, embedded in a paraffin block, cut into 4-μm sections, and mounted on glass slides. The slides were maintained at RT for 12 h and deparaffinized. After hydration, sections were stained with hematoxylin and eosin for evaluation of structures.
For immunohistochemical analysis of ER and FSHR, sections of cell transplant specimens were immunostained using an automated immunostainer (Ventana®, BenchMark XT; Roche, Tucson, AZ). Rabbit monoclonal anti-ER (1:1000; Neomarkers, Lab Vision Corp., Fremont, CA) and rabbit polyclonal anti-FSHR (1:200; MyBioSource, San Diego, CA) were used as primary antibodies for analysis of ER and FSHR expression. The slides were incubated at 37°C for 20 min in a humidity chamber and then primary antibodies against ER and FSHR were allowed to react at 35°C after blockage of endogenous peroxidase activity via the administration of hydrogen peroxide. After 32 min, the glass slides were treated with a biotinylated polyvalent secondary antibody solution, and incubated with horseradish peroxidase-conjugated avidin-biotin complex, followed by treatment with 3,3-diaminobenzidine and hydrogen peroxide. Finally, the nucleus was counterstained with hematoxylin.
Statistical analysis
For each set of data, independent experiments were repeated at least three times, with data representing the mean±SEM of all repeats. The statistical differences between experimental groups were determined by one-way analysis of variance (ANOVA) followed by Tukey test for multiple comparisons or unpaired t-test for single comparison of experimental data to the control using GraphPad Prism analysis software. Results were considered significant at P<0.05, and these differences were denoted by an asterisk or different letters.
Results
Previous works by others have shown that extracellular matrices such as Matrigel can have beneficial effects on ovarian follicle culture, granulosa cell communication, proliferation, survival, and steroidogenesis in vitro in various species [24]. We first studied the differentiation of SSCs into ovarian-cell-like cells in the presence of Matrigel. Figure 1A shows spheres of stem cells before inducing differentiation. These cells were then cultured in differentiation medium for 12 days (D12) before Matrigel was added to the culture for an additional 6 days (D18). Clusters of primordial germ cell (PGC)–like cells (PLCs) were observed at 12 and 18 days of differentiation, respectively (Fig. 1B, C). PLCs showed typical PGC characteristics: round, larger than somatic cells, and a blebbed appearance (Fig. 1D). At a late stage of differentiation (day 30 and later), OLCs with zona-pellucida-like morphology were also observed (Fig. 1E), suggesting a subset of our stem cells have differentiated into female germ-cell-like cells in this differentiation medium. The inclusion of Matrigel in the media enhanced RNA levels of the germ cell markers Dazl, Oct4, and Mvh, as determined by RT-qPCR (Supplementary Fig. S1A, B). Using the nuclear germ cell marker STELLA, we observed dual expression with the cytoplasmic proteins DAZL and MVH (Supplementary Fig. S1C). The expression and cellular location of these germ cell markers in our differentiated PLCs is similar to that of PGCs isolated from 12.5 dpc mice (PGCs; the top panels of Supplementary Fig. S1C). We next sought to investigate whether other ovarian cells, such as granulosa and theca cells, had also been generated in our differentiation system. FSH receptor (FSHR) and aromatase are markers of granulosa cells. As shown in Fig. 2A, Fshr and aromatase were expressed at D18 and D24 of differentiation. The expression of these two granulosa cell markers was significantly increased in the presence of Matrigel, although their expression levels were still 3–20-fold lower than that of ovarian cells. ICC also revealed aromatase-positive cells after induced differentiation (Fig. 2B) with the percentage of aromatase-positive cells significantly higher in the Matrigel-treated group (Fig. 2C). In an ovarian follicle, the production of estradiol requires the theca cell to generate androgen, which is then transported to granulosa cell and aromatized there to become estradiol. Estradiol was detected in our induced differentiation culture, and its level is significantly increased in a time-dependent manner (Fig. 2D), further suggesting that functional ovarian follicular somatic cells were also generated in our differentiation system. The decreased estradiol levels at D21 and D24, compared with D15 and D18, of differentiation is possibly the result of the degradation of estradiol in the spent media by factors or enzymes from the prolonged in vitro differentiation system.
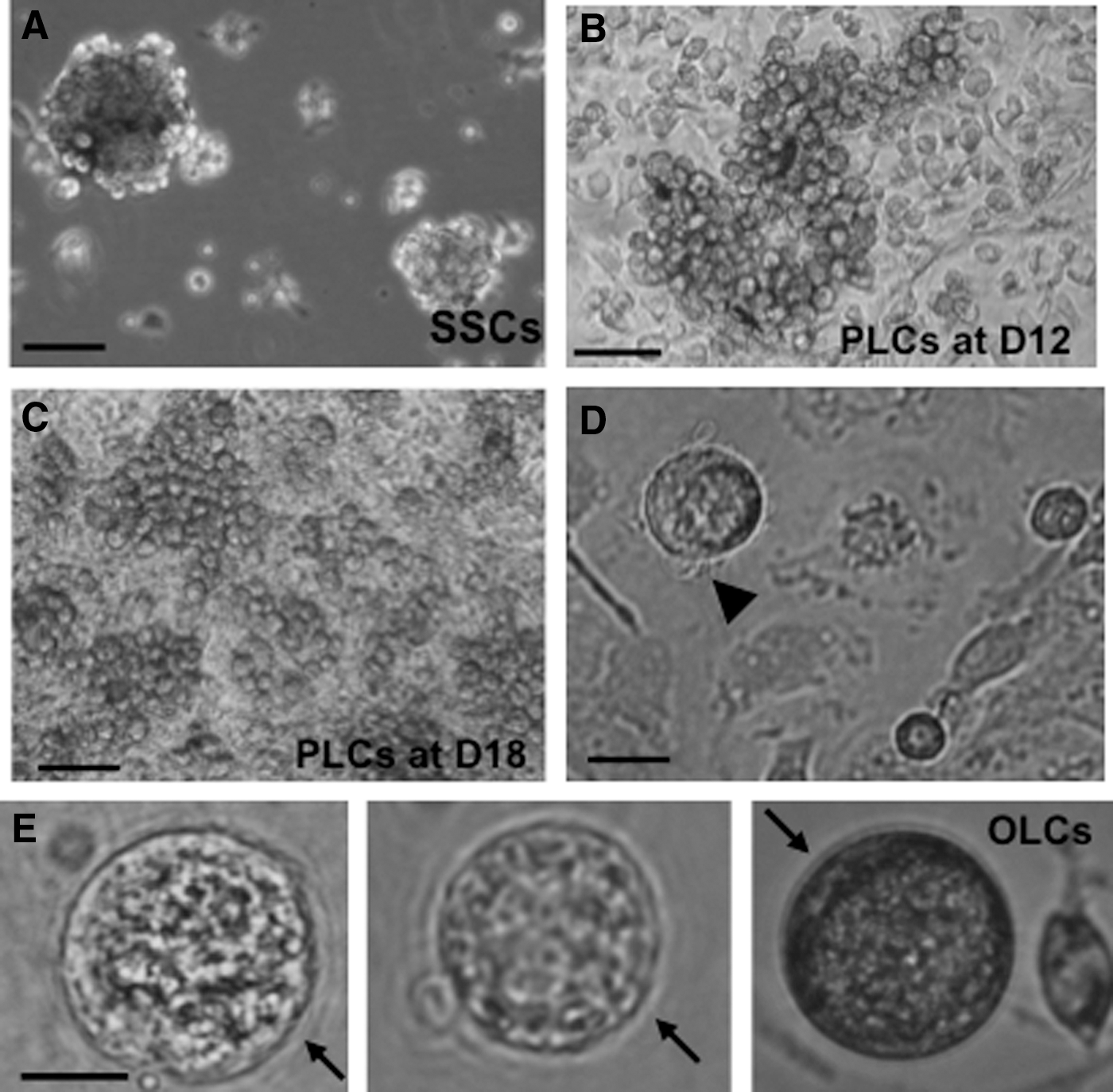
Differentiation of germ-cell-like cells from mouse skin-derived stem cells (SSCs). SSCs were differentiated into PGC-like cells (PLCs) for 12 days (D12) in media containing pig follicular fluid, and then cultured with Matrigel for an additional 6 days (D18).
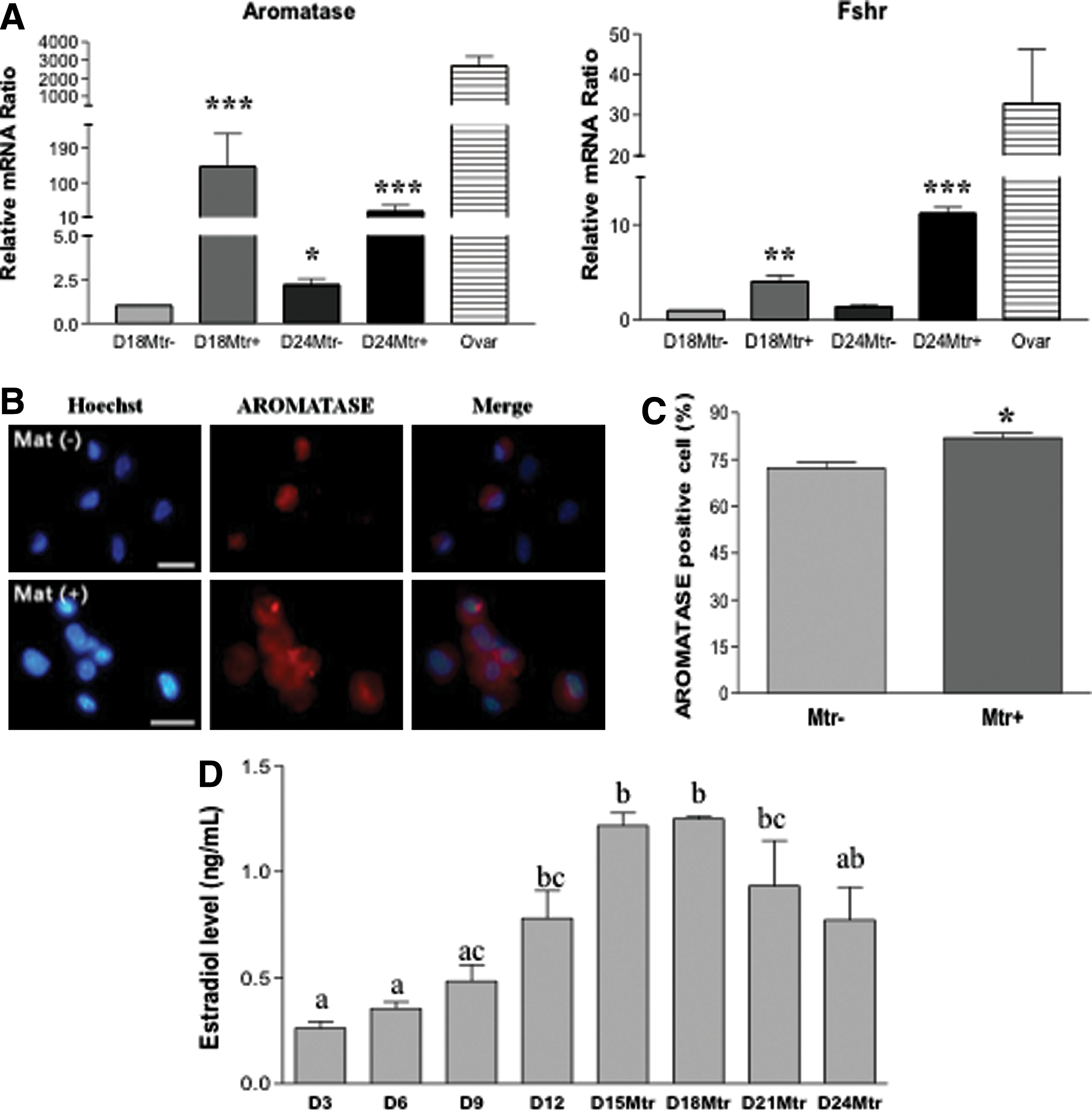
Matrigel enhances the expression of germ cell markers during SSC differentiation to ovarian-cell-like cells. SSCs were differentiated into PLCs and OLCs until D12 before being cultured with or without Matrigel (Mtr) to D18 or D24.
We next investigated whether the mixture of these ovarian-cell-like cells can produce estradiol following transplantation into ovariectomized mice, a widely used postmenopausal model [25,26]. After induced differentiation and culture with Matrigel, these ovarian-cell-like cells were transplanted (in some of the experiments, cells were labeled with PKH26 before transplantation) into ovariectomized mice. Mice receiving induced ovarian-cell-like cell transplant are denoted as Cell Trans. The control mice had bilateral ovariectomies without cell transplantation (OVX), while the sham operation mice underwent the same surgical procedures as the OVX group, but their ovaries were not removed (Sham OP). Estrus cycles were determined via vaginal smear analysis 8 weeks after surgery. It was revealed that the estrus cycle of all the OVX mice was arrested at the metestrus/diestrus stage (Fig. 3A), whereas 13 of the 19 Cell Trans animals (68%) showed more than one estrus cycle between 8 and 16 weeks after in vivo transplantation of differentiated ovarian-cell-like cells (Fig. 3A, B). Additionally, those Cell Trans mice in which estrus cycling was restored displayed increased vaginal patency and coloration during estrus and proestrus (data not shown). The responding Cell Trans mice showed dynamic cycling, although this was not regular and varied by mouse.
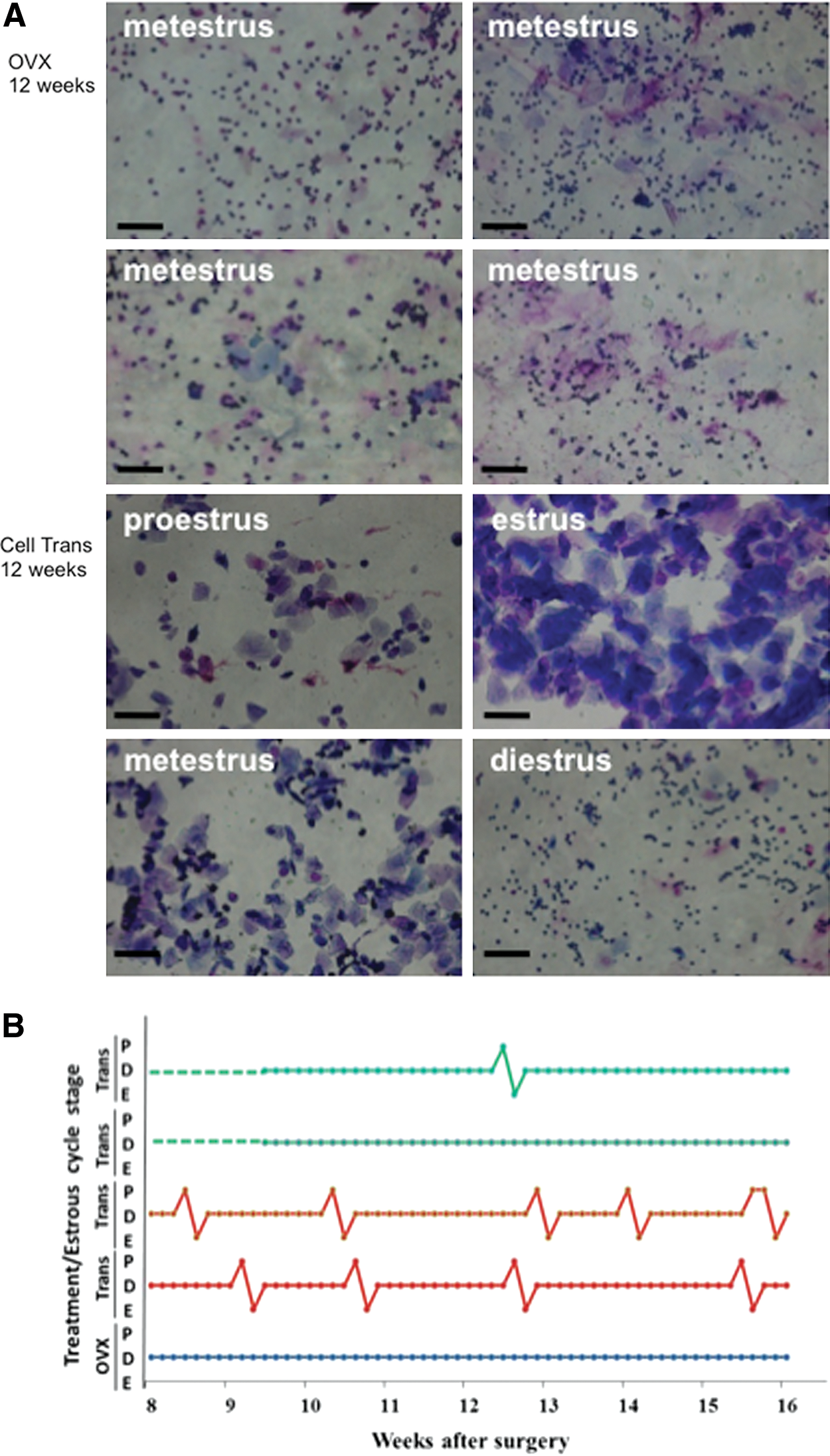
Ovarian-cell-like cell transplantation restored estrus cycling in ovarectomized mice. Vaginal cytology was analyzed between weeks 8 and 16 after surgery.
Twelve to fourteen weeks after surgery, serum estradiol levels were assayed via ELISA, when cycling mice were in the proestrus/estrus stage. As shown in Fig. 4A, serum estradiol in the OVX group decreased to 31% of that of the Sham OP group (59.0±4 pg/mL vs. 188.9±29 pg/mL; P<0.05). In contrast, the estradiol level in the Cell Trans mice remained almost the same (178.0±35 pg/mL) as that of the Sham OP mice. Patients with premature ovarian failure have increased levels of FSH in the blood due to the depletion of growing follicles within the ovary [27]. To study whether the ovarian-cell-like cell transplants have any influence on pituitary gonadotropin levels, serum FSH levels were also analyzed. FSH was 0.39±0.2 ng/mL in the Sham OP group; its level was significantly increased in OVX mice (1.62±0.14 ng/mL; P<0.05). Cell transplantation resulted in a decreased serum FSH concentration (0.94±0.1 ng/mL; Cell Trans group) compared with the OVX mice (Fig. 4B) that was similar to that of the Sham OP group. Twelve to fourteen weeks after surgery, some experimental mice were euthanized and a severely atrophic uterus was observed in the OVX group (Fig. 4C), with a significantly lower weight (48.3±3.5 mg) compared with that of the Sham OP group (156.0±6.6 mg; P<0.05). In Cell Trans mice, uterus morphology was similar to that of Sham OP mice in their thickness, color, and texture. Moreover, the uterine weight (126.7±12.5 mg) was higher than that of the OVX mice, and comparable to the uterus weight of Sham OP mice (Fig. 4C, D). RT-qPCR revealed a fourfold increase in expression of estrogen receptor (ER) in the uterus of OVX mice compared with that of the Sham OP group (P<0.05). The expression of ER in the Cell Trans mice was comparable to that of the Sham OP mice (Fig. 4E).
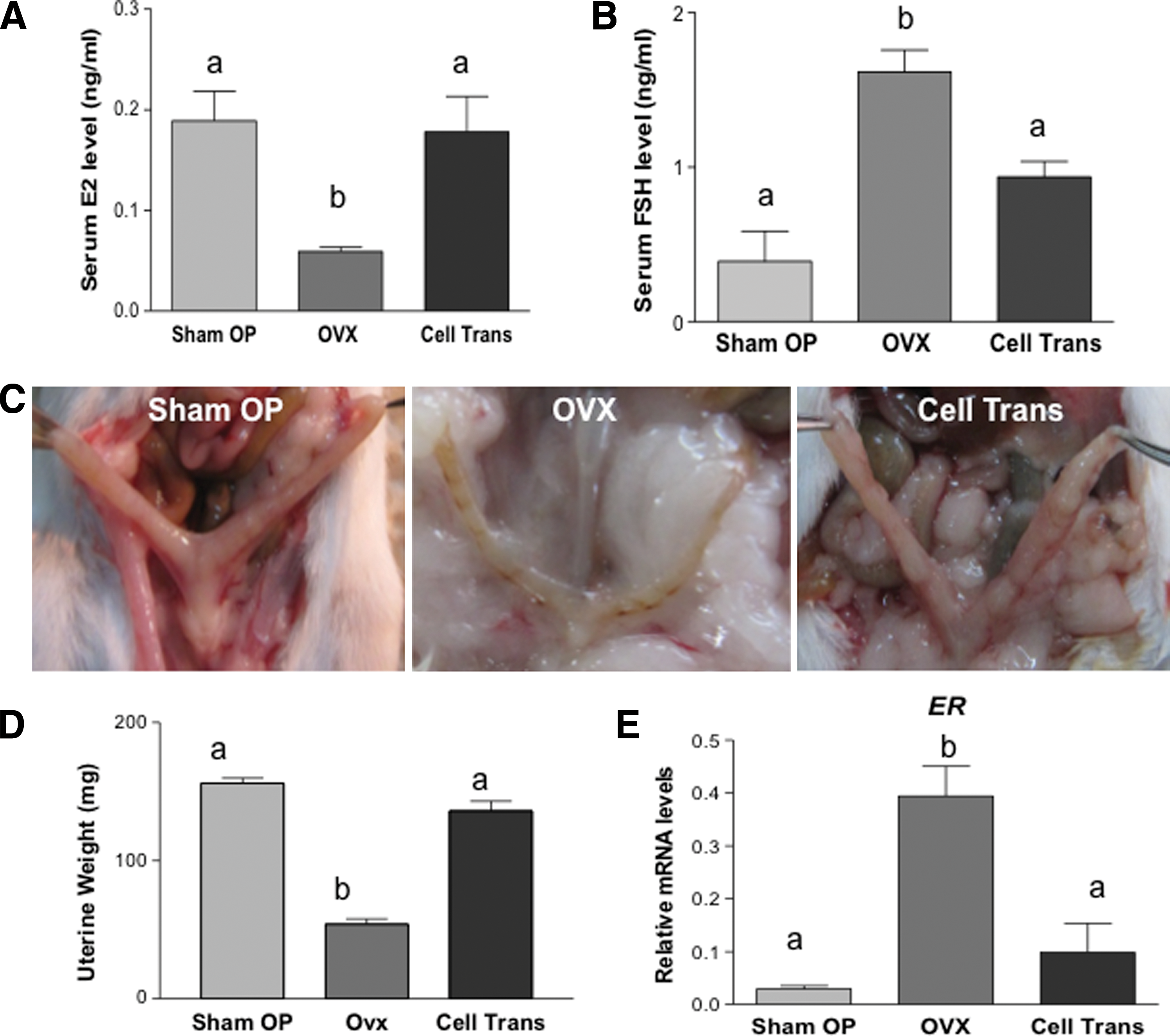
Influence of an ovarian-cell-like cell transplantation on serum estradiol (E2), follicle-stimulating hormone (FSH), uterus morphology, weight, and estrogen receptor (ER) expression.
At 12–14 weeks of transplantation, transplants were recovered in the kidney capsule fat or under the kidney capsule where they were originally transplanted. Figure 5 shows histological sections of the transplants. Increased blood vessel formation was also observed around or within the transplants (Fig. 5A–C). PKH26-positive (red fluorescent) cells were detected in the transplants (Fig. 5D–F), and some of them formed ring-like structures (Fig. 5D–F), similar to an ovarian follicle. PKH26-positive cells were detected in the transplanted kidney fat, suggesting the formation of follicle-like structures from our transplanted cells (Fig. 5E). The number of PKH26-positive cells detected is likely an under-representation of the actual transplanted cells as some of the transplanted cells may have lost their fluorescent label after 3 months in vivo.
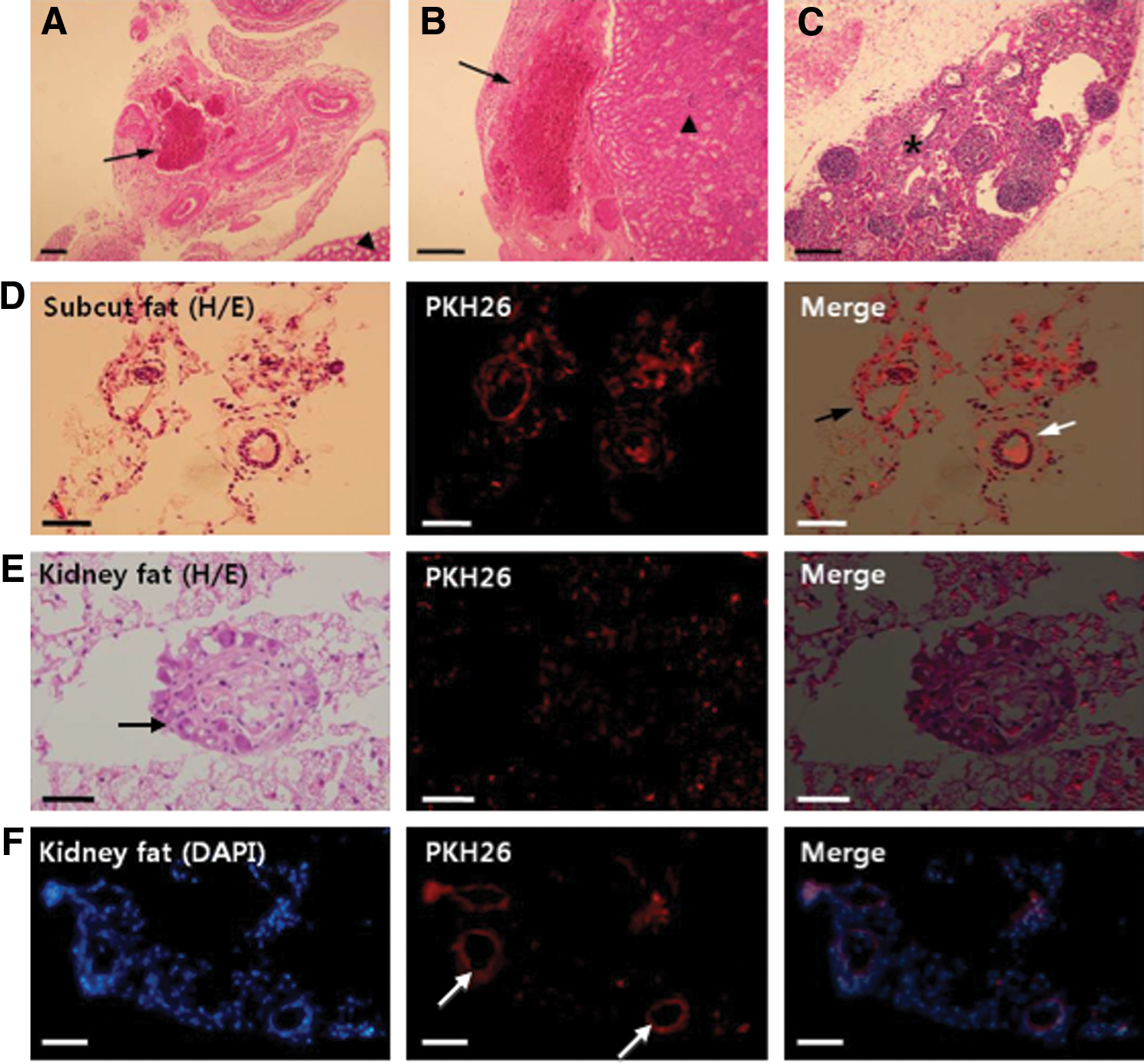
Transplanted ovarian-cell-like cells formed blood vessels and follicle-like structures in vivo.
Immunohistochemical analysis was performed to detect FSHR expression in the cell-transplanted tissues. FSHR is a marker of granulosa cells. FSHR-positive cells were detected in the transplant under the kidney capsule (Fig. 6A), and in the subcutaneous fat (Fig. 6B). Especially strong expression of FSHR was observed in some round follicle-like structures in transplants within the kidney fat (Fig. 6C). Figure 6D shows a subpopulation of FSHR-expressing cells in a cell cyst that were positive for PKH26, indicating they were transplanted cells. Figure 6E is non-cell-transplanted control fat around the kidney and only very weak signal was detected, likely reflecting the background of the assay. Figure 6F shows an FSHR-positive kidney capsule section in which cells were transplanted. Immunohistochemistry of the consecutive sections with antibodies against DAZL and FSHR (Fig. 6G, H, respectively) revealed that a small, distinct population of DAZL-positive cells was present among the FSHR-positive cells. This data suggests that in vivo, both germ cells and somatic cells are present in the transplant.
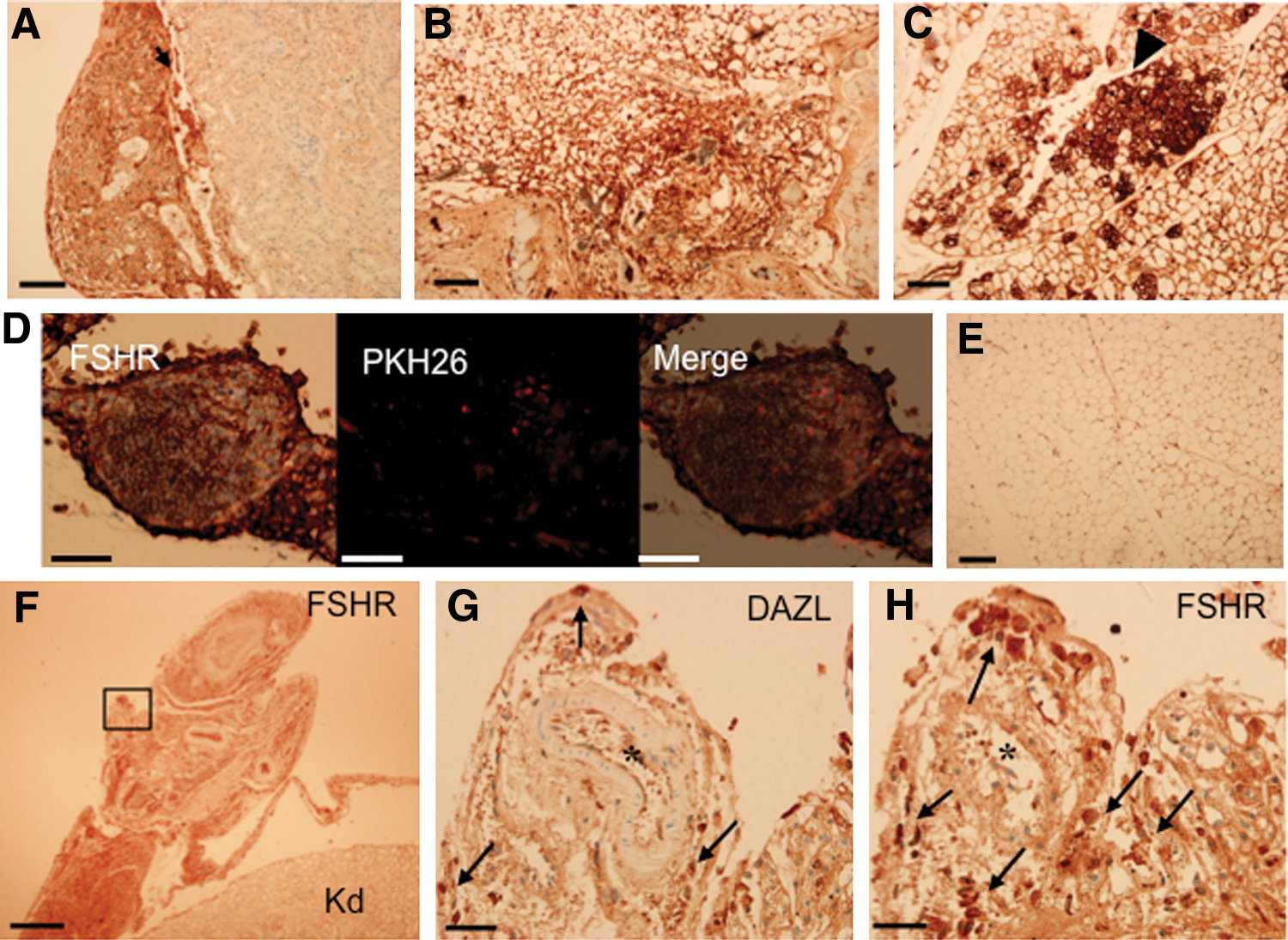
FSH receptor (FSHR) and DAZL are expressed in the cells transplanted into fat and the kidney capsule as detected by immunohistochemistry. Transplants under the kidney capsule
Discussion
We report on the in vivo function of ovarian granulosa- and theca-like cells differentiated from SSCs. Our conclusion is based on our findings that transplantation of these cells into ovariectomized mice resulted in the (1) restoration of serum estradiol level, (2) suppression of serum FSH levels similar to that of mice with intact ovaries, (3) resumption of estrus cycling, and (4) prevention of uterus atrophy following ovariectomy.
Premature ovarian failure or premature menopause is a primary ovarian disorder characterized by premature depletion of ovarian follicles before the age of 40 years, affecting ∼1% of women of this age group. In addition to infertility, the associated endocrine disorder may cause metabolic, cardiovascular, or neurological disorders [27,28]. Due to the marked decline or absence of ovarian follicles, decrease of circulating estradiol levels, and increase of serum FSH levels (reviewed in Goswami and Conway [29]). The rise in FSH concentration is a consequence of the failure to suppress pituitary FSH secretion via negative feedback due to low levels of ovarian steroids and peptides [30]. This hormone profile (low estradiol and high FSH) is well mirrored in ovariectomized mice [31], as in our current investigation. Transplantation of stem-cell-derived ovarian-cell-like cells into ovariectomized mice reversed the FSH and estradiol changes in these mice. These data suggest that the transplanted cells exhibit endocrine functions of ovarian follicular cells, and contributed to the changes in circulating estradiol. They were thus able to communicate with the pituitary gland to regulate FSH levels. Our finding that follicle-like structures in the transplants were often surrounded by blood vessels is consistent with this notion of their endocrine function.
Uterus hypotrophy in ovarectomized mice was reported previously, where the wet weight of the uterus of 3-month-old mice decreased to ∼50 mg compared with that of a sham control that was ∼150 mg [32], a range that is very similar to our findings in this current study (Sham OP 156.0 mg vs. OVX 48.3 mg). The same study also showed that administration of estradiol at a concentration of 5 μg/(kg·day−1) for 32 days resulted in uterus hypertrophy (mean uterus weight increased to 175–190 mg) [32]. It is worth noting that no uterus hypertrophy was observed in our ovarian-cell-transplanted mice even four months after transplantation. This is possibly due to the fact that the estradiol supply in our Cell Trans mice is more “physiological” in a sense that it is not released in a constant manner, as would be expected of an implanted therapeutic estradiol-release pellet. Ovarian-cell-like cells differentiated from porcine skin stem cells have been reported to respond to FSH by increasing estradiol production [8]. Our current study showed FSHR expression in the granulosa-like cells both in vitro and in vivo. It is tempting to speculate that the transplanted ovarian-cell-like cells secreted estradiol into circulation in response to the stimulation by serum FSH, and that this elevated serum estradiol then suppressed FSH production in the pituitary gland, similar to pituitary-gonadal axis endocrine communication [33]. Consistent with this hypothesis, the elevated FSH level in OVX mice was decreased after cell transplantation, and near physiological estrus cycling was restored in two-thirds of the Cell Tran mice as determined by changes in hormone levels, vaginal smear cytology, and external genitalia. However, estrus cycle progression in the Cell Trans mice was not perfectly normal, with only a few complete cycles observed in the 60 days of monitoring (Fig. 3B). In addition, no cycling was observed in most Cell Tran mice more than 5 months after transplantation (data not shown). The former reflects the notion that the transplant is not functional to the extent of a real ovary, and the latter is most likely due to exhaustion of OLCs, and thus follicles, in the transplant after an extended period of time in vivo. This is consistent with the findings that the follicle-like structures were atypical in the transplants recovered 12–14 weeks after surgery (Figs. 5 and 6), and almost no transplants were recovered after 20 weeks (data not shown). It is likely that transplanted ovarian-like cells degenerated or were reabsorbed after an extended period of time in vivo.
We have previously reported the generation of OLCs from postnatal mouse SSCs when differentiated in a defined medium [12]. With this differentiation system, estradiol was not detected in the spent media, and aromatase was not expressed (our unpublished data), suggesting that granulosa and theca cells were not fully induced. In the current study a differentiation media that included 5% follicular fluid and 5% fetal bovine serum, which was previously shown to stimulate follicle-like structure formation and estradiol production [8], was employed. This resulted in both aromatase expression and estradiol production. The addition of Matrigel to the system further increased the level of aromatase expression and estradiol production, suggesting that the extracellular matrix mixture may have enhanced the cell–cell communication in our differentiation system containing germ-cell- and granulosa-like cells, as well as enhancing granulosa function. This finding agrees with previously reported results where the aggregation of cells together with a biomaterial supported the development of follicles from the preantral to antral stage, responsiveness to FSH stimulation, and secretion of estradiol [34,35]. Although follicular fluid was used to induce differentiation of stem cells into ovarian-cell-like cells, the possibility that nonovarian cells are also present in our differentiated cell population cannot be excluded. Transplanting SSCs that had been intentionally differentiated into other cell types could shed light on what effect these cells may have on restoring ovarian hormonal function and what influence the ovary-deficient in vivo environment may exert on these cells. The mechanisms behind the generation and differentiation of ovarian granulosa and theca cells from skin stem cells have yet to be revealed.
Primordial follicle formation in the developing ovary is generally thought to be coordinated by growth factors in a paracrine and autocrine manner. Ovarian factors, such as kit ligand, leukemia inhibitory factor, bone morphogenic proteins, keratinocyte growth factor, and basic fibroblast growth factor (bFGF), are known to promote the primordial to primary follicle transition (reviewed in Skinner [36]). Follicular fluid contains factors that are secreted from granulosa cells, theca cells, and oocytes within the follicles. Many growth factors, such as growth differentiation factor (GDF) 9, GDF9b, stem cell factor (SCF), and bFGF, are known to be present in follicular fluid. SCF is known to facilitate PGC survival and proliferation [37,38], and enhances the interaction between PGCs and gonadal somatic cells [38]. Whether and how these factors direct the skin stem cells to become ovarian cell is currently unclear. Interestingly, previous studies indicate that the development of the ovarian follicle is dependent on machinery intrinsic to the oocyte [13,14]. It was suggested that some oocyte-specific factors may be responsible for folliculogenesis. Factor in the germline alpha (FIGα), a germ-cell-specific transcription factor, may play an important role in the formation of early stage of follicle [39], and we have previously found FIGα to be expressed in OLCs differentiated from mouse skin stem cells [12]. Further, although we used only female SSCs as a proof-of-concept study, it has been previously demonstrated that both XY and XX mouse ES cells have the ability to form oocytes [10]. Whether SSCs from male mice have a similar potential for generating ovarian-cell-like cells warrants future investigation.
Young patients with premature ovarian failure, who develop the disease before a woman achieves peak adult bone mass, sustain a sex steroid deficiency for considerably more years than naturally menopausal women. This may cause a significantly higher risk for osteoporosis and cardiovascular disease [40]. Therapies for the relief of symptoms and for decreasing the associated disease risks are currently based on hormone replacement, particularly estradiol [41]. A WHO study published in 2002, however, showed an increased risk of breast cancer, stroke, and heart disease associated with the use of estradiol-based hormone therapy and this has led many women to discontinue or choose not to receive treatment, although hormone-based therapy is still the most effective therapy available to date [42]. Thus, a more comprehensive and safe means for ovarian failure control is needed. It is known that the negative feedback mechanisms needed to regulate FSH production by estradiol are maintained for years after the onset of menopause [43,44]. Previous work using ovariectomized mice as a model to study the effect of ovarian transplantation has shown that pituitary-ovarian communication can be re-established after the transplant [45]. In a recent study, the ovaries of recipient mice were removed early in life and, at 11 months of age, the mice underwent ovarian transplantation with 60-day-old ovaries. It was found that these recipients were able to start cycling again, and their life span was enhanced compared with control mice with their original ovaries intact. These results indicate that ovarian transplants can not only maintain ovarian endocrine function, but could also increase longevity [46]. Our study is the first attempt to investigate the feasibility of restoring estradiol production in an ovariectomized animal. Before answering the question of whether ovarian-cell-like cells differentiated from stem cells could be a means of tissue therapy for endogenous estradiol production to alleviate symptoms of ovarian hypofunction, many further studies must be done. These include the following: (1) identifying factors responsible for inducing granulosa cell differentiation in the current follicular fluid containing media so a more defined media can be used, (2) specifically selecting differentiated cells for transplants to avoid potential tumor formation, and (3) testing for potential systemic side effects after transplantation. The feasibility of differentiating stem cells obtained from the skin of postnatal animals has been shown in the current study; further experiments will be needed to demonstrate that this potential exists for human SSCs.
Footnotes
Acknowledgments
The authors would like to thank Dr. Arthur Leader from the Ottawa Fertility Center, University of Ottawa, for helpful discussion of the project.
Acknowledgement of funding: Canada Institutes for Health Research (CIHR); National Science and Research Engineering Council (NSERC); the Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA); and National Research Foundation of Korea grant (NRF-2012R1A1A4A01009879).
Author Disclosure Statement
No competing financial interests exist.