
Abstract
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and is closely linked with autism. The genetic basis of FXS is an expansion of CGG repeats in the 5′-untranslated region of the FMR1 gene on the X chromosome leading to the loss of expression of the fragile X mental retardation protein (FMRP). The cause of FXS has been known for over 20 years, yet the full molecular and cellular consequences of this mutation remain unclear. Although mouse and fly models have provided significant understanding of this disorder and its effects on the central nervous system, insight from human studies is limited. We have created human induced pluripotent stem cell (iPSC) lines from fibroblasts obtained from individuals with FXS to enable in vitro modeling of the human disease. Three young boys with FXS who came from a well-characterized cohort representative of the range of affectedness typical for the syndrome were recruited to aid in linking cellular and behavioral phenotypes. The FMR1 mutation is preserved during the reprogramming of patient fibroblasts to iPSCs. Mosaicism of the CGG repeat length in one of the patient's fibroblasts allowed for the generation of isogenic lines with differing CGG repeat lengths from the same patient. FXS forebrain neurons were differentiated from these iPSCs and display defective neurite initiation and extension. These cells provide a well-characterized resource to examine potential neuronal deficits caused by FXS as well as the function of FMRP in human neurons.
Introduction
F
FXS is caused by a trinucleotide repeat expansion in a single gene, FMR1, resulting in lack of the fragile X mental retardation protein (FMRP) [4,5]. Expansion of a CGG triplet repeat in the 5′ untranslated region of the FMR1 gene leads to hypermethylation of the FMR1 promoter and subsequent transcriptional silencing of the gene and thus loss of expression of FMRP [3 –7]. FMRP is expressed in many tissues, but is most abundant in neurons of the brain and in the testes [8 –11]. FMRP is an RNA binding protein that binds to specific mRNAs to control the location and protein translation of these mRNAs, which play a key role in neuronal synaptic plasticity [12,13]. About 4% of mRNAs in the brain are bound by FMRP [13], and over 400 potential mRNA targets have been identified in mouse brain [14,15], many of which are implicated in autism [16,17].
Efforts to understand the effect of FMRP loss on brain development and function have been aided by animal models. Because FXS is caused by the lack of a single, well-conserved [4,5] gene product, FMRP-knockout models [18 –24] in the mouse [25], fly [19,21,22,24], and zebrafish [23] have existed for some time. These models exhibit many of the phenotypes typical of the human syndrome [18 –25] and thus have been valuable to the field. Yet, animal models created by knockout of the FMR1 gene cannot recapitulate the mechanism of FMR1 silencing nor the potential regulatory mechanisms that may be in play during neural development [26]. Therefore, to fully represent the FXS phenotype, the human mutation needs to be studied in the context of human neural development. Though human studies have traditionally been difficult due to a lack of available tissue, human pluripotent stem cells enable FXS disease modeling. Human embryonic stem cells (hESCs) that carry the FXS mutation have been generated from embryos shown to carry the FMR1 gene mutation by preimplantation genetic diagnosis [27,28]. Induced pluripotent stem cell (iPSC) technology has also enabled the generation of pluripotent stem cell lines to model FXS [27,29]. In contrast to hESCs, iPSCs are generated by forced expression of pluripotency genes in adult somatic cells, most commonly skin fibroblasts [30,31]. Here we describe the establishment of iPSCs from three young boys with FXS. Fibroblasts were obtained from skin biopsies from these individuals and reprogrammed using retroviral vectors. The FXS mutation is preserved during the reprogramming process from fibroblasts from these individuals to iPSCs. Further, we report the differentiation of these cells into forebrain neurons that enable insight into the properties of human FXS neurons. As a proof of principle, we show that these human FXS neurons exhibit neurite outgrowth deficits.
Materials and Methods
Fibroblasts from FXS and control subjects
Skin biopsies were obtained from selected patients at the Waisman Center at the University of Wisconsin-Madison per Institutional Review Board–approved human subject protocols. Fibroblasts were isolated from skin biopsies.
Reprogramming
Fibroblasts were reprogrammed to iPSCs according to previously published methods [30]. Clones from each cell line were chosen based on morphology, growth, and pluripotency for continued analysis. Karyotype analysis (G-banding) was carried out at the WiCell Research Institute Cytogenetics Lab, following standard protocols.
Cell culture and neuronal differentiation
iPSCs were maintained on mouse embryonic feeder layers according to established protocols. iPSCs were differentiated according to our previously established methods [32 –34]. After separation from feeder cells and maintenance in suspension culture for 7 days with dual-SMAD inhibition for the first 3 days [35], aggregates of iPSCs were differentiated to primitive neuroepithelial aggregates (NEAs) in an adherent culture in neural induction medium (NIM) consisting of DMEM/F12 (Life Technologies, Carlsbad, CA), N2 supplement (1:100; Life Technologies), and heparin (2 μg/mL; Sigma, St. Louis, MO). NEAs were mechanically detached at days 14–16, and cultured in suspension as neurospheres in NIM with B27 supplement (1:100; Life Technologies). For neuronal differentiation, neurospheres were plated or dissociated and plated on poly-ornithine/laminin-coated coverslips (Sigma/Life Technologies) in neural differentiation medium containing DMEM/F12, N2 (1:100), B27 (1:100), 10 ng/mL BDNF (Peprotech, Rocky Hill, NJ), 10 ng/mL GDNF (R&D Systems, Minneapolis, MN), cAMP (Sigma), and ascorbic acid (Sigma) for an additional 2–4 weeks. For three-germ-layer analysis, aggregates of iPSCs lifted from feeder cells were maintained in hESC media without FGF2 for 15–30 days.
RT–polymerase chain reaction and quantitative reverse transcription–polymerase chain reaction
RNA was isolated from cells using the E.Z.N.A. total RNA Kit I (Omega Bio-Tek, Norcross, GA) and reverse transcribed to cDNA using the qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). Polymerase chain reactions (PCRs) containing 2XGoTaq Green Master Mix (Promega, Madison, WI), forward and reverse primers, 5 ng cDNA, and water were amplified using a G-Storm thermocycler. PCR products were resolved on a 2% agarose gel and visualized using ethidium bromide (Promega) under UV light. Quantitative PCRs were performed in triplicate with SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) and run on an Applied Biosystems 7500 Real-Time PCR System. Beta-actin was used to normalize gene expression between runs and cell lines unless otherwise listed. Analysis of results was performed using the comparative CT method to determine fold change for a given primer [36].
Forward and reverse primers used for specific genes:
Beta-actin: GCGAGAAGATGACCCAGATC CCAGTG GTACGGCCAGAGG
cMyc-endo: CGGGCGGGCACTTTG GGAGAGTCGC GTCCTTGCT
cMyc-exo: GGGTGGACCATCCTCTAGAC CCTCGTC GCAGTAGAAATAC
FMR1: GCAGATTCCATTTCATGATGTCA ACCAC CAACAGCAAGGCTCT
KLF4-endo: AGCCTAAATGATGGTGCTTGGT TTGA AAACTTTGGCTTCCTTGTT
KLF4-exo: GGGTGGACCATCCTCTAGAC GGAAGT CGCTTCATGTGG
OCT4-endo: AGTTTGTGCCAGGGTTTTTG ACTTCA CCTTCCCTCCAACC
OCT4-exo: GGGTGGACCATCCTCTAGAC CCAGGT CCGAGGATCAAC
SOX2-endo: CAAAAATGGCCATGCAGGTT AGTTG GGATCGAACAAAAGCTATT
SOX2-exo: GGGTGGACCATCCTCTAGAC GGGCTG TTTTTCTGGTTG
AFP: AGCTTGGTGGTGGATGAAAC CCCTCTTCAG CAAAGCAGAC
ACTA2: CAGGGCTGTTTTCCCATCCAT GCCATGT TCTATCGGGTACTTC
NCAM: ATGGAAACTCTATTAAAGTGAACCTG; TA GACCTCATACTCAGCATTCCAGT
OCT4: CGAGCAATTTGCCAAGCTCCTGAA; TTCGG GCACTGCAGGAACAAATTC
Immunoblot
Cells were harvested in lysis buffer [20 mM Tris (pH 8), 137 mM NaCl, 1% NP-40, 10% glycerol, and protease inhibitors] and cleared by centrifugation. Protein extracts were size fractionated with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Bio-Rad, Hercules CA) and immunoblotted with antibodies to FMRP and actin. Proteins were visualized by incubation with horseradish-peroxidase-coupled secondary antibodies (Bio-Rad) and enhanced chemiluminescence (GE Healthcare Bio-Sciences, Piscataway, NJ).
Repeat length assay
The number of FMR1 CGG repeats was determined for all samples using a PCR-based protocol previously described [37]. The protocol combined gene-specific primers that flank the CGG repeat region of the FMR1 gene with gender-specific primers, a polymerase mixture, and a reaction buffer that is optimized for amplification of GC-rich DNA. PCR was performed on an ABI Veriti thermal cycler (Applied Biosystems, Grand Island, NY). Samples were subsequently denatured at 93°C for 30 s before undergoing capillary electrophoresis on an ABI 3730xl DNA Analyzer with POP-7® polymer using a 50-cm array. The PCR products were also verified on agarose gel electrophoresis. Capillary electrophoresis is capable of defining exact CGG repeat number on the samples with 200 CGG repeats or less. The CGG repeat number was estimated by comparing to DNA sizing ladder visualized on agarose gel electrophoresis.
Methylation assay
Methylation patterns of the fibroblast and iPSC lines were analyzed at 22 CpG sites in the FMR1 promoter region by EpigenDx (Hopkinton, MA).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde (Fisher Scientific, Waltham, MA) in phosphate-buffered saline (PBS) for 10–30 min. Cells were permeabilized and blocked for 30 min in 5% normal goat or donkey serum and 0.2% Triton X-100 and incubated in primary antibody (Table 1) and 5% serum at 4°C overnight. Cells were washed and incubated with Alexa Fluor (Life Technologies) secondary antibodies in 5% serum for 30 min before being washed and stained with Hoechst for 5 min and mounted to glass slides with Fluoromount-G mounting media (Southern Biotech, Birmingham, AL).
FMRP, fragile X mental retardation protein; IB, immunoblot; IF, immunofluorescence.
Neurite outgrowth assays
Forebrain neurospheres after 20 days of differentiation from iPSCs were used for neurite outgrowth analysis. Neurospheres were plated onto acid-washed coverslips coated with 50 μg/mL poly-
Results
Generation of iPSCs from clinically defined FXS individuals
Fibroblasts were derived from skin biopsies obtained from three boys diagnosed with FXS and one apparently healthy individual at the Waisman Center at the University of Wisconsin-Madison (Fig. 1A). The boys with FXS were recruited from a large ongoing project on language development in children with FXS. Boys in the language development study were recruited nationally and eligibility criteria included the following: (1) between 4 and 10 years of age at enrollment, (2) English was the primary language spoken in the child's home, (3) spoken language was the child's primary means of communication, and (4) the child lacked any significant uncorrected motor or sensory impairments, with all criteria assessed through parent report. The boys all also entered the language development study with positive results on molecular genetic testing for FXS. Finally, standardized testing conducted as part of the language development study, which included administration of a nonverbal test of intelligence, confirmed that the boys in the study were functioning in the IQ range expected for FXS, with most meeting IQ criteria for intellectual disability. The C603 control line was derived from a healthy male (Fig. 1A).
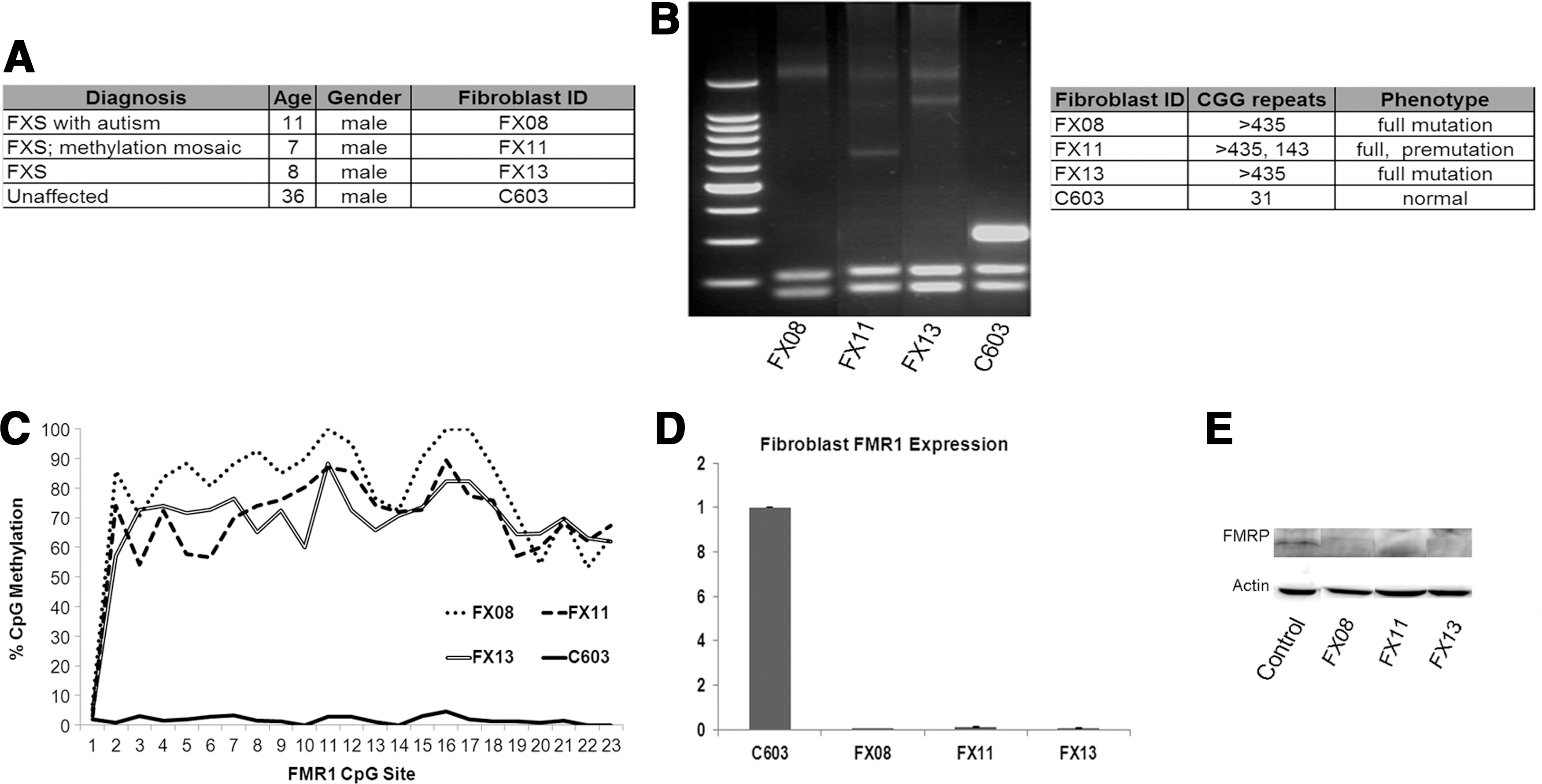
Fibroblasts from fragile X syndrome (FXS) individuals.
We first confirmed that fibroblasts from FXS individuals had the characteristic expansion of a CGG triplet repeat in the 5′ untranslated region of the FMR1 gene and hypermethylation of the FMR1 promoter (Fig. 1B, C). Using a CGG repeat analysis assay [39], we show that fibroblasts derived from all three FXS individuals had CGG repeat lengths >435, well above the 200-repeat threshold required for promoter methylation [3 –7]. Control cells (C603) had CGG repeat length in the normal range. Interestingly, the FX11 fibroblasts had two different repeat lengths for FMR1: one that fell in the full-mutation range and one in the premutation range. Thus, this individual was mosaic for FXS, meaning that there are different sizes of the repeat expansion in different cells [40]. Assessment of CpG methylation in the FMR1 promoter showed that all FXS fibroblasts had methylated FMR1 promoters, while control cells did not (Fig. 1C). The cells from the mosaic fibroblasts (FX11) have equal methylation compared with full-mutation cells, perhaps indicating a higher representation of full-mutation cells in this individual's cell population.
Methylation of the FMR1 promoter leads to transcriptional silencing of the FMR1 gene and loss of FMRP. Evaluation of FMR1 transcript level in the fibroblasts by quantitative PCR confirms that full-mutation FXS fibroblasts FX08 and FX13 lack FMR1 expression, consistent with the FXS diagnosis (Fig. 1D). The FX11 line shows an intermediate expression of FMR1, consistent with a mosaic FXS mutation (Fig. 1D). Immunoblot for FMRP confirms that the FXS lines do not express FMRP whereas the controls have robust FMRP expression (Fig. 1E).
The characterized patient-derived fibroblasts were reprogrammed to iPSCs using retroviral vectors for OCT4, SOX2, KLF4, and cMYC [30]. The resulting cells were analyzed for the expression of endogenous transcription factors associated with pluripotency—OCT4, SOX2, KLF4, and cMYC—and silencing of exogenous genes (Supplementary Fig. S1A; Supplementary Data are available online at
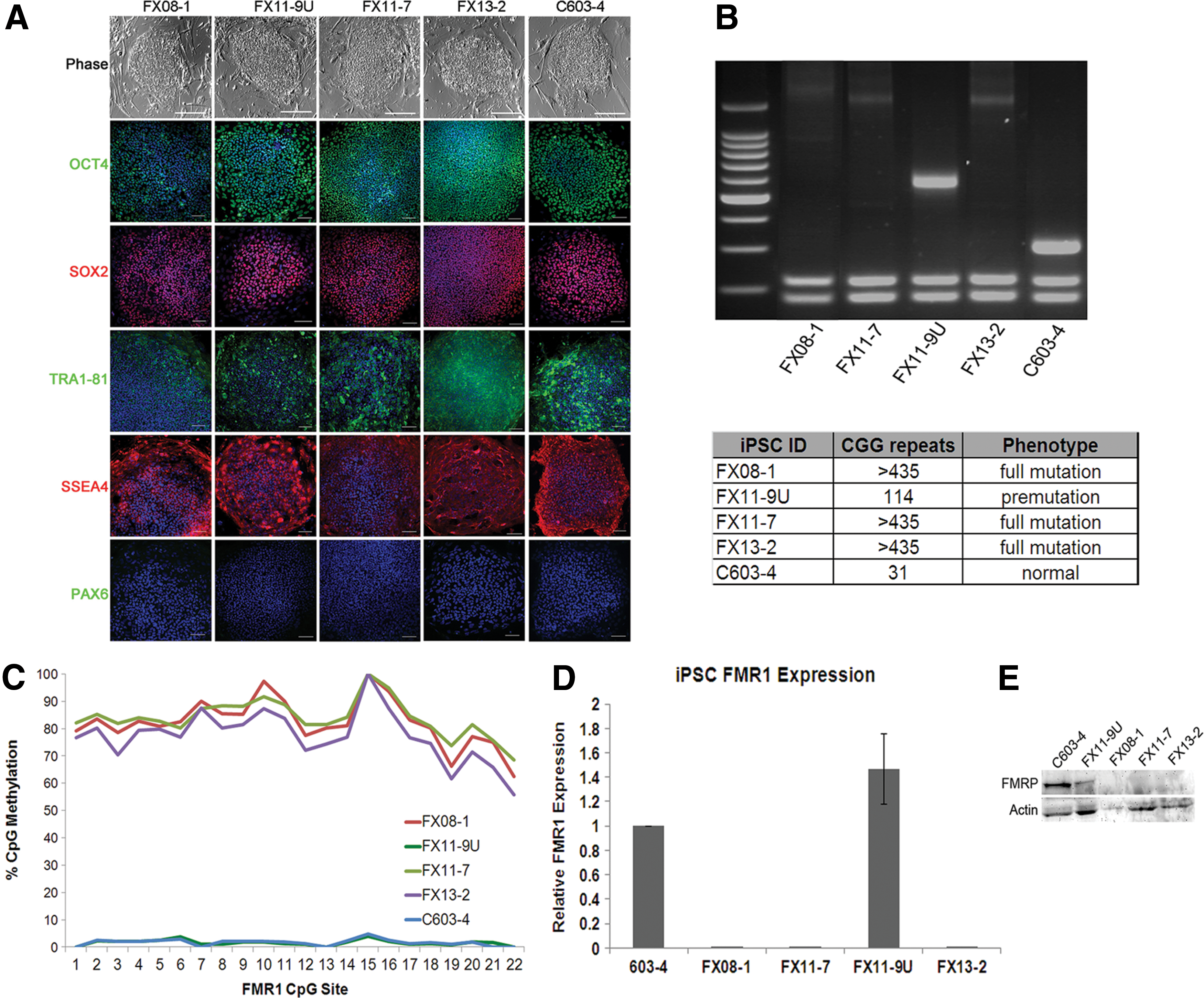
FXS induced pluripotent stem cells (iPSCs) preserve characteristic genetic and epigenetic marks of FMR1 silencing. iPSCs were generated from individual fibroblast lines.
CGG repeat length analysis revealed that reprogramming of control cells resulted in iPSCs that retained the CGG repeat length that was defined in the parent fibroblasts (Fig. 2B). Reprogramming of full-mutation FXS fibroblasts (FX13 and FX08) yielded iPSCs that retained the full-mutation repeat length (>435) in all cells (Fig. 2B). Reprogramming of the mosaic FX11 fibroblasts resulted in the generation of two isogenic lines: one with full-mutation CGG repeats (FX11-7, >435 repeats), and another with <200 repeats (FX11-9U, 114 repeats) (Fig. 2B). One clone from each fibroblast cell line was chosen for further characterization (FX08-1, FX13-2, and C603-4). It is interesting that, unlike the control cells, the exact repeat length in premutation cells was not perfectly preserved upon reprogramming. This result may reflect the instability of CGG repeats in the premutation range. CpG methylation in the FMR1 promoter in all cells was consistent with expectations based on repeat length and fibroblast results. CpG sites in the full-mutation iPSCs (FX08-1, FX11-7, and FX13-2) had high levels of methylation (>75%); the control line (C603-4) and premutation (FX11-9U) line had almost no methylated CpG sites in the FMR1 promoter region (<2%) (Fig. 2C). Evaluation of FMR1 transcript level and FMRP protein level in the resultant iPSCs confirms that lines FX08-1, FX11-7, and FX13-2 lack FMR1 and FMRP expression consistent with the FXS diagnosis, whereas the C603-4 lines show normal levels of FMR1 and FMRP expression (Fig. 2D, E). The premutation FX11-9U line expressed more FMR1 transcript, consistent with previous reports [41], although the results were not statistically significant. Taken together, these results indicate that the FMR1 gene mutation is preserved during reprogramming in these iPSCs.
FXS iPSCs differentiated into human forebrain neurons
Although FMRP is expressed in many cell types, it is concentrated in neurons and the intellectual disability associated with FXS supports the idea that neuronal function is most sensitive to FMRP loss. Structural abnormalities, as well as cognitive and behavioral abnormalities associated with FXS, suggest the critical role of FMRP in the forebrain. In fact, the highest levels of FMR1 mRNA are found in developing neocortical structures in human [42]. Therefore, the generation of human FXS forebrain neurons will enable insight into the properties of human FXS neurons and their development. iPSCs from FXS individuals and controls were differentiated into forebrain neurons according to previously published methods [32 –34]. This procedure takes advantage of developmental principles to generate neural progenitor cells with forebrain identity, unless patterned to other neuronal subtypes [43]. Neurons differentiated from control and FXS iPSCs for 5–6 weeks in culture were identified by βIII-tubulin immunofluorescence (Fig. 3). Immunofluorescence for FMRP confirmed that neurons differentiated from control iPSCs express FMRP while FMRP is absent in FXS-iPSC-derived neurons (Fig. 3A). To confirm that these neurons were forebrain neurons, the cells were immunostained for the forebrain-specific transcription factor FOXG1. Both control and FXS neurons express FOXG1, confirming that the neurons are of forebrain identity (Fig. 3B). Taken together, these results show that forebrain neurons can be generated from FXS iPSCs that are comparable to controls.
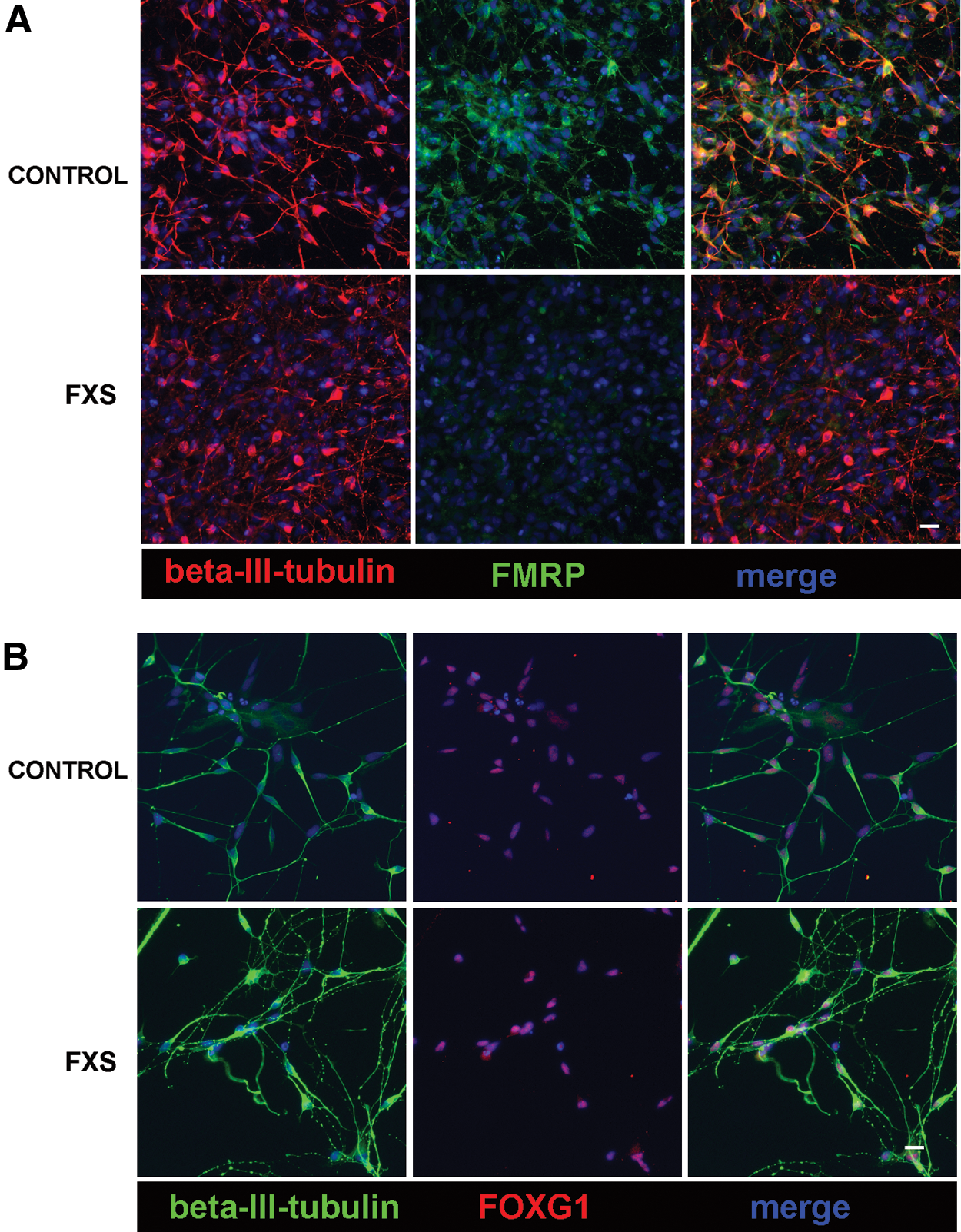
FXS and control iPSCs generate forebrain neurons in vitro.
FXS-iPSC-derived forebrain neurons exhibit neurite outgrowth defects
Studies from FMR1-knockout animals have shown that neurite length and branching is reduced when FMRP is absent [44 –46]. However, there is conflicting in vitro data on whether human FXS neurons have neurite outgrowth defects [29,47 –49]. All these reports, including our own, rely on the description of morphological characteristics, which can vary based on cell source, neuronal subtype, culture conditions, and method of analyses. To resolve this discrepancy, we evaluated our FXS and control iPSC-derived neurons during the initial steps of neurite initiation and outgrowth using live cell imaging and analysis. In developing mouse hippocampal neurons, FMRP is localized to growth cones [44], suggesting that FMRP regulates growth cone motility and axon guidance [44 –46]. To investigate whether FMRP plays a similar role in human neurons, we first analyzed neurite growth defects in fixed forebrain neurosphere explants after 2 days in vitro. Forebrain spheres from control and FXS iPSCs were fixed and immunolabeled for neural-specific βIII-tubulin together with F-actin. Within 24–48 h in culture, forebrain neurons from control iPSCs extend a profuse array of processes tipped by growth cones. Remarkably, neurite outgrowth by forebrain neurons from two independent lines of FXS spheres was dramatically reduced as compared with control forebrain neurons (Fig. 4A, B). We compared both the number and length of neurites that emerge from forebrain spheres and found that FXS forebrain spheres extend significantly fewer processes that are significantly shorter relative to control neurons. Interestingly, βIII-tubulin-positive neurons in the FXS forebrain spheres appear to be more motile, as they often migrate away from the sphere, unlike control neurons, which typically stay aggregated within the sphere. These results suggest that FXS neurons may have defects in both neurite initiation and extension. We further probed for possible neurite growth defects using live cell time-lapse imaging of individual growth cones from control and FXS forebrain spheres (Fig. 4C). Although developing control forebrain neurites are tipped by dynamic growth cones that exhibit robust forward translocation, growth cones from FXS forebrain neurons exhibit restricted motility and a slow rate of neurite extension (Fig. 4D). Together, these data suggest that neurite initiation and extension are defective in human forebrain neurons derived from FXS iPSCs.
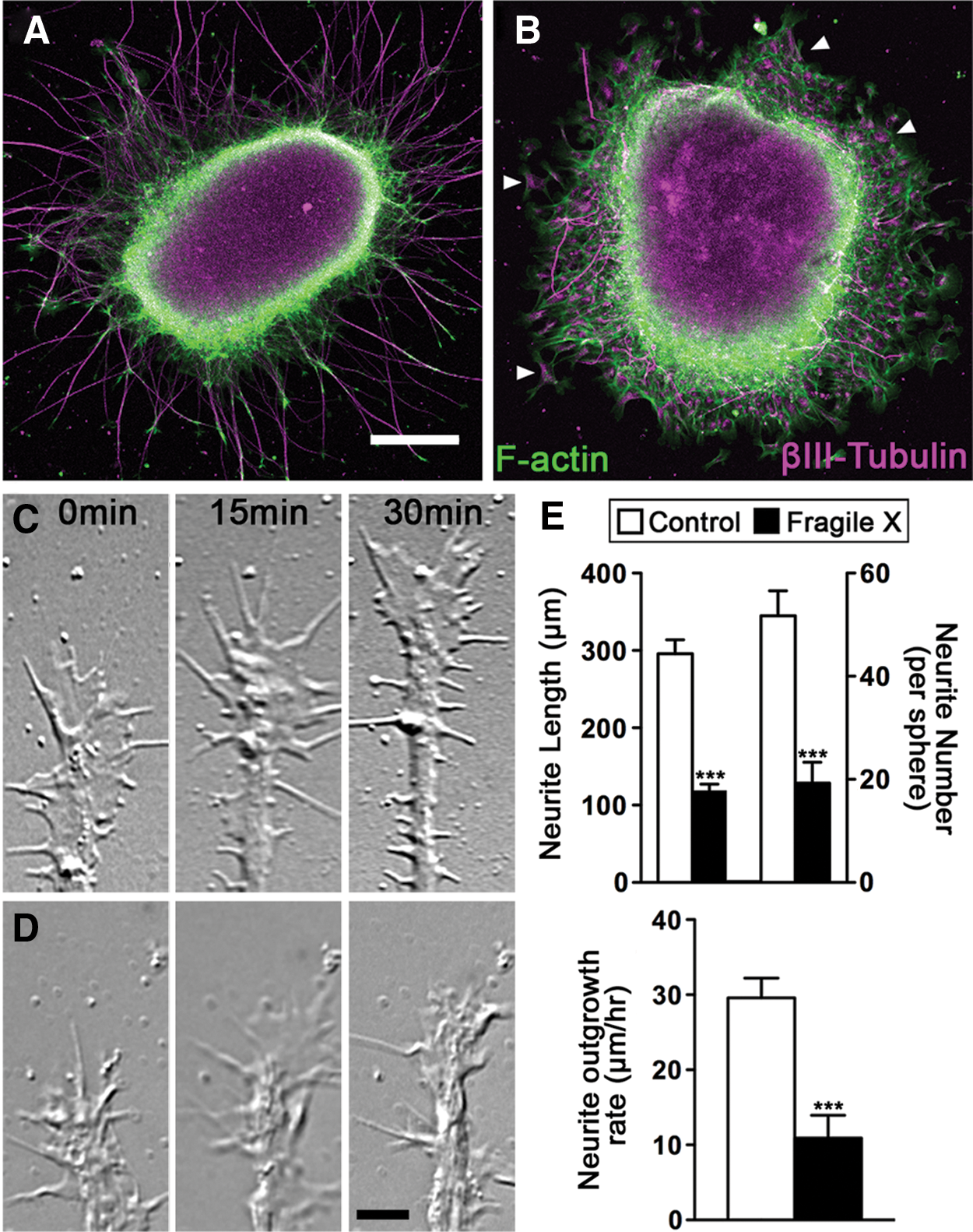
Human FXS forebrain neurons have reduced neurite initiation and outgrowth. Low-magnification images of human forebrain neurospheres from control
Discussion
Fragile X human pluripotent stem cells
Species-specific differences in molecular and neurodevelopmental aspects of FXS necessitate the need for a human FXS model. While human studies have traditionally been hampered due to a lack of available tissue, pluripotent stem cells enable disease modeling. Both hESCs and iPSCs have been reported [27 –29].
hESCs that carry the FXS mutation have been generated from male embryos shown by preimplantation genetic diagnosis to have methylated full-mutation length repeats in the FMR1 gene [28]. FXS hESCs maintain the naive epigenetic pattern of the FMR1 gene and the gene does not become methylated and silenced until the pluripotent cells differentiate [27]. However, derivation of cells from embryos precludes the behavioral characterization of the subjects. In addition to the ethical issues these cells raise, the cells have limited accessibility to many researchers as there are relatively few cell lines in existence and fewer are on the NIH human stem cell registry. Thus, working with these cells is difficult for most researchers interested in studying the underlying mechanisms of FXS.
iPSC technology has enabled the generation of pluripotent stem cell lines from patients with disease-causing mutations to model diseases [30,31,50]. iPSCs can be generated from individuals with known biological and behavioral characteristics, thereby potentially providing a link between behavior and biology. These cells do not have ethical issues beyond standard human subject considerations and can theoretically be made from hundreds of individuals.
FXS iPSCs have been generated from banked fibroblasts from FXS individuals [29]. While banked fibroblasts provide a useful source of cells, the severity of behavioral problems of individuals, including autism status, from which the fibroblasts were obtained, is often not known. For many disorders, including FXS, where the behavior of FXS individuals varies, this uncertainty can lead to an inability to find consistent differences between sets of diseased and healthy pluripotent stem cell lines. Variability between patient phenotypes can obscure robust differences between FXS and controls as well as identify differences that are not relevant to all patients. In addition, banked fibroblasts have generally been passaged numerous times that may hinder reprogramming, differentiation, or the mutation. In fact, the FMR1 repeat lengths have been shown to change upon reprogramming from these cells [29]. These problems can be avoided by using patient-derived cells.
Potential drawbacks in iPSC studies must be kept in mind. Several factors introduce variability and affect the ability to compare data from multiple studies, including patient differences, iPSC reprogramming methods, and neuronal differentiation paradigms. Inherent genetic variation among individuals due to genetic diversity, variability of iPSC clones from a single individual, as well as disease presentation presents major challenges to iPSC disease modeling [51 –53]. Epigenetic and copy number diversity adds another layer of complexity [54] that may plague iPSCs to a greater extent than other samples. iPSC reprogramming methods have evolved from integrating retrovirus to defined chemicals and nonintegrating plasmids, yet little data exist to suggest that there are major differences in iPSCs and their differentiated progeny based on the reprogramming method.
The data presented here address shortcomings in current FXS pluripotent stem cell research by minimizing both patient phenotype variability and cell line variability. First, we present iPSCs generated from three boys who were drawn from a large, nationally recruited cohort displaying the range of behavioral impairments typical for individuals with FXS, thereby potentially providing a link between behavior and biology. Second, we minimize cell line variability through isogenic lines. The isogenic lines generated from the FXS mosaic fibroblasts allow for a set of lines that vary genetically only at the FMR1 promoter region, thus minimizing confounding random variability between cell lines. Finally, these iPSCs preserve the FMR1 mutation found in parent somatic cells. As the field of disease modeling using iPSCs progresses, it is becoming clear that having appropriately defined and robust sets of disease-specific lines and controls is essential for accurate and reproducible comparisons. Differentiation of these cells into functional forebrain neurons will provide an invaluable tool to examine potential neuronal deficits caused by FXS as well as the function of FMRP in human cells.
FMR1 repeat length and silencing
The causal mutation in FXS is a trinucleotide CGG repeat expansion in the FMR1 gene. When the mutational expansion of the CGG repeats exceeds 200, it leads to methylation of the repeats and the FMR1 promoter, chromatin condensation, and a loss of FMRP protein expression. The FXS iPSCs that we present preserve the full-mutation CGG repeat lengths and consequent methylation and silencing of the parental somatic cells. All patient-derived cells in this study had CGG repeat lengths >435, well above the 200-repeat threshold required for promoter methylation [3 –7]. The mechanism of FMR1 methylation and silencing in relation to repeat length (and specifically the 200-repeat threshold) is not well understood. Further, the presence of interspersing AGG within the CGG repeats, which were not assessed in our study, has an effect on CGG repeat stability and may also affect epigenetic silencing [55,56].
FXS hESCs and iPSCs differ in the epigenetic state of the FMR1 gene. In FXS hESCs with full-mutation repeats, the FMR1 gene is unmethylated and expressed in the pluripotent stage and only become methylated and silenced upon cell differentiation [27]. In contrast, FXS iPSCs do not return to the naive epigenetic state found in FXS hESCs. The FMR1 gene remains methylated and silenced during reprogramming [27,29]. These data suggest that the mechanism of FMR1 silencing may be more similar to X-inactivated genes than dynamic pluripotency genes. iPSCs with the methylated FMR1 can be exploited to address the mechanism of FMR1 silencing and for gene reactivation strategies. Because the FMRP coding sequence is normal, one potential therapeutic strategy for FXS is to demethylate the gene promoter and restore expression of FMRP. In fact, there are men with FMR1 full-length CGG expansion mutations who show no or only mild symptoms because their CGG and FMR1 gene are unmethylated [57 –60]. Animal models, particularly mouse models, cannot address the mechanism of FMR1 silencing because they are generated by knocking out the FMR1 gene and analogous CGG repeats in these animals do not cause epigenetic modifications or transcriptional silencing of the gene [26].
Human FXS neurons
The ability to generate neurons from disease-specific iPSC lines ideally allows for examination of both neuronal development and function of mature neurons. The role of FMRP in human neuronal development and function has not been well defined, hindering the translation of mouse discoveries into human therapies.
FMRP binds to and traffics specific mRNAs to control the timing and location of local protein translation in developing neurons and during synaptic plasticity [12]. Loss of FMRP function leads to elevated translation of a number of mRNAs in animal model neurons. Moreover, in developing mouse hippocampal neurons, FMRP is localized to growth cones [44 –46]. We show that the process of neurite outgrowth is altered in human FXS neurons, confirming that FMRP regulates growth cone motility and axon guidance in human neurons and sets the foundation to investigate how lack of FMRP changes the guidance of axonal growth cones to their targets during development and plasticity.
Functional studies on human FXS neurons are needed to elucidate a clear mechanistic role for FMRP in the processes, including neurotransmission and synaptic plasticity, which are thought to be the primary deficits in FXS. Further, FXS can be used as a platform to understand the molecular and cellular defects that occur in a defining form of autism. Such an endeavor requires subtype-specific neurons and functional maturation of neurons. The first step is to differentiate PSCs into specific neuronal subtypes affected in FXS. Lack of FMRP likely affects different subtypes of neurons differently, indicating that the neuronal subtype being analyzed is important. Structural abnormalities as well as cognitive and behavioral abnormalities associated with FXS suggest a critical role of FMRP in the forebrain. We differentiated FXS iPSCs, which like hESCs are inherently primed toward the neural lineage, by exposure to minimally supportive media that is sufficient to allow differentiation along a “default” program to primitive neural progenitor cells and on to become dorsal forebrain cortical-like neurons [43,61,62]. Alternatively, hESCs and iPSCs can be directed to ectoderm by inhibition of the Smad pathway using inhibitors of the transforming growth factor beta and activin/nodal signaling [35]. These neuronal populations typically include robust numbers of excitatory glutamatergic and inhibitory GABAergic cells [61]. Recent reports suggest that a combination of retinoic acid and dual Smad inhibition can enhance glutamatergic projection neuron differentiation [63].
Analysis of neurotransmission defects in FXS requires maturation of human neurons to a point at which they possess mature dendritic spines and mature electrophysiological activity reminiscent of mature neurons. An increased density of immature, long, and thin neuronal dendritic spines is consistently found in the brains of FXS patients [64 –66], providing clues to defective synaptic transmission in FXS. However, functional maturation of pluripotent-stem-cell-derived neurons has proven to be a challenge in the PSC field. With these iPSCs in hand, we are now poised to address these challenges to uncover numerous roles of FMRP in human brain development and function.
Footnotes
Acknowledgments
The authors thank Xinyu Zhao and members of the Waisman Center Fragile X Focus Group for helpful discussions. We thank Xiaoqing Zhang, Jianfeng Lu, and Yingnan Yin in the Waisman Center iPSC core for reprogramming services. We also thank Jason P. Weick for assistance with neuronal differentiation and Anne Antkins and Rebecca Reese for technical assistance. The project was supported by grant UL1TR000427 to the University of Wisconsin Institute for Clinical and Translational Research by the National Institutes of Health National Center for Advancing Translational Sciences (NCATS) (A.B.), a FRAXA Research Foundation Grant (A.B.), NIH R01 HD054764 (L.A.) and NIH R01 NS41564 (T.M.G.). This work was also funded in part by a Heckrodt Family Foundation Grant to the Waisman Center and a core grant from the NIH-NICHD (P30 HD03352).
Author Disclosure Statement
Leonard Abbeduto has received financial support to develop and implement outcome measures for Fragile X syndrome clinical trials from F. Hoffman-LaRoche, Ltd, Roche TCRC, Inc., and Neuren Pharmaceuticals Limited.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.