
Abstract
In mammals, the permanence of many forms of hearing loss is the result of the inner ear's inability to replace lost sensory hair cells. Here, we apply a differentiation strategy to guide human embryonic stem cells (hESCs) into cells of the otic lineage using chemically defined attached-substrate conditions. The generation of human otic progenitor cells was dependent on fibroblast growth factor (FGF) signaling, and protracted culture led to the upregulation of markers indicative of differentiated inner ear sensory epithelia. Using a transgenic ESC reporter line based on a murine Atoh1 enhancer, we show that differentiated hair cell-like cells express multiple hair cell markers simultaneously. Hair cell-like cells displayed protrusions reminiscent of stereociliary bundles, but failed to fully mature into cells with typical hair cell cytoarchitecture. We conclude that optimized defined conditions can be used in vitro to attain otic progenitor specification and sensory cell differentiation.
Introduction
A
It has previously been shown that murine embryonic stem cells (ESCs) are capable of differentiating toward the otic lineage in vitro [1 –3]. All these strategies are based on the generation of the non-neural ectoderm from ESCs, which is promoted by the suppression of endo- and mesodermal lineages [2,3]. This leads to presumptive preplacodal cells competent of responding to otic-inducing fibroblast growth factor (FGF) signals with upregulation of early otic lineage markers, which reflects the in vivo situation [4,5]. ESC-derived otic precursors are thought to attain a commitment toward the otic lineage that enables differentiation into major inner ear cell types, including hair cells and supporting cells [2]. Commitment of progenitors present in the native inner ear primordium, also known as the otocyst, is in agreement with cell grafting studies in chicken embryos [6 –8]. The concept of otic lineage commitment of murine ESC-derived otic progenitor cells has been elegantly demonstrated by the ability of self-guided differentiation of these cells when cultured in a three-dimensional (3D) system [3].
The first reports of otic guidance with monolayer cultured human ESCs (hESCs) revealed a propensity to differentiate along an otic neurogenic lineage, giving rise to neurons with ability to functionally reinnervate cochlear hair cells in a gerbil model of auditory neuropathy [9,10]. Although cells generated with a monolayer strategy expressed hair cell makers, they only displayed a rudimentary resemblance to sensory hair cells [9].
In this study, we present an embryoid body (EB)-based guidance protocol for generation of human otic progenitor cells in defined culture conditions. We further show that self-guided differentiation of human otic progenitor cells in protracted cell cultures leads to generation of hair cell-like cells that display many features of nascent hair cells, but fail to mature into bona fide hair cells. Our experiments reveal the potential as well as the limitations of current culture methods for the human otic lineage.
Materials and Methods
Cells
An institutional stem cell research oversight committee of the Stanford institutional review board approved the human stem cell research conducted in this study. Besides overseeing scientific and ethical considerations, the approval involves verification that the research complied with the United States, State of California, and the California Institute for Regenerative Medicine guidelines and regulations.
Human H9 ESCs, passage 40–67, were maintained on mitomycin C-treated or irradiated mouse embryonic fibroblasts (MEF) in knockout DMEM/F12 supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin, 1× nonessential amino acid solution, 2 mM
Otic induction and cell differentiation
EBs were cultured in ultralow attachment surface plates in the hESC medium supplemented with 100 ng/mL recombinant human Dickkopf-related protein 1 (DKK-1; R&D Systems), specific inhibitor of Smad3 (SIS3) at 3 μM (Sigma), and IGF1 at 10 ng/mL (Sigma). Half of the medium was replaced every day. On day 15, the EBs were transferred into poly-
On day 21, the medium was replaced with the advanced DMEM/F12 supplemented with 15% KSR, N2, and B27, and ampicillin (50 μg/mL). The concentration of KSR was reduced to 10% on day 27 and to 5% on day 33. SU5402 (a gift from Pfizer R&D) was used at 10 μM for control samples from day 15 to 21.
Quantitative RT-PCR
Total RNA was isolated (RNeasy Plus Micro Kit; Qiagen) and 5 ng was used per sample for reverse transcription (High Capacity Reverse Transcription Kit; Applied Biosystems). Quantitative RT-PCR (qRT-PCR) was performed with the CFX96 Touch™ Real-Time PCR Detection System (BioRad) using the SsoFast EvaGreen Master Mix (BioRad). qRT-PCR results presented are mean values for Supplementary Fig. S1a (Supplementary Data are available online at
Immunocytochemistry
The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature. Nonspecific binding sites were blocked for 1 h in 0.2% Triton X-100 and 1% bovine serum albumin in PBS. Cells were incubated overnight at 4°C with diluted antibodies: 1:200 polyclonal rabbit antibody to PAX2 (Covance), 1:50 polyclonal goat antibody to PAX8 (Santa Cruz), 1:50 polyclonal goat antibody to DLX5 (Santa Cruz), 1:1,000 polyclonal guinea pig antibody to MyosinVIIA, 1:500 polyclonal rabbit antibody to MyosinVIIA (Proteus Biosciences), 1:1,000 polyclonal rabbit antibody to espin (ESPN), 1:100 polyclonal rabbit antibody to p27Kip1 (NeoMarkers). 1:1,000 polyclonal chicken antibody to GFP (Abcam), 1:200 polyclonal goat antibody to SOX2 (Santa Cruz Biotechnology), and 1:200 monoclonal rabbit antibody to OCT4 (Life Technologies). The FITC-, TRITC-, and Cy5-conjugated species and subtype-specific secondary antibodies were used to detect primary antibodies. Nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI) or SYTO60 red fluorescent nucleic acid stain. Images were acquired using a Zeiss Axioimager LSM 5 Exciter epifluorescence/confocal microscope. Cell counting was done by analysis of five representative areas containing between 500 and 3,000 cells each per data point using the ImageJ particle count function, as well as manual confirmation of the software's accuracy.
Scanning electron microscopy
The cells were fixed for 4 h with 2% glutaraldehyde/4% paraformaldehyde with 50 mM CaCl2 and 20 mM MgCl2 in 0.1 M HEPES buffer (pH=7.4), postfixed with 1% aqueous OsO4, dehydrated in a graded ethanol series, and dried by critical point drying with liquid CO2 (Autosamdri-815; Tousimis). Specimens were sputter coated with 100Å Au/Pd using a Denton Desk II Sputter Coater and viewed with a Hitachi S-3400N variable pressure SEM operated under high vacuum at 5–10 kV at a working distance of 7–10 mm. Chemicals were supplied by Electron Microscopy Sciences.
Generation of H9 ATOH1-nGFP hESCs
A 2,566 bp Atoh1 reporter DNA fragment consisting of the murine Atoh1 enhancer placed upstream of the human β-globin basal promoter followed by the enhanced green fluorescent protein equipped with an amino-terminal nuclear localization signal was PCR amplified from J2XnGFP plasmid DNA (a gift from Dr. Jane Johnson, UT Southwestern). Gateway attB1 and attB2 sequences were added to the 5′ and 3′ primers to generate an amplicon compatible with the gateway cloning system (BP Clonase II and pDONR221 vector; Life Technologies). Further recombination into pJTI-CHS4R4 DEST (Life Technologies) resulted in a plasmid carrying the attB-flanked Atoh1 reporter fragment and a hygromycin resistance gene with the thymidine kinase promoter. This plasmid was co-electroporated with a phiC31 integrase expression plasmid into pseudo-attP sites of H9 hESCs [11] using program B-16 of the Amaxa Biosystems nucleofector in 100 μL of nucleofector solution 1 (Lonza). Transfected cells were selected on hygromycin-resistant MEFs (Millipore) in the presence of the ROCK inhibitor (Millipore). Colonies were picked, subcloned, and ultimately led to three independent H9 ATOH1-nGFP hESC lines. The continued expression of pluripotency markers OCT4, NANOG, Tra-1-80, and SOX2 was verified and one of the three H9 ATOH1-nGFP lines was used for all the experiments.
Flow cytometry
H9 and H9 ATOH1-nGFP hESCs were differentiated until day 42 and dissociated after a 40-min incubation in the Accutase solution (Innovative Cell Technologies), filtered with a 70-μm strainer (BD Biosciences), and incubated with 50 μg/mL propidium iodide (Sigma) for dead cell labeling. Sorting was conducted with an ARIA II flow cytometer (Becton Dickinson). Debris, doublets, and cell clumps were excluded with two consecutive gating steps (forward-scatter height vs. forward-scatter area, side-scatter area vs. side-scatter width), followed by rejection of propidium iodide-positive cells. Cells were deposited individually into 96-well plates.
Single cell qRT-PCR
RNA was extracted, directly reverse transcribed (Superscript III RT; Life Technologies), and preamplified for 20 cycles with Platinum Taq polymerase (CellsDirect One-Step qRT-PCR kit; Life Technologies) using amplicon-specific DELTAgene Assays (Fluidigm). For qRT-PCR, the exonuclease (NEB)-treated samples were diluted 5× and analyzed on 96.96 Dynamic Array Integrated Microfluidic Circuits using SsoFast EvaGreen Supermix with Low ROX (Bio-Rad) on a Biomark HD (Fluidigm) multiplex qRT-PCR system. Log2Ex values were calculated for each sample for each gene, where Log2Ex is equal to the Ct value of the gene subtracted from the limit of detection (LoD) Ct value for the set of assays (LoD-Ct=22). The higher the Log2Ex value, the higher the expression level. Only cells that produced Ct values above the LoD-Ct threshold for both reference genes (ACTB, GAPDH) were included in the analysis. Amplicon-specific assay primers (DELTAgene Assays) were validated with adult human utricle cDNA and human fetal cDNA (Clontech).
Statistical analysis
Data are presented as mean value±standard deviation (SD) with the number of independent experiments (n) indicated. Statistical differences were determined with paired one-tailed t-tests using Aabel (Gigawiz) or Excel (Microsoft). P values 0.01 to 0.05 are indicated with *, 0.001 to 0.01 with **, and <0.001 with ***. Graphs were created using Aabel (Gigawiz).
Results
Systematic screening for guidance conditions leading to upregulation of early otic markers in hESC-derived cultures
The transcriptional regulators PAX2, PAX8, and DLX5 are markers for the otic lineage in vivo and have been utilized in previous stem cell guidance experiments [1,2,5,9]. Although not specific individually, coexpression of these genes has been valued as a strong indication for early otic lineage identity [2,3,9]. We used PAX2 expression as a primary indicator for potential generation of early otic progenitors from hESCs. We began with generating EBs and maintaining them in conditions aimed to suppress the formation of endo- and mesodermal lineages for periods ranging from 5 to 30 days. This was done through inhibition of WNT and TGFβ signaling and promotion of the cranial ectoderm with IGF, which is similar to our previous murine ESC otic guidance protocol [2]. We verified that adding the WNT inhibitor DKK-1 and the TGFβ-signaling inhibitor SIS3 is sufficient for suppressing mesodermal and endodermal differentiation during EB formation (Supplementary Fig. S1a). The generation of presumptive cranial ectoderm was followed by a 3–20-day otic induction phase during which, we systematically tested the activation and inhibition of FGF-, BMP-, Notch-, and WNT-signaling pathways. The highest number of PAX2 expressing cells, identified immunocytochemically, occurred after 15–20 days of EB formation followed by a 6-day period of inductive FGF signaling, divided into an initial 3-day period of WNT activation with R-spondin1 and BMP inhibition with Noggin, followed by 3 days of BMP4 treatment (Supplementary Fig. S1b and Supplementary Table S2). These experiments led us to devise a differentiation protocol consisting of an initial 15-day period to generate a presumptive non-neural/preplacodal cell population, followed by a 6-day otic induction period (Fig. 1a). We hypothesized that the product of this differentiation is a population of presumptive otic progenitor cells that coexpress marker genes generally found in the early otic anlagen such as the otic placode and otocyst. Coimmunolabeling for PAX2 and PAX8 (Fig. 1b) and for PAX2 and DLX5 (Fig. 1c) supports this hypothesis. About 38.6%±7.2% of PAX2-expressing cells coexpressed PAX8, and a similar number of PAX2-positive cells (37.3%±13.0%) colabeled with antibodies to DLX5. Reciprocally, 66.1%±3.6% of PAX8-expressing cells were immunopositive for PAX2, and 58.3%±7.4% of DLX5-positive cells colabeled with antibodies for PAX2 (n=4). These results suggest that a substantial portion of the PAX2-expressing cells also expressed PAX8 and DLX5, which indicates an early otic lineage phenotype. In further support of this hypothesis, we confirmed that the PAX2-expressing cell population was distinct from PAX6-expressing presumptive lens and trigeminal placodal, retinal, and neural progenitor cells, which have been reported previously to differentiate in conditions similar to the ones utilized in our guidance protocol [12,13]. Of the 32.2%±5.8% of PAX6-expressing cells present at day 21 of the guidance protocol, 6.7%±2.9% cells coexpressed PAX2 and vice versa, 6.4%±2.0% PAX2-positive cells were also labeled with antibodies for Pax6 (n=4, Fig. 1d).

Otic lineage guidance of human embryonic stem cells (ESCs).
In a parallel experiment, we investigated whether upregulation of otic lineage marker genes in the presumptive progenitor cell population at day 21 was dependent on FGF signaling. Inhibition of FGF signaling with SU5402 [14] between day 15 and 21 (Fig. 2) led to a significant reduction of expression of mRNA encoding PAX2, PAX8, and the dorsal otocyst marker Oc90 [15] by 96%, 93%, and 100%, respectively (Fig. 2a).
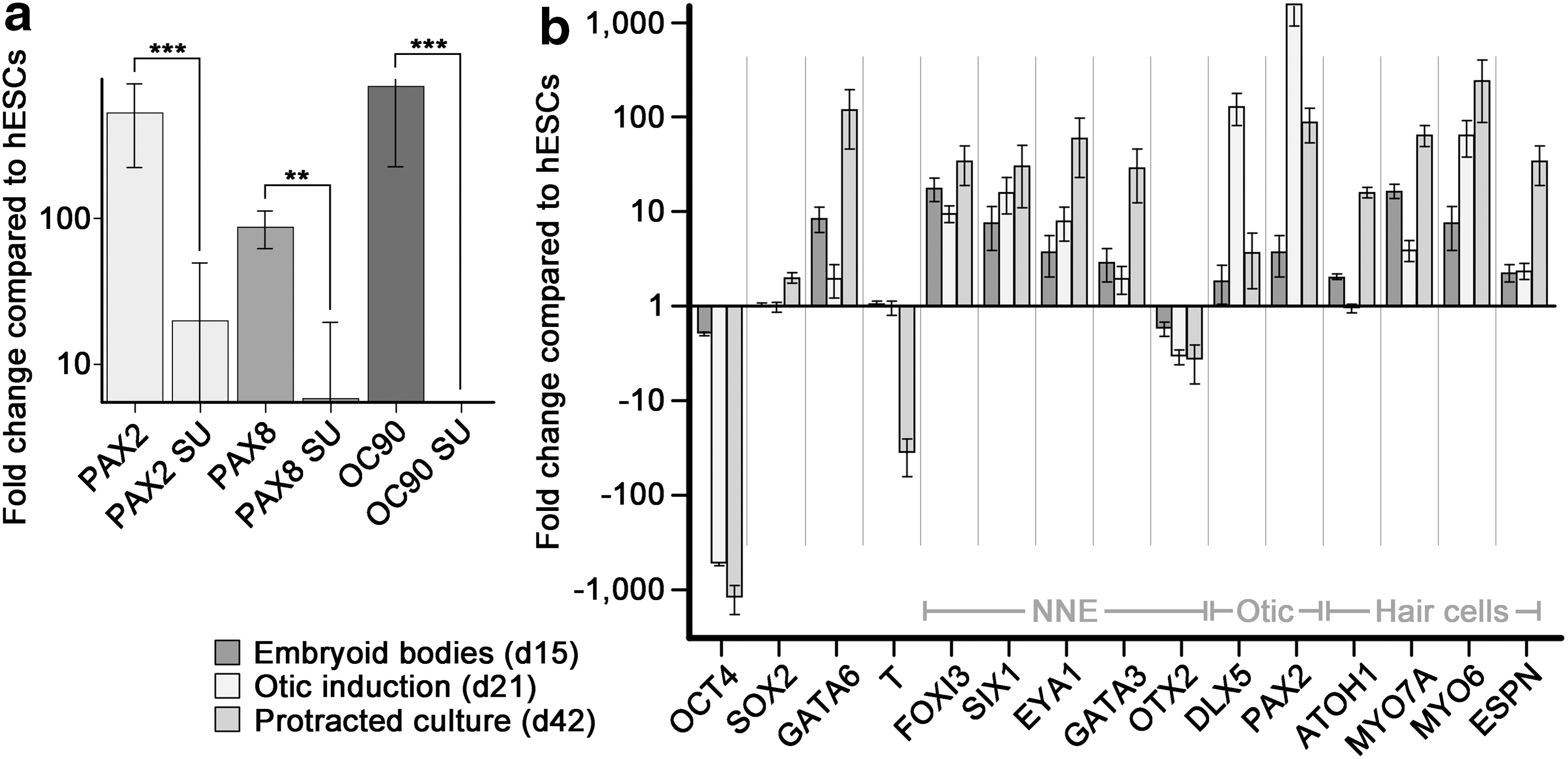
Quantitative RT-PCR for otic lineage marker genes.
Early otic marker-expressing progenitor cells differentiate into cells expressing sensory hair cell genes
Grafting studies showed that committed otic progenitor cells differentiate independently of the surrounding tissue and do not require external signaling for proper differentiation [7,8]. This feature also applies to stem cell-derived murine progenitor cells; although in these previous cases, other cell types present in the embryonic chicken inner ear after grafting, in the 3D aggregates, or in the presumptive otic progenitor cells required coculture with mesenchymal stromal cells from the chicken utricle for proper cytomorphological maturation [1 –3]. For differentiation of human inner ear cell types, we found that protracted culture in decreasing concentrations of KSR resulted in upregulation of genes indicative of hair cell differentiation (Figs. 1a and 2b). Quantitative RT-PCR revealed a downregulation of the hESC pluripotency marker OCT4 during this successive differentiation process. SOX2, a transcription factor important for self-renewal and pluripotency of hESCs as well as a marker for otic lineage cells that adopt prosensory identity [16,17], was continuously expressed at the EB stage and subsequently upregulated as the guided cell population differentiated. The endodermal marker GATA6 was upregulated during EB formation, indicative of the heterogenic differentiation process, but subsequently downregulated at the presumptive otic progenitor stage of differentiation. Whereas expression of the mesodermal marker Brachyury (T) was downregulated after protracted differentiation, GATA6 was upregulated, suggesting the presence of endodermally derived cells at this stage.
The formation of the preplacodal ectoderm that is competent to give rise to cranial placodes is an important step during cranial development [5,18]. Studies in various model organisms have put forward a number of marker genes expressed in the preplacodal ectoderm, such as FOXI3 [19,20], SIX1 [21,22], EYA1 [23,24], GATA3 [23,24], and DLX5 [24,25]. All these genes were upregulated during the successive differentiation process. OTX2, a gene that is important for the development of anterior cranial placodes [26], was expressed at low levels throughout the course of in vitro differentiation. OTX2 has been shown to be important during chicken inner ear development, specifically during patterning of sensory patches and cochleovestibular ganglion formation [27,28], and was previously found upregulated in cultures derived from murine ESCs that were differentiated into hair cell-like cells [1]. In contrast, OTX2 expression did not increase in hESC-derived presumptive otic lineage cells.
Upregulation of PAX2 was detectable already during EB formation and was much more prominent, as expected, following induction with FGF at the presumptive otic progenitor cell stage. Finally, sensory hair cell markers such as ATOH1, MYO7A, MYO6, and ESPN were upregulated after protracted cell differentiation, indicative of cells in the culture adopting a hair cell-like phenotype.
Sensory epithelium markers are coexpressed after protracted differentiation
The observed upregulation of hair cell markers suggests that some cells in the protracted cultures may have started to differentiate into sensory hair cells. We hypothesized that sensory hair cell-like cells would be associated with cells that express prosensory marker genes, supporting cell markers, and epithelial marker genes. Our cultures harbored MYO7A-positive cells surrounded by cells expressing the prosensory and supporting cell marker SOX2 (Fig. 3a). SOX2-expressing cells, in turn, coexpressed the supporting cell marker P27kip1 [29,30]. We observed a qualitative difference in the apparent immunostaining intensity where strongly expressing SOX2-positive cells were either immune negative for P27kip1 or exhibited minimally detectable levels (arrowheads in Fig. 3b). Cells with more intense P27kip1 staining, on the other hand, generally expressed SOX2 at moderate-to-low levels (asterisks in Fig. 3b). In developing murine cochlear sensory epithelia, SOX2 is initially strongly expressed in the prosensory domain and attains moderate levels in differentiating supporting cells that concurrently upregulate P27kip1 [17,31]. Differentiating hair cells downregulate SOX2 and do not express P27kip1 [32,33]. MYO7A-positive cells occurred in regions expressing the epithelial marker gene EpCAM, which is expressed in inner ear sensory epithelia [34,35]. Overall, in protracted cultures of hESC-derived presumptive otic progenitor cells, we found hair cell- and supporting cell-markers expressing cells in regions with epithelial character.
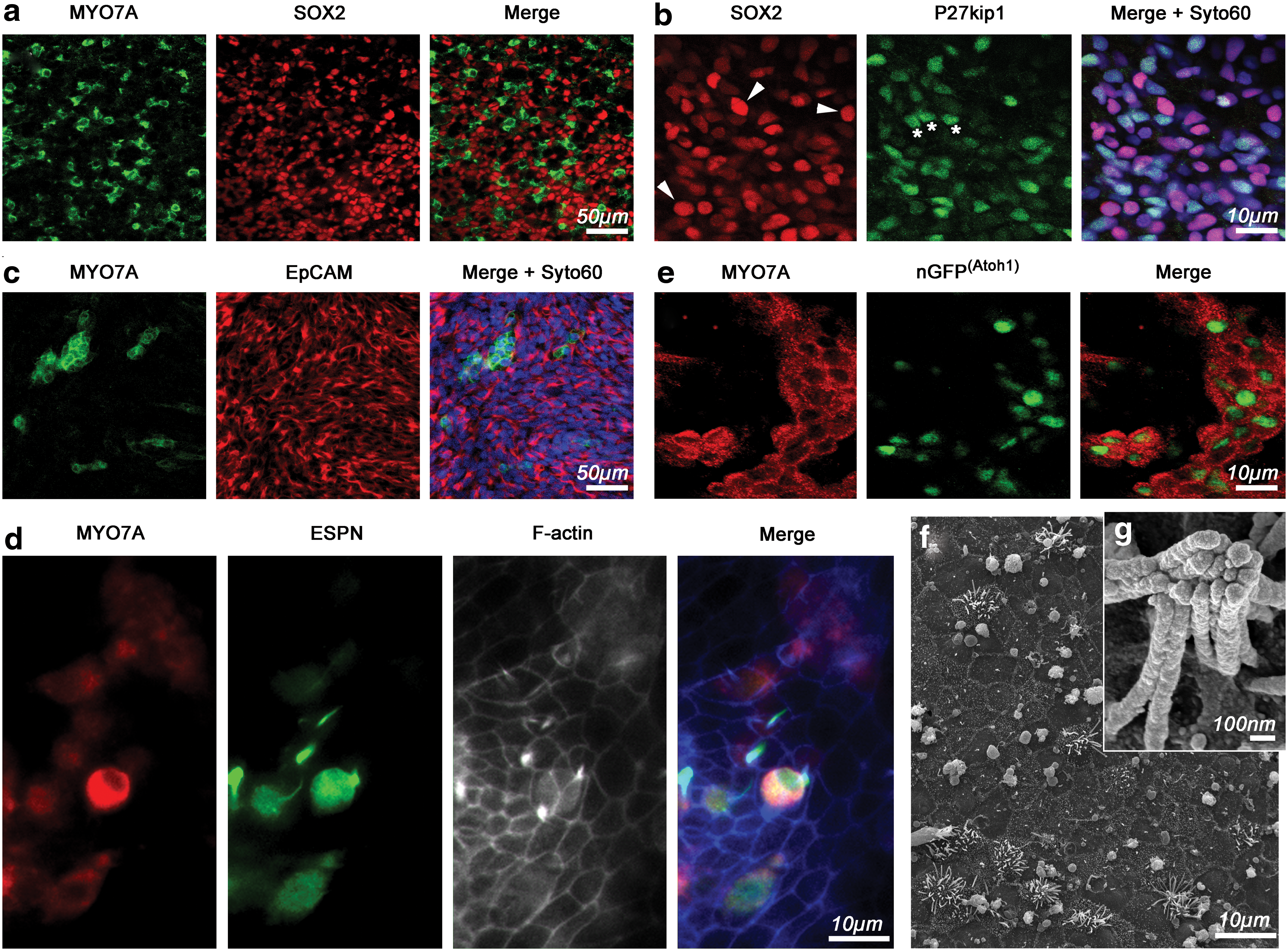
Sensory epithelium marker expression and nascent hair cell-like cytomorphology.
Sensory hair cell-like cells fail to acquire mature cytoarchitecture
MYO7A is not a definite marker for hair cells because it is also expressed in the retina and kidney [36]. When we performed double immunostainings for the hair bundle marker ESPN [37,38], we found that a minority of MYO7A-expressing cells coexpressed ESPN (Fig. 3d). In cells, where ESPN immunoreactivity was asymmetrically distributed and enriched in apparent protrusions emerging from MYO7A-positive cells, we observed coassociation with filamentous actin (Fig. 3d). These results suggest that a finite number of cells in protracted cultures of hESC-derived progenitor cultures are able to express multiple hair cell markers. Nevertheless, multiple hair cell marker-positive cells were rare, on the order of a few dozen per culture dish, which made it virtually impossible to identify cells for scanning electron microscopy (SEM) for higher-resolution imaging or even for physiology as shown in previous studies [2].
We sought to overcome this limitation by generating a transgenic H9-derived hESC line, where we utilized integrase-mediated integration of a murine Atoh1 enhancer/minimal promoter-driven nuclear eGFP (nGFP) reporter gene [39]. Because ATOH1 is expressed in hESCs as well as in mouse ESCs [2,40], we hypothesized that the resulting H9 ATOH1-nGFP hESC line would express nGFP in the undifferentiated state, which was indeed the case (Supplementary Fig. S2). As previously observed with murine Atoh1-nGFP ESCs [2], the nGFP reporter was downregulated when hESCs started to differentiate and it became expressed again in protracted cultures of progenitor cells, where MYO7A-positive cells displayed nGFP expression (Fig. 3e). Expression of nGFP allowed us to scan culture dishes after protracted differentiation and to mark potential hair cell-like cells (also see Sinkkonen et al. [35]), to process the sample for SEM and to image hair cell-like cells. In the nGFP-positive areas, we found protrusions extending from occasional cells that were localized in areas where the cellular organization displayed epithelial characteristics (Fig. 3f). In most cases, these protrusions were splayed and did not closely resemble the typical morphology of sensory hair cell bundles. In rare instances, we encountered more organized protrusions, which were more reminiscent of the typical coherent morphology normally associated with stereociliary hair bundles (Fig. 3g).
The lack of typical hair bundle morphology suggested that human hair cell-like cells are at a nascent state of development and fail to fully mature in the conditions provided in our in vitro culture system. This situation has been reported before in cultures of mouse otic progenitors derived from the neonatal inner ear [35], and even in cases where the bundle morphology was developed well enough to display mechanosensitivity, the murine ESC-derived hair cell-like cells did not show mature physiological maturation likely due to missing environmental cues [2]. When we increased the time of differentiation up to 3 months, we found that hair cell marker-expressing cells disappeared from the cultures, indicating that the cells died instead of further differentiating. The fact that some nGFP-positive cells coexpressed hair cell markers after protracted differentiation (Fig. 3e) spurred our interest to investigate whether it would be possible to quantify marker gene expression at the single cell level. Qualitatively, we observed cells with a strong nGFP expression and cells with midlevel nGFP expression in our cultures. MYO7A-positive cells and epithelial organization appeared to be more correlated with cells that expressed nGFP at medium intensity. Flow cytometry confirmed this qualitative impression and revealed that, in protracted differentiation cultures, 19.8% of cells expressed nGFP at medium levels and 3.1% of cells displayed a strong nGFP expression (Fig. 4a). We sorted 144 nGFPhigh-level, 144 nGFPmidlevel, and 192 nGFPnegative cells and quantitatively analyzed at the single cell level the expression of hair cell marker genes, markers associated with ATOH1 expression in other cell types distinct from sensory hair cells, and control genes (Fig. 4b and Supplementary Fig. S3). An initial analysis of hair cell marker gene expression in all 480 cells revealed that the nGFPmidlevel group was indeed the population where most hair cell marker-positive cells were found, although MYO7A, in particular, was also detectable in the majority of nGFPnegative cells (Fig. 4b). Within the nGFPmidlevel population, 9.0% of the cells coexpressed at least three or more hair cell markers with 10 of 13 cells triple positive for ATOH1/MYO7A/MYO15A, whereas the number of cells coexpressing three or more hair cell markers was 2.1% and 5.2% in the nGFPhigh-level and nGFPnegative population, respectively (Fig. 4c). It is noteworthy that all hair cell marker-positive cells, independently of their nGFP expression state, also expressed ATOH1 mRNA, based on single cell RT-PCR.
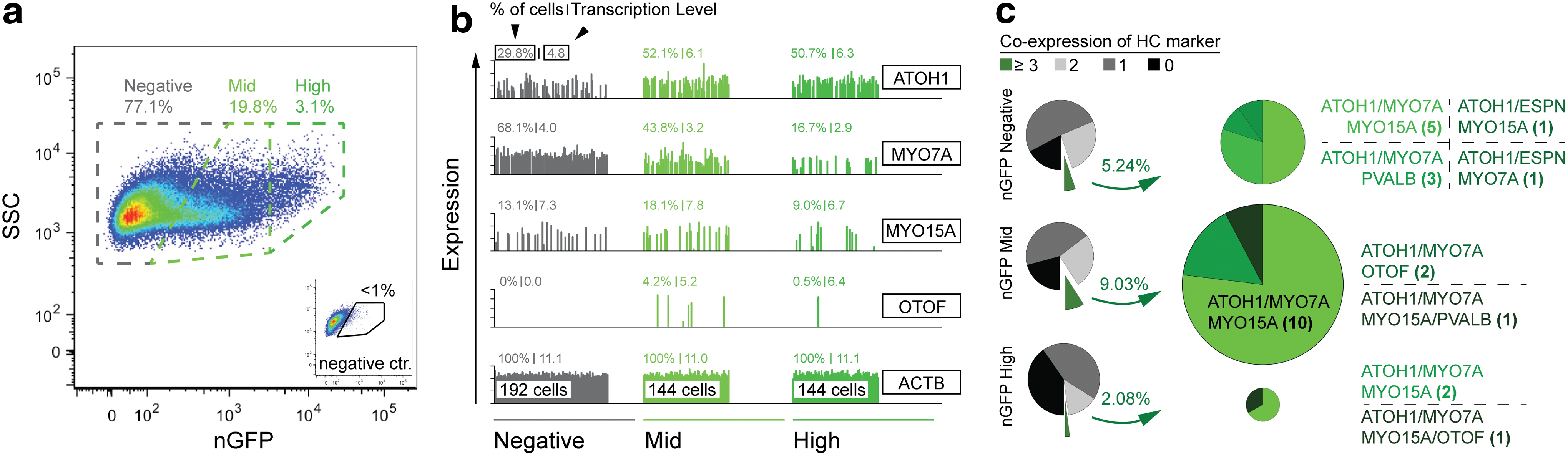
Single cell analysis of three different sorted H9
ATOH1-nGFP
populations after 42 days in vitro.
Discussion
In this study, we used an EB-based guidance protocol to generate a non-neural ectoderm cell lineage defined by expression of multiple marker genes (FOXI3, GATA3, DLX5, SIX1, and EYA1) and by its ability to further differentiate into cells expressing preplacodal and otic placode marker genes (PAX2 and PAX8). We further showed that the expression of otic marker genes depends on the FGF signaling activity, which is an indication that human otic induction from ESCs is an FGF-dependent process, as previously demonstrated [9] and suggested by the lineage guidance of mouse ESCs [1,2]. Placodal development and otic induction is a complex process involving multiple signaling steps during which, specification and competence of individual cells become successively more restricted until the progenitor cells are capable of differentiation into the lineage-defining cell types without additional external guidance [5,6]. Our empirically determined guidance protocol reflects some of these previously described principles. For example, attenuation of BMP signaling in hESC-derived cultures expressing non-neural ectoderm markers results in the upregulation of markers indicative of anterior placodes [41]. In our cultures, we blocked BMP signaling and promoted WNT signaling, followed by activation of BMP signaling, all during a concurrent period of treatment with FGFs. The transient promotion of WNT signaling is based on results showing that activation of canonical WNT signaling in competent ectodermal precursors promotes the generation of murine otic placode tissue at the expense of epidermis [42]. The same study reports that activation of β-catenin in native otocyst cells promotes a dorsal otic phenotype, which might explain the robust upregulation of the dorsal otic marker OC90 observed in our cultures [15]. Although it is not clear whether the expression of dorsal otic markers indicates the potential tendency of hESC-derived otic progenitors to differentiate into nonsensory otic cells, it is a possibility that will require attention in subsequent studies.
Previous studies with murine ESC- and iPSC-derived otic progenitors generated in culture suggested that proper hair bundle formation requires an inducing signal provided through coculture with stromal feeder cells isolated from the embryonic chicken utricle nonsensory epithelium [2]. Coculture with stromal feeder cells was technically not feasible in protracted differentiation cultures because mitotically inactivated chicken feeders did not survive long enough to allow for efficient and reliable experimental exploration. Conditioned media from inactivated chicken stromal cells also failed to promote presumptive otic progenitors to differentiate into hair cells, suggesting the potential necessity of direct cell-to-cell interactions. We found, however, that successive reduction of serum replacement in a feeder-free culture system during the differentiation period resulted in upregulation of hair cell markers. The incidence of hair cell marker expression in these protracted differentiation experiments was low, which is likely an indication of the high heterogeneity of the presumptive otic progenitor cell population, a potential tendency of the otic cells to differentiate into nonsensory cells, and a sign that only a few regions of the culture dishes were able to evolve a local microenvironment capable of initiating differentiation of hair cell-like cells. Our flow cytrometric cell sorting and single cell qRT-PCR results give insight into the efficiency of the differentiation protocol: about 20% of all cells encountered after protracted differentiation expressed nGFP Atoh1 at midlevels, which we determined to be an indicator of a potential hair cell phenotype. Of these nGFP-expressing cells, 9% of cells coexpressed at least three hair cell markers. Our single cell analysis revealed a correlation of ATOH1 expression at the level of cell numbers and expression levels with the presence of nGFP, which supports the notion that the reporter activity was coinciding with ATOH1 expression.
Nevertheless, cells that expressed multiple hair cell markers were also encountered in nGFPhigh-level and nGFPnegative cell populations, suggesting that the generated H9 ATOH1-nGFP hESC line is probably not an absolutely optimal reporter for human hair cell differentiation. On the other hand, murine Atoh1 is downregulated as hair cells mature and several of the selected hair cell markers that we used are not necessarily coexpressed in native hair cells because they appear sequentially. ESC-derived hair cell-like cells consequently do not necessarily have to express all hair cell markers simultaneously.
The murine Atoh1 enhancer used in our study is activated in many additional Atoh1-dependent cell types, including neural progenitors in the spinal cord and cerebellum, Merkel cells, and secretory cells of the gut [39]. Furthermore, likely due to the lack of native regulatory/inhibitory elements, the Atoh1-nGFP reporter is inaccurate in the reported mouse model and is expressed in other tissues and organs such as the apical ectodermal ridge of developing limbs, the developing cortex, spinal cord, dentate gyrus, retina, and olfactory epithelium [39]. We detected markers for cerebellar granule progenitors and Merkel cells in individual nGFP-positive cells (Supplementary Fig. S3), but we also found enrichment of the glial and cochlear supporting cell marker GFAP [43] in about 49% of all nGFPhigh-level cells. Considering that the reporter cell line employs a murine enhancer in combination with a basic promoter that has been previously shown to have some degree of unfaithful activity [39], it is feasible to consider that the lack of other regulatory elements in combination with possible integration site-specific effects in human H9 ESCs might contribute to the observed diversity.
We conclude that although the generation of nascent hair cell-like cells from hESCs occurs in vitro, the efficiency of the procedure is limited. More applied utilization of the method will require further optimization, such as generation of more homogeneous populations of otic progenitors with prosensory phenotype, enrichment methods, as well as testing the competence of prosensory progenitors to generate sensory epithelia.
Proper differentiation of hair cell-like cells generated from various progenitors does not generally happen in substrate-attached cultures unless some signals are provided through the use of cocultured cells, often taken from the developing or neonatal inner ear [2,9,35,44]. In 3D environments, however, generated through aggregation, grafting into developing inner ears, mesenchymal-to-epithelial transition, and self-organized sphere formation, cytomorphologies highly reminiscent of sensory hair cells have been reported [1,3,45,46]. Our study suggests that similar to murine hair cell-like cells, human hair cell-like cells very likely require an enabling microenvironment for proper differentiation. Our goal was to develop a substrate-attached culture system in chemically defined conditions. Within these confined parameters, we have shown that otic lineage cells and sensory epithelial cells can be differentiated. Because the contributing factors leading to proper hair cell differentiation in 3D environments are unknown, we suggest that the culture system reported here can be used to screen for factors that ultimately lead to human hair cell-like cells with typical hair bundle morphologies and functional features such as mechanotransduction.
Footnotes
Acknowledgments
The authors thank all members of the Heller laboratory for helpful discussions and comments on the manuscript, Dr. Anthony Oro and Dr. Susie Lee (Stanford) for advice and materials for the hESC transgenics, and Dr. Caroline Desponds (Inception 3, Inc.) for helpful discussions. This work was supported by the National Institute on Deafness and Other Communication Disorders grants DC006167 and DC012250 to S.H., by a P30 core grant (DC010363), by the Stanford Initiative to Cure Hearing Loss, and in part by the FP7-Health-2013-Innovation cooperative grant by the European Commission. J.W. is supported, in part, by a postdoctoral fellowship (WA3420/1) from the Deutsche Forschungsgemeinschaft. G.D. is supported by CIRM TB1-01194 and a PSM grant from the National Science Foundation.
Author Disclosure Statement
Parts of the method described in the manuscript have been patented and are licensed to Inception 3, Inc. M.R., K.O., and S.H. are scientific advisors to the licensee.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.