
Abstract
The canonical Wnt/β-catenin pathway has long been associated with self-renewal and expansion of embryonic stem cells (ESCs). Recent studies have brought into question some earlier assumptions concerning the functional role that canonical Wnt signaling plays in self-renewal mechanisms by demonstrating clear effects on differentiation. In addition, Wnt is crucial for cell fate determination during embryogenesis. The seemingly contradictory data compiled over several years now point to a more complex system of organismal development by which the downstream effects of Wnt activity are largely determined by the context in which signaling occurs. This review will assess seemingly contradictory findings regarding Wnt signaling in either self-renewal or differentiation in ESCs and then explicate cellular scenarios that determine the context by which Wnt ligands exert their differential effects. Both physiological embryonic development and pathogenic adult carcinogenesis provide opportunities to gleam illuminating insights from stem or stem-like progenitor cells that place Wnt signaling at the center of important cellular decisions to expand or differentiate. The impact of heterogeneity, concentration, metabolic state, and the array of accessible interacting cofactors from various signaling pathways that regulate these functions in ESCs will be critically examined, and implications for early embryonic development and cancer biology will be explored.
Introduction
I
Ligands in the Wnt family of secreted, cysteine-rich glycoproteins were first discovered over 30 years ago, and to this day their precise roles in development remain controversial. Pleiotropic effects in cells make them enigmatic proteins to study. Adding to this problem is a great deal of redundancy in signaling, with 19 Wnt ligands and 10 Frizzled (Frz) receptors identified, exhibiting promiscuity and low specificity in binding for high cross-reactivity [1]. This pleiotropy in binding kinetics is largely conferred by a highly conserved lipid-modified serine residue in one pocket and a hydrophobic stretch at a separate pocket for forming tight contacts with corresponding Frz clefts [2]. In addition to redundancy in receptor activation, the downstream effects conferred by Wnt signal transduction are incredibly diverse, confounding efforts to unmask mechanisms of activity under different cellular contexts.
Much effort has been made to elucidate the complex signaling mechanisms downstream of Wnt over the past several years. The noncanonical Wnt pathways are β-catenin-independent and mediate planar cell polarity and calcium-dependent signaling [3]. Canonical Wnt signaling through β-catenin (Fig. 1) has been implicated in self-renewal but also differentiation [4], and will be the focus of this review. Briefly, Wnt ligands bind to Frz receptors and recruit co-receptor low-density lipoprotein receptor-related proteins (LRP) 5 or 6. This prevents phosphorylation, ubiquitination, and proteolysis of β-catenin by a destruction complex consisting of glycogen synthase kinase (GSK)-3β, adenomatous polyposis coli (APC), casein kinase (CK)1α, and Axin1. Axin1 is present at the lowest concentrations of all destruction complex members [5], making it the rate-limiting component and thus an important regulatory target. Axin1 is inhibited by direct contact with Disheveled (Dvl)1, which is recruited to activated Frz upon Wnt stimulation [6]. In the absence of Wnt, the scaffolding protein, APC, has been shown to bind Axin1 in a manner mutually exclusive with Dvl1, causing displacement of Dvl1, thus preventing Axin1 inactivation [7].
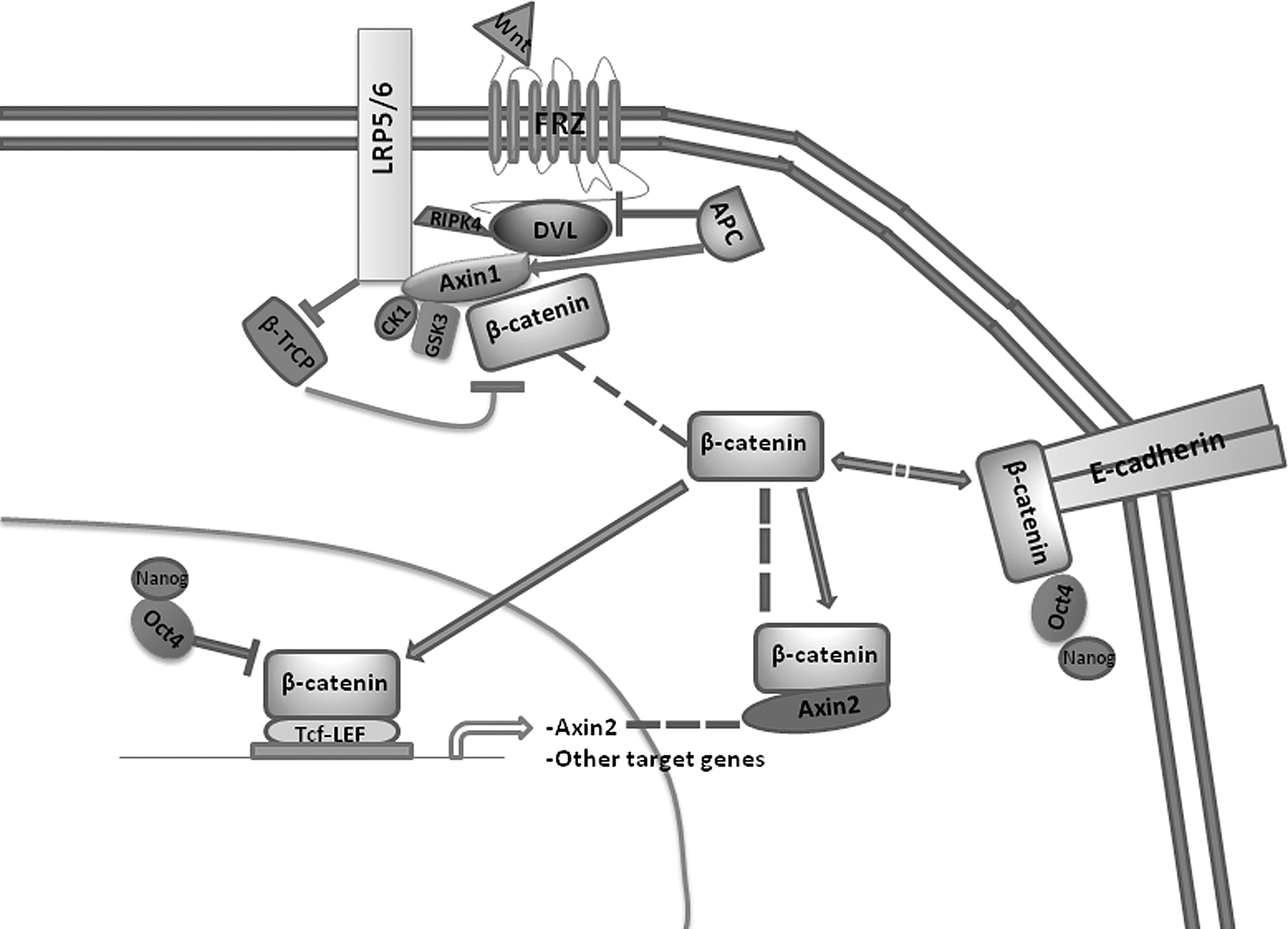
Expanded view of canonical Wnt signaling in stem cells. Following binding of certain Wnt ligands to a Frz cell surface receptor, β-catenin becomes stabilized in the cytoplasm by inhibition of phosphorylation by casein kinase 1α (CK1α) and glycogen synthase kinase (GSK)-3β. This blocks ubiquitination of β-catenin by beta-transducin repeat containing E3 ubiquitin protein ligase (β-TrCP) and subsequent proteosomal degradation [176]. Stabilized β-catenin can saturate the destruction complex and accumulate in the cytoplasm. This leads to nuclear translocation where it binds to T-cell-specific transcription factor (Tcf )/lymphoid enhancer-binding factor (LEF) transcription factor complexes, and in the absence of Oct4, efficiently drives transcription of target genes, including other Tcf/Lef components [49] and Axin2 [65]. Cytoplasmic β-catenin can form a stable complex with Axin2 in the cytoplasm, possibly providing a mechanism of negative transcriptional feedback and maintaining self-renewal functions [24]. Conversely, β-catenin associates with membrane-bound E-cadherin for roles in cell adhesion and cytoskeletal processes that also has implications for embryonic stem cell (ESC) self-renewal [78 –80]. These membrane and cytoplasmic stores can re-enter the cytoplasm in the presence of sufficient regulatory signals [24,72]. Oct4, a core pluripotency factor, has been implicated in modulating both the transcriptional and non-transcriptional effects of β-catenin following Wnt stimulation [37,72].
The result of Wnt signaling is the stabilization of β-catenin, which may then translocate to the nucleus. Despite the fact that β-catenin lacks a DNA-binding domain, nuclear β-catenin binds with high affinity to T-cell-specific transcription factor (Tcf ) and lymphoid enhancer-binding factor (Lef ) transcription factors and drives the activation of target gene transcription [8,9]. Wnt signaling is crucial in development and also in certain adult progenitor cell populations, most notably those in intestinal crypts [10], hair follicles [11], and the hematopoietic system [12].
Wnt in Self-Renewal and Differentiation
Embryonic stem cells (ESCs) are characterized by indefinite expansion in cell culture and pluripotency, the ability to differentiate into all cell lineages present in adults. Differences in culturing mouse (m)ESCs and human (h)ESCs have confused attempts to decipher genes and pathways necessary and sufficient for maintenance of self-renewal and pluripotency. mESCs are readily maintained in a naïve pluripotent state by addition of leukemia inhibitory factor (LIF) and, in the absence of feeder cells or serum, either BMP4 or a cocktail consisting of a GSK-3 inhibitor plus an antagonist of mitogen-activated protein kinase kinase-1/2 (2i) [13 –16] to the culture medium. Treatment of hESCs with these factors alone leads to rapid precocious differentiation in serum-free conditions [17]. In contrast to mESCs, hESCs contain genetic and epigenetic modifications characteristic of cells in a later stage in development, poising them for differentiation [17,18]. These include X chromosome inactivation in female ESCs, increased DNA methylation, and bivalent domain formation composed of active histone 3 trimethylation on lysine residue 4 (H3K4me3) and concomitant H3K27 trimethylation on the promoters and bodies of many critical developmental genes [17]. hESCs more closely resemble mouse post-implantation epiblast stem cells (EpiSC), which compose the extraembryonic ectoderm. EpiSCs retain pluripotency in that they can form all three germ layers yet are more genetically and epigenetically primed for differentiation, as opposed to naïve pluripotent mESCs [18 –20]. Maintenance of EpiSCs and hESCs relies on a combination of transforming growth factor β (TGF-β) or Activin A signaling through Smad2 [21,22] and fibroblast growth factor (FGF)2 [23]. A combination of a GSK-3 inhibitor (GSK-3i) and an Axin-stabilizing tankyrase inhibitor is also sufficient in sustaining EpiSCs and hESCs in serum- and feeder-free conditions [24]. Establishment of naïve pluripotent hESCs using a combination of LIF, TGF-β1, FGF2, 2i, and inhibitors of p38 and c-Jun N-terminal kinase has made possible the amelioration of the many clear genetic and phenotypic discrepancies otherwise distinguishing hESCs from mESCs [25], and should allow for more trustworthy data extrapolation between the two cell types in future studies. Due to the extensive usage of primed hESCs and naïve pluripotent mESCs for analysis of canonical Wnt signaling, differences between the cell types must be taken into consideration.
Many studies in mESCs have implicated canonical Wnt signaling in the control of self-renewal [14,16, 26 –29], although necessity in maintaining the pluripotent state has been disputed [30 –32]. Recently, differentiation of mESCs to EpiSCs was demonstrated to be preventable by Wnt signaling, which strongly disposed the cells to a state of indefinite self-renewal [33]. Studies in hESCs have yielded ambiguous results, possibly owing, in part, to the intermediate “poised” state of hESCs already discussed. Some studies support a role in self-renewal [14,34,35], others in differentiation [36 –38], and yet others show initial effects on self-renewal but loss of colony-forming ability after several days [38 –40]. Exogenous factors, such as choice of culture medium, can have substantial effects. For example, the use of feeder cells is strongly associated with maintenance of self-renewal in hESCs [39,41], possibly owing in part to the mitogenic bioactive lipid, lysophosphatidic acid [41]. Clearly, many factors must be considered when assessing the role Wnt signaling plays in cell fate decisions, including artificial modifications from use of certain in vitro models.
A strong case for Wnt signaling being associated with differentiation rather than self-renewal of hESCs was made by Moon and colleagues, who demonstrated that canonical Wnt signaling was silent during hESC self-renewal and became activated in sorted cells expressing genes indicative of commitment toward a mesodermal lineage [37]. Transcriptional effectors of β-catenin were negatively regulated by Oct4, a core pluripotency factor responsible for maintenance of symmetric division in self-renewing, pluripotent ESCs [37]. Oct4/β-catenin complexes have been demonstrated to form in GSK-3α/β-ablated or Wnt3a-treated mESCs [42]. Direct Oct4/β-catenin interaction has also been implicated in the upregulation of another core pluripotency factor, Nanog [35]. While Oct4 knockdown in hESCs increased β-catenin transcriptional effects [37], Wnt3a or GSK-3i treatment, or genetic ablation of GSK-3 in mESCs led to increased transcription downstream of Oct4, implicating Tcf/Lef-independent effects of Wnt/β-catenin signaling under conditions favoring self-renewal [42].
The Tcf/Lef family of transcription factors is composed of three transcriptional activators, Tcf1, Tcf4, and Lef1, through which β-catenin exerts its downstream effects [8,9,43]. A fourth family member, Tcf3, mainly serves as a transcriptional repressor [44]. β-catenin is a major co-activator of Tcf1 in ESCs [45], as Tcf4 and Lef1 are expressed at much lower basal levels [46]. Expression of Tcf1 is associated with resistance to differentiation and expansion of mESCs in the absence of Tcf3, while Tcf3 deletion phenocopies the self-renewal effects observed with Wnt 3a treatment [47]. Direct transcriptional targets of Tcf1 involved in self-renewal include Oct4, Sox2, and liver receptor homolog-1 [29,48], implicating canonical Wnt/β-catenin signaling in ESC self-renewal via the Tcf1 effector.
Tcf3 is a potent repressor of many Wnt target genes, including Lef1 [49]. Wnt3a signaling in mESCs relieved Tcf3-mediated inhibition of Lef1 and other β-catenin/Tcf-family target genes through a direct β-catenin/Tcf3 interaction, permitting downstream transcriptional activation [49]. Tcf3 is phosphorylated in a β-catenin-dependent manner, leading to its displacement from DNA [50]. Therefore, Wnt/β-catenin signaling converges on genes bound by the Tcf3 repressor for dis-inhibition of transcription in addition to the direct co-activator functions of β-catenin.
Tcf3 also inhibits the core pluripotency network of Oct4/Nanog/Sox2, while Wnt3a promotes transcription [47,51]. Importantly, GSK-3 inhibition in mESCs is sufficient for dis-inhibition of Tcf3 repression of pluripotency factors in a β-catenin-dependent manner [32]. Increased expression of pluripotency factors following Tcf3 de-repression can further facilitate direct Oct4/β-catenin contact in the context of the interactome generated by the pluripotency network [42,47]. This activity is still partially dependent on β-catenin interactions with Tcf1 and Tcf3, however, as β-catenin binding to Oct4 was greatly reduced in Tcf3-null mESCs, suggesting that Tcf proteins function in the recruitment of β-catenin to chromatin [47]. ChIP-Seq data confirmed that nuclear β-catenin binding was similarly spread out between Oct4/Sox2 and Tcf/Lef motifs in GSK-3i-treated mESCs [52]. Tcf1 and Tcf3 have conserved DNA-binding domains [53], and increased activation of Tcf1 following Wnt stimulation has been postulated to drive ESC self-renewal by outcompeting Tcf3 for consensus sequence binding [47]. However, this conflicts with the minimal Tcf/Lef reporter activity measured in pluripotent as opposed to differentiating cells [24,37,38] and the Oct4-mediated repression of Tcf/Lef reporters [37,42]. Transcriptional activation of several target genes downstream of Tcf1 or Lef1 is thus likely inhibited in an environment favoring self-renewal, even if nuclear β-catenin is present. Nuclear β-catenin would instead aid in self-renewal by Oct4-dependent effects and/or through de-repressing key transcriptional targets of pluripotency factors normally inhibited by Tcf3 to synergize with signaling of the core pluripotency circuitry independent of Tcf/Lef transactivation (Fig. 2A) [42,52]. Functional significance of direct binding to Oct4 is as yet unknown and would be aided by the generation of mutant proteins with this binding region deleted. If, as posited by Yi et al., Tcf1 acts to recruit β-catenin to chromatin [47], then it is possible that subsequent binding of Oct4 may structurally favor activation of larger transcription networks through proximal interactions with promoters and enhancers of functionally related genes controlled by a common set of transcription factors within discrete chromosomal segments. This would be amenable to chromosomal conformation capture techniques and in line with the consolidation of a transcriptome favoring self-renewal and resistance to differentiation despite dispensability for the process as a whole as long as core pluripotency factors remain highly expressed.
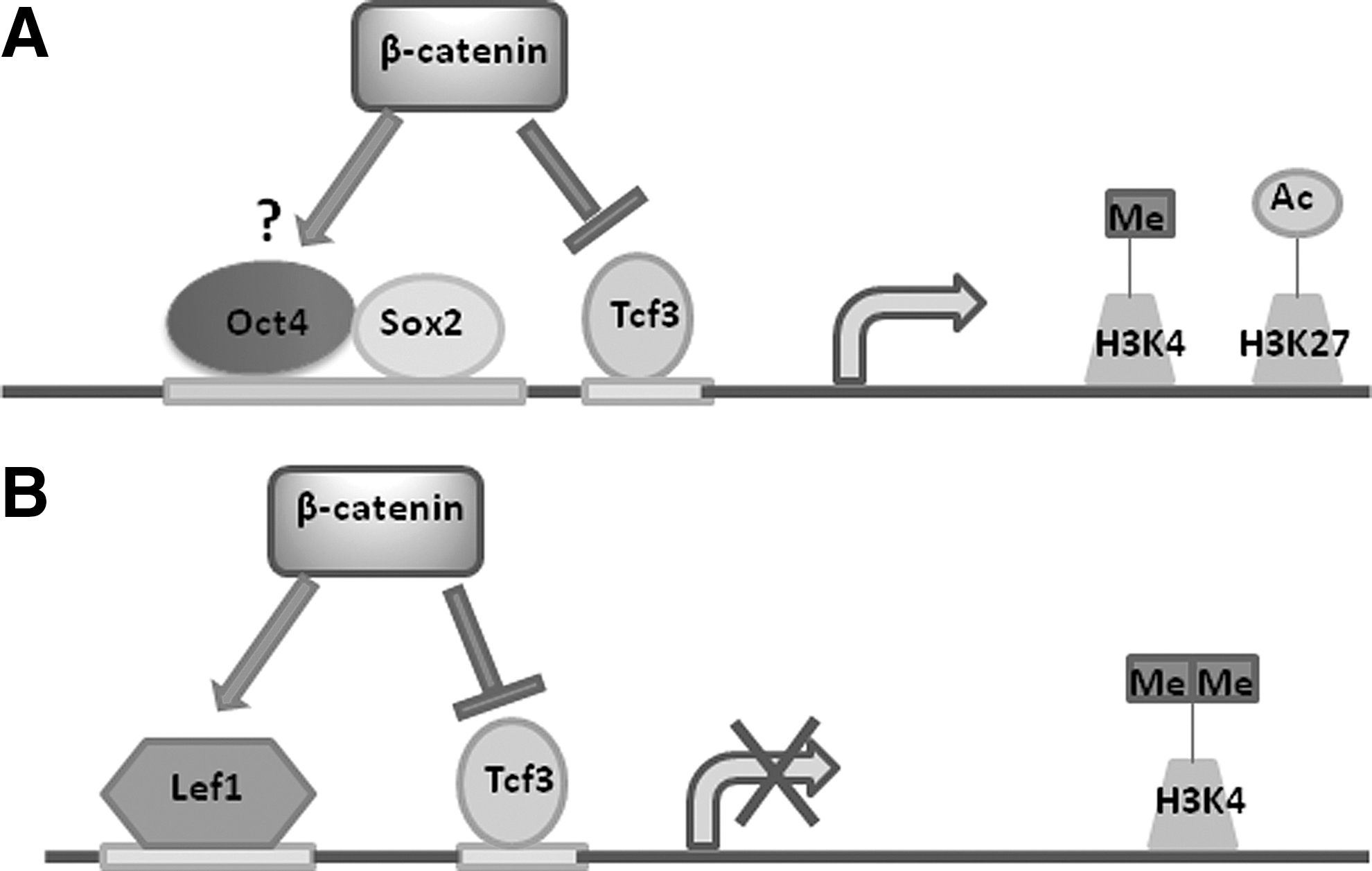
Nuclear β-catenin transcriptional regulation in ESCs.
Of course, many factors in the pluripotency network are likely involved that favor the self-renewal effects of β-catenin in ESCs, with the context of signaling being altered in the absence of Oct4/β-catenin interactions as cells begin to differentiate and Tcf/Lef transcriptional activity rises to allow for expression of genes involved in cell fate decisions and differentiation. It is important to note that Tcf3 additionally exerts β-catenin-independent effects, as Tcf3-null transgenic mice are embryonic lethal prior to gastrulation [54], while embryos expressing Tcf3 mutated at β-catenin-binding regions did not suffer any defects until after gastrulation [49]. This links Wnt-dependent transcriptional activity with cell fate determination in vivo and is supported by phenotypic evidence from mice deficient in essential components of the canonical Wnt signaling pathway, which exhibit substantial defects in mesendoderm formation [31,55,56]. When the β-catenin/Tcf3-interaction was ablated in Tcf-positive mESCs, only a partial inhibition of Wnt3a signaling was observed, implying redundancy at this stage [47]. Tcf/Lef-dependent transcriptional signaling for ESC self-renewal would thus appear to be dispensable in light of the above findings that alleviation of Tcf3 repression constitutes a major mechanism of Wnt-stimulated transcriptional effects in self-renewing cells but yet no overt phenotypic defects are observed in vivo in the absence of Tcf3/β-catenin interaction prior to gastrulation. These results also support the data showing Tcf/Lef reporter activity is diminished in self-renewing pluripotent ESCs compared to those undergoing differentiation [36 –38].
With the advent of new technology to probe genomic organization at trancriptionally robust super-enhancers [57,58] as well as the emerging role of regulatory long non-coding RNA and enhancer RNA in developmental processes [59], the mechanisms governing the nuclear effects of canonical Wnt/β-catenin signaling should become more clearly elucidated in coming years. Although β-catenin binds to both Oct4 and Tcf/Lef transcription factors in ESCs, there is an enrichment of active H3K4me1 and H3K27ac chromatin marks in enhancers of Oct4/Nanog/Sox2 target genes in the presence of core pluripotency factors [52]. In contrast, enhancers of traditional Tcf/Lef target genes were enriched with H3K4me2 modifications in the presence of both β-catenin and core pluripotency factors in mESCs, suggestive of chromatin in a poised yet inactive state (Fig. 2B) [52]. These data point toward an added layer of regulation beyond control of expression levels of different Tcf/Lef factors. Essentially, inhibition of Tcf3 by increased Wnt signaling in ESCs could overcome one barrier hindering the expression of genes silenced in the pluripotent state, but is only sufficient for genes that have permissive chromatin marks, preventing premature expression of cell fate-associated genes. The epigenetic state of transcriptional targets of β-catenin thus plays a crucial role in the expression of lineage-specific genes downstream of Wnt signaling, enabling both temporal and spatial regulation partly through regulation of enhancer activity. These effects may be gene-specific, as chromatin interactions with certain Tcf1 targets important for self-renewal, but not lineage specification, may be permissive for β-catenin-dependent transcription, offering an explanation for the apparently contradictory results correlating Tcf1 activation with self-renewal [47]. Since knockdown of Tcf1 in wild-type mESCs did not cause overt cellular defects [47], Tcf1-dependent transactivation would appear to be non-essential for pluripotency despite being wired in ESCs to fortify the state of pluripotency and self-renewal.
Overall, tight epigenetic regulation of ESCs during pluripotency maintenance, as well as low expression levels of Tcf4 and Lef1 in concert with gene-specific Tcf3-mediated repression, act synchronously to silence differentiation genes in ESCs that would otherwise become activated by association of β-catenin with Tcf/Lef components. This is in line with the findings by Osei-Sarfo and Gudas identifying dynamic alterations in the transcriptional activation of Wnt target genes in mESCs following treatment with the differentiation-inducing morphogen, all-trans retinoic acid (RA), despite unchanged β-catenin promoter binding in both the presence and absence of RA [48]. Aside from direct transcriptional effects on primary response genes via retinoic acid receptor (RAR) isoform binding, RA causes massive changes in chromatin structure via interactions with various components of the RA/RAR transcriptional complex, inducing transcription of larger sets of secondary response genes to inexorably drive the differentiation process [60]. Intriguingly, RA was shown to decrease canonical Wnt signaling and simultaneously increase non-canonical Wnt signaling [47], supporting the idea of existence of a counter-regulatory feedback circuit between canonical and non-canonical Wnt pathways [61,62]. Furthermore, the decrease in Tcf/Lef-orchestrated transcription in ESCs observed with either Oct4 under conditions of self-renewal [37], or RA under conditions favoring terminal differentiation [48], are indicative of a temporal window when canonical Wnt signaling is highly active. Although these experiments are conducted in vitro, they point toward a meta-stable period of high plasticity in the process of cell fate determination where specific developmental genes become switched on by precisely controlled genetic, microenvironmental and positional cues in the process of physiological development, truly making this pathway a central pillar in the transition between states of self-renewal and differentiation.
In summary, accumulating evidence appears to point away from a necessary role for downstream Wnt/β-catenin transcriptional activation in ESC self-renewal and toward the existence of a larger regulatory circuit controlling β-catenin-dependent effects on self-renewal and differentiation that allows flexibility in maintaining the necessary phenotypes required for important time-dependent events to unfold in their proper sequence. Dysregulation of any key process within this system would have disastrous consequences at the level of organismal development. Therefore, complicated machinery to safeguard against such events would likely be highly evolutionarily conserved, and the result is that even a key mediator of self-renewal and cell fate decisions, such as β-catenin, would fit in the larger scheme in ways that would alter the final outcome depending on the model being used for investigation. To shed light on additional ways in which the canonical Wnt pathway can orchestrate events involved in self-renewal or differentiation in stem cells, non-transcriptional effects must also be taken into consideration.
The Effects of β-Catenin Localization on Self-Renewal and Differentiation
Sequestration of β-catenin into different cell compartments or under the control of different cellular signals is becoming increasingly appreciated as a key component of its regulation once stabilized. The traditional view of Dvl-mediated dissociation from the destruction complex as the sole mechanism of β-catenin activation has recently been challenged by Clevers and colleagues, who have demonstrated that inhibition of β-catenin ubiquitination occurs independently of dissociation from the destruction complex [63]. Under physiological conditions, endogenous β-catenin may remain bound and still become stabilized without initial nuclear translocation. Once saturation of binding to limiting amounts of Axin1 occurs, cytoplasmic accumulation would result to exponentially increase free β-catenin available for signaling. Kinetic analyses of Wnt inhibition of β-catenin degradation at or upstream of phosphorylation by both CK1α and GSK-3β hint at interference of the assembly of, or restructuring of the parts of the destruction complex independent of mere sequestration or saturation of the entire complex [64]. How this complex is inhibited or modified is a pertinent question up for debate, wherewith the resolution thereof may lead to a more precise understanding of the functioning of the machinery regulating such pleiotropic effects.
Axin2, like Axin1, is a key adaptor for Wnt-dependent signaling, and it is associated with transcriptional repression of the β-catenin pathway [65]. Binding of stable β-catenin to Axin2, a direct transcriptional target of β-catenin, was shown to be necessary and sufficient for the promotion of self-renewal in EpiSCs and hESCs, through cytoplasmic sequestration and inhibition of nuclear translocation [24]. Nuclear translocation and subsequent Tcf/Lef reporter activation was instead associated with precocious differentiation. Addition of a GSK-3i alone was not sufficient for maintenance of self-renewal in EpiSCs and led to transactivation of Tcf/Lef-dependent targets while addition of an Axin-stabilizing tankyrase inhibitor prevented nuclear translocation of β-catenin and maintained self-renewal in an Axin2-dependent manner [24]. This implicates Axin2 in acting separately from traditional Axin1-mediated degradation through the destruction complex. In this model, it can instead mediate the construction of a cytoplasmic scaffold for stabilized β-catenin to act in a transcription-independent manner for sustaining self-renewal conditions. In line with these effects in EpiSCs, release of Wnt for autocrine canonical signaling and accumulation of cytoplasmic Axin2 is a hallmark of adult epidermal skin cells to maintain populations of stem cells capable of self-renewal following injury [66]. Axin2 is restricted to epidermal stem cells and not expressed in slow-cycling hair follicle bulge cells, which do not require Wnt signaling for self-renewal [67]. It would appear that negative regulation of Wnt transcriptional signaling by Axin2 promotes self-renewal mechanisms, but the sequestration of stabilized β-catenin in a cytoplasmic complex independent of destruction complex-mediated degradation events opens the possibility that stable β-catenin may serve heretofore unknown roles in cell fate decisions and stem cell expansion.
A potential bottleneck in the restriction of stable β-catenin to cytoplasmic and membrane compartments is limited binding to nuclear import factors, including Nup62, Nup153, and RanBP2/Nup358 [68]. This may come into play when using pharmacological inhibitors of members of the destruction complex, such as the frequently used GSK-3 inhibitors. Some of the differential effects between the use of GSK-3 antagonists and other methods for increasing β-catenin transcriptional activity may partly be explained by this phenomenon [69]. However, caution should be exercised when assessing the transcription profile of GSK-3 inhibitor-treated cells due to Wnt-independent effects, including stabilization of both c-Myc [70] and Snail1 [71], which are also targets of GSK-3-mediated degradation with common effects on signaling.
The hypothesis that localization of β-catenin is critical for self-renewal and differentiation was additionally corroborated by Faunes et al. in mESCs [72]. Here, it was demonstrated that, despite low downstream transcriptional activity of β-catenin following exogenous Wnt administration under self-renewal conditions and increased transcription during differentiation, β-catenin accumulated in the plasma membrane in a complex bound to Oct4 and E-cadherin in pluripotent cells but not differentiating cells [72]. These data raise the possibility that β-catenin activity can become uncoupled from Wnt-stimulated nuclear translocation and downstream transcriptional activation in ESCs when Oct4 is expressed, potentially betokening added layers of nucleocytoplasmic regulation and challenging prior reports suggesting that membrane localization is inconsequential to Wnt signaling [73]. Excess β-catenin accrued in the plasma membrane following Wnt overexpression could instead fortify cell adhesion processes in a transcription-independent manner. Membrane bound β-catenin is an essential component of adherens junctions, largely through its contacts with E-cadherin, and establishes cell–cell contacts by linking the plasma membrane with the actin cytoskeleton [74,75]. β-catenin binds with lesser affinity to α-catenin [75], which links membrane activity to nuclear effects by stabilizing APC and promoting β-catenin degradation and inhibition of canonical Wnt transcriptional activation [76]. Intriguingly, knockdown of α-catenin in hESCs increased transcriptional activity of β-catenin and primed the cells for endodermal differentiation through interference with the destruction complex [76].
In contrast to the differentiation-inducing effects linked to increased transcriptional activity of β-catenin following α-catenin knockdown, loss of self-renewal ancillary to deletion of β-catenin in mESCs can be rescued by addition of Tcf/Lef-signaling defective β-catenin [31], further arguing against the absolute requirement for canonical Wnt/β-catenin-mediated transcriptional events in self-renewal. Loss of β-catenin in mESCs is compensated for by an increase in plakoglobin (γ-cadherin), which is able to bind to E-cadherin in β-catenin-deficient cells [31]. Stable plakoglobin expression is also sufficient for retention of gene expression and morphological phenotypes of mESCs under differentiation conditions [77]. Interestingly, loss of self-renewal in β-catenin knockout mESCs can be rescued by administration of an E-cadherin-α-catenin fusion protein to maintain cell contacts and enhance LIF signaling through stabilization of a Lifr/Gp130 complex [78].
Loss of self-renewal potential in mESCs has been recapitulated by E-cadherin deletion alone, independent of β-catenin status [79]. Naïve hESCs show more prominent E-cadherin membrane localization than primed hESCs that are poised to lose their pluripotency [25]. In addition, mouse fibroblasts have been reprogrammed to inducible pluripotent stem cells (iPSC) using E-cadherin in addition to traditional reprogramming factors Sox2, Klf4, and c-Myc and in the absence of Oct4 [80]. Opposite effects were observed in murine pluripotent cells displaying characteristics of EpiSCs, in which E-cadherin deletion and loss of E-cadherin/β-catenin contact was demonstrated to instead preserve self-renewal [81]. Caution should be exercised in interpreting these results, however, as established protocols for identifying and culturing EpiSCs had not yet been established and LIF supplementation was used for most experiments. Similarly, experiments showing that ablation of β-catenin in EpiSCs caused defects in self-renewal were also performed in standard mESC media [82]. It is plausible that the effects of Axin2 observed by Kim et al. [24] were a consequence of maintaining specific complexes having functions in cell adhesion and cytoskeletal dynamics that includes β-catenin retained in the cytoplasm and membrane in the undifferentiated state. This may later shift toward increased nuclear signaling as contextual alterations in the cell favor cell fate determination and differentiation.
Taken together, these data argue that the effects of β-catenin on cell adhesion processes, particularly through E-cadherin binding, have a functional role in preserving self-renewal in ESCs by maintaining cell integrity and cell–cell contacts. The finding that plakoglobin can substitute for β-catenin to avoid loss of self-renewal in mESCs points toward the existence of compensatory circuits for self-renewal maintenance. The differences in the findings between disparate studies likely reflect interference by some variables mentioned beforehand, such as ESC stage (primed or naïve) and cell culture conditions used. Similarly, while the canonical Wnt/β-catenin pathway promotes transcription of several genes involved in stem cell maintenance and de-represses Tcf3 inhibition of target genes for pluripotency [49], this dimension of Wnt/β-catenin signaling also appears to be dispensable for the self-renewal effects in the presence of adequate LIF/Stat3 signaling. Instead, there appears to exist a more complicated regulatory system in place by which canonical Wnt signaling contributes to both self-renewal and differentiation based on contextual cues that change as different signaling networks become activated or repressed temporally during the procession through developmental stages. The difficulty in recapitulating these temporal regulatory processes in cell culture remains a major challenge.
Although the exact role of β-catenin-mediated cell adhesion in ESC and EpiSC self-renewal remains controversial, some studies suggest it may be an important factor [24,72]. Alternatively, regulatory mechanisms may exist in pluripotent cells that sequester β-catenin within the cytoplasm and plasma membrane for the purpose of preventing excessive nuclear translocation of high concentrations of β-catenin that would otherwise overwhelm the genetic and epigenetic safeguards against Tcf/Lef-dependent transcription of genes involved in lineage specification. In either case, as cells lose expression of pluripotency factors during developmental processes in vivo and begin the differentiation process, commitment toward a specific lineage at such early stages requires extremely precise regulation to avoid defects that could compromise proper development and ultimately threaten survival. Because of the fine-tuning required for proper cellular responses following exposure to Wnt signals during different stages of development, the implications of possible gradient effects in the orchestration of cell fate decisions should be considered.
Establishment of Wnt Gradients for Dose-Dependent and Cell-Specific Signaling
A burgeoning area of research in Wnt biology is the study of Wnt gradients in the determination of cell fate. This idea is not new, as Wnt gradients in fly embryos were associated with anterior-posterior axis patterning nearly 20 years ago [83]. However, detailed mechanistic insights have been lacking until recently.
Evidence for the existence of Wnt concentration gradients
The existence of Wnt concentration gradients in vivo is mechanistically supported by the biphasic actions of the machinery available for components of the canonical Wnt signaling pathway. Wnt is dynamically regulated by its endogenous antagonist, Dickkopf-1 (Dkk1) [84]. Biphasic actions by the Dkk1 receptor Kremen (Krm) on LRP localization permit differential effects based on concentrations of Wnt proteins or Dkk1 [85]. Recruitment of LRP co-receptors is critical for silencing of the destruction complex during β-catenin activation by Wnt [86,87]. High Dkk1 concentrations cause internalization of LRP to terminate Wnt signaling via Krm [88], while absence of Dkk1 results in Krm-mediated co-localization of LRP on the cell surface to interact with Frz during Wnt signaling [89].
In vitro support for differential Wnt gradient effects has been garnered in mESCs, suggesting a possible role in asymmetric cell division [90]. The use of live cell imaging to track dividing cells showed that Wnt3a-coated beads caused cells to divide asymmetrically with the bead-proximal cells remaining undifferentiated [90]. The effect was not seen in beads coated with the non-canonical signaling transducer, Wnt5a. This was the first study documenting a direct role of Wnt signals on the asymmetric differentiation of ESCs, opening up intriguing possibilities for further research. Microfluidic devices have been developed that can accurately control Wnt gradients in real time for in vitro experiments [91].
Stem cell heterogeneity
In culture, hESCs are highly heterogeneous, and sorting reveals populations that have high or low Wnt signaling activity [41]. The importance of concentration effects in these populations was evinced, as Wnthi populations matched the profile for cells in the primitive streak (PS) primed for differentiation toward mesodermal or endodermal commitment. Conversely, Wntlo populations were maintained in the pluripotent state with proclivity to form neuroectodermal cells upon differentiation (Fig. 3B) [41]. Effects in embryoid body formation reveal similar patterns [92], and the PS is unable to form in mice depleted of either Wnt3 [93] or β-catenin [94]. Consistent with these findings is severe impairment of mesendodermal and neuroectodermal differentiation in mESC cell lines deficient in Ctnnb-1, the murine gene that encodes β-catenin (Fig. 3A) [31].
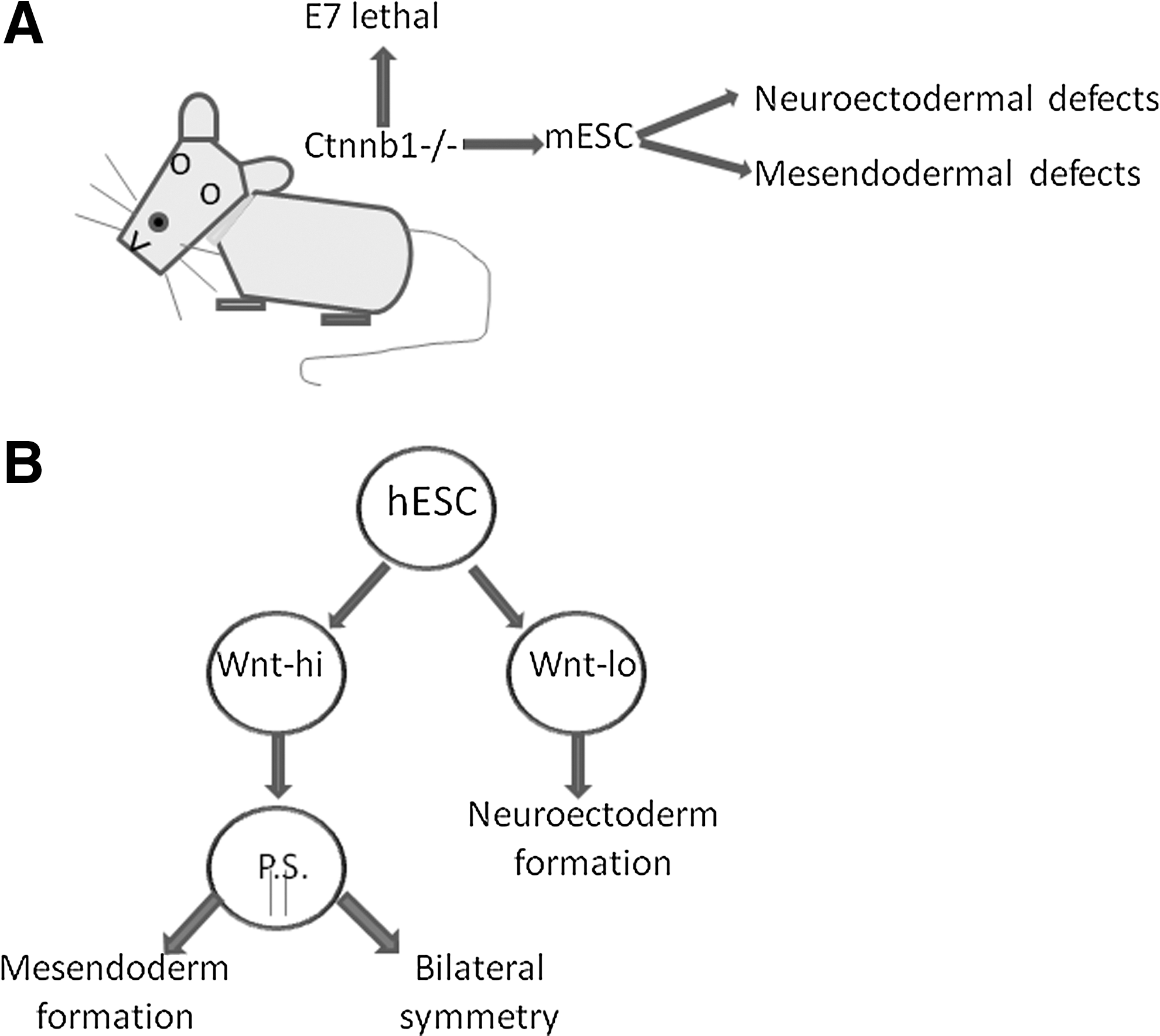
Wnt concentration gradients in embryonic development.
Notably, activin/nodal signaling in combination with canonical Wnt signaling recapitulated PS formation and mesendodermal differentiation in hESCs [36,95 –97]. Convergence of Wnt and TGF-β-family pathways at the PS stage of embryogenesis is logical, as mesendoderm formation from the PS requires disruption of cell adhesion processes for ingression of the primitive surface epithelium to form the basis of internal organs. This involves an initial epithelial-to-mesenchymal transition (EMT) during primitive endoderm formation. This pattern is repeatedly observed in extravasation of cancer cells, of which Wnt signaling at the leading edge of tumors is a potent driver of dedifferentiation [98] and EMT [99 –102] in many tumor types. These stem cell-like phenotypes are often accompanied by a dramatic shift in TGF-β-family signaling from a pro-differentiation toward an EMT-inducing state capable of extensive crosstalk with Wnt and additional reactivated stem cell pathways, including Notch and Hedgehog [103].
Wnt signaling in the regulation of metabolic networks
Wnt signaling is associated with different lineage fates, but how Wnt gradients directly affect differentiation and patterning mechanistically in development is unresolved. Metabolomic analyses of ESCs under pluripotent and differentiating conditions have evinced stark contrasts in metabolic flux through energetic pathways and in redox environment [104 –106]. Studies mechanistically linking key regulatory genes downstream of Wnt/β-catenin signaling in ESCs with shifts in metabolic flux associated with naïve, primed and differentiating cellular phenotypes are lacking, but clues may be ascertained from studies using adult cells and tissue exhibiting high Wnt signaling. A strong effect on metabolic homeostasis has been inferred in liver tissue, with a shift toward glycolytic reprogramming and lactate overproduction [107]. This occurs in the context of a larger phenomenon called metabolic zonation. Metabolic zonation is the adaptability of the liver to compensate for different nutritional environments within the body through appropriate spatial organization of hepatocytes with differing expression profiles along the same axis [108]. Wnt signaling is crucial for establishment of such zones, and alterations in β-catenin signaling lead to different expression profiles along this axis [109]. Metabolite signaling is also essential for embryonic patterning [110,111] and cell fate decisions [104,112,113].
Stem cells undergoing differentiation experience a shift toward oxidative redox status and altered metabolic requirements [104,106,114]. β-Catenin is a likely candidate to mediate some of these transitory effects due to its central involvement in cell fate decisions. Indeed, during extraembryonic endoderm differentiation, metabolic reprogramming associated with β-catenin transcriptional activity was observed in F9 teratocarcinoma stem cells in response to either elevated hydrogen peroxide or from RA administration [115]. The redox state of nucleoredoxin (NRX) was shown to play a key role in β-catenin-mediated transcriptional events [116]. Under reducing conditions in undifferentiated F9 cells, NRX binds and sequesters Dvl2, while oxidizing conditions cause displacement of Dvl2 from NRX. This increases downstream transcription of Tcf/Lef target genes to enable the cells to undergo a trajectory of differentiation characteristic of primitive endoderm formation [116].
Nutritional status may also play a role in coupling Wnt signaling with metabolic activity. Inhibition of both GSK-3 and mTOR in human hematopoietic stem cells (HSCs) led to increased self-renewal accompanied by increased β-catenin transcriptional activity [117]. This effect could theoretically be related to abnormal protein production in the artificial cell culture state with aberrant nutrient level exposure compared to the physiological niche [118]. However, conformable with these results was the finding that mTor inhibitor rapamycin increased HSC self-renewal and hematopoiesis in aged mice, although the role of Wnt signaling remains unclear [119]. These examples highlight the importance of the metabolic and nutritional state of the cell and crosstalk with other pathways acting simultaneously with Wnt/β-catenin in transcriptional activation self-renewal or differentiation pathways. Additional research is needed to asseverate any potential implications of Wnt signaling acting in concert with nutritional pathways in ESCs in the maintenance of pluripotency and during differentiation.
As disparate niche systems provide unique metabolic requirements for cells, the ability of Wnt to fine-tune nutritional and metabolic pathways may at least be partially responsible for guiding the transition of differentiating cells toward specific terminal lineages best fit for the niche being created in part from the gradient effects. Involvement in microenvironmental metabolic control may help explain how a single pathway could steer cells toward differentiation and maintain grounds for self-renewal, depending on cellular context.
Lessons from Neoplastic Transformation
Cancer cells exhibit many characteristics of stem cells, as evidenced by their reactivation of stem cell genes and pathways [120], metabolic profile [121,122], and genomic structural organization [123,124]. Wnt signaling is frequently activated in many types of tumors by various mechanisms [100]. For this reason, characterization and analysis of the effects of canonical Wnt/β-catenin signaling in different cancer types may provide important clues concerning contextual signaling. Expression of β-catenin in cancer is confined to specific tissues and cell types, but many parallels can be found that offer insights into mechanisms of self-renewal and cell fate decisions that are mediated by Wnt/β-catenin activity. Cancers alter the balance between cell division and differentiation by targeting pathways essential to cell fate determination [125]. Because of the central role of canonical Wnt signaling in orchestrating the finely tuned transition between self-renewal and differentiation during normal physiological development, Wnt-expressing cancers are well-equipped to utilize the bidirectional ability of differentiated cells to reactivate some of these pathways, generating a subset of cancer stem cells (CSC) in the process [126]. These cells can serve as tumor-initiating cells (TIC) or generate subclones independent of the initial TICs yet entirely capable of facilitating tumor growth, dissemination, and colonization of new regions. The dynamic nature of tumor evolution in recapitulating both developmental and regenerative processes dependent in part on canonical Wnt signaling [100,119,126 –128], and the interest in targeting this pathway for cancer therapeutics [129 –131], underscore the importance of elucidating common themes behind physiological and pathological activation of canonical Wnt/β-catenin signaling.
Mechanisms of Wnt pathway activation in cancers
Constitutive Wnt/β-catenin signaling is oncogenic in certain cancer models (Fig. 4A, B), including, but not limited to, colorectal [132], head and neck [133,134], and liver [135,136] cancers. The canonical Wnt/β-catenin pathway is additionally associated with tumor suppression in melanomas (Fig. 4C) [137,138]. Intestinal crypt cells and oral keratinocytes are two examples of cells subject to transformation accompanied by increased Wnt signaling. Constitutive β-catenin activation in concert with enhanced nuclear factor kappa-B signaling in an inflammatory microenvironment is sufficient for transformation of differentiated intestinal epithelial cells to tumor-initiating cells in mice [98]. Wnt enhances oncogenic Ras signaling in colorectal cancers by inhibiting GSK-3-dependent degradation of Ras [139] while negatively regulating Ras in melanomas [138]. Inactivating mutations in APC are the most frequent genetic alteration in colorectal cancers [140,141], and are an early event [142], providing a mechanism for enhanced β-catenin signaling in driving neoplastic transformation. In contrast, the antagonistic effects on Ras oncogenic signaling and its function as a tumor suppressor in melanocytes further signify that the nature of Wnt signaling is determined by cellular context and not by intrinsic functional activity.
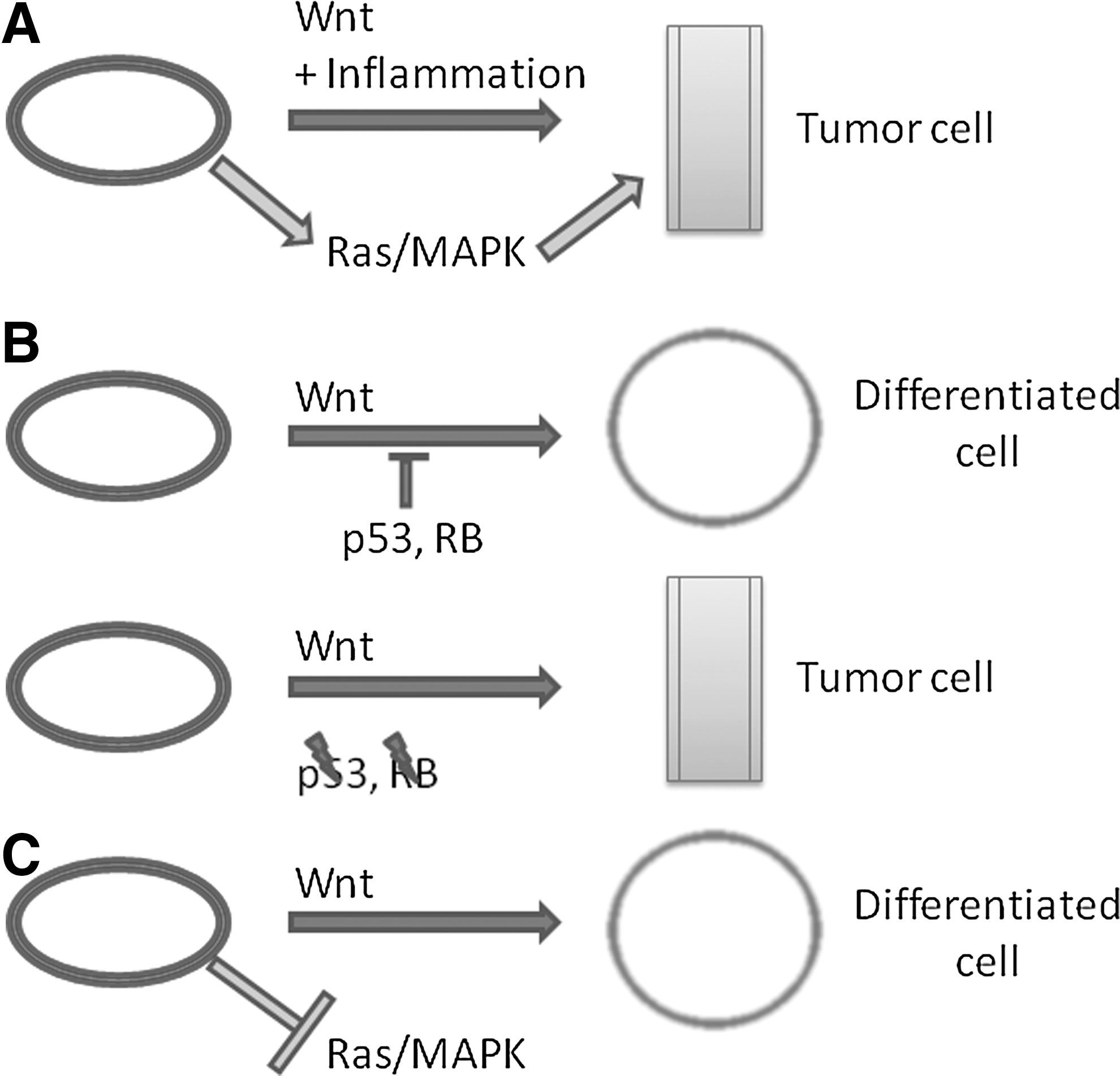
Proposed model of Wnt oncogenic and tumor suppressor activity in cancer.
Adult stem cell niches provide an ideal environment for unraveling mechanisms of heterologous Wnt signaling. They can also be informative in the endeavor to understand how dysregulation of this pathway can drive neoplastic transformation. Context-dependent responses to the canonical Wnt/β-catenin pathway have been elucidated to a large extent in the leucine-rich repeat-containing heterotrimeric guanine nucleotide-binding protein-coupled receptor (Lgr)5-expressing stem cells of intestinal crypts. Lgr5 is a member of the Wnt signaling pathway and is encoded by a Wnt target gene [143]. Paneth cells adjacent to Lgr5+ stem cells in intestinal crypts provide one source of Wnt for stem cell signaling [144]. Deletion of APC in crypt stem cells, but not their Lgr5− progeny of transit-amplifying cells, leads to a dramatic expansion of crypt cells and rapid adenoma formation in mice [145]. These adenomas are then sustained by additional Lgr5+ cells within the adenoma, as determined by lineage retracing using a tamoxifen-inducible multicolor “Confetti” Cre-reporter system [146]. While these data strongly support a hierarchical model of cancer initiation from one transformed progenitor population, a multiclonal origin of colorectal cancer is also possible, as differentiated Lgr5− cells can spontaneously give rise to Lgr5+ cells that can also substantially contribute to tumor development in the presence of inflammation and constitutive β-catenin signaling [98]. Several inferences can be made from these studies: (1) heterologous signaling within a stem cell niche allows precise regulation of Wnt signaling in normal crypt stem cells to maintain homeostasis; (2) dependence on Wnt signaling for proliferative function is not conserved between crypt stem cells and their progeny, as transit-amplifying cells with identical APC mutations rarely give rise to adenomas; (3) loss of paracrine regulatory control by an adjacent cell population due to mutation(s) permitting cell-autonomous Wnt effector signaling disrupts the balance between cell proliferation and differentiation sufficient to initiate precancerous lesions; and (4) crosstalk between Wnt signaling and signaling secondary to additional physiological factors such as inflammation may allow lateral conversion of differentiated cells to CSCs that can contribute to tumor growth.
Activation of the canonical Wnt pathway is driven by APC mutations in up to 83% of colorectal cancers [141], but this mutation is less common in other cancers. Mutations unrelated to the β-catenin destruction complex are also targeted in some cancers. The β-catenin binding partner, Fat1 cadherin, paradoxically exerts either oncogenic or tumor suppressive properties in glioblastomas and head and neck squamous cell carcinomas (HNSCC) [147 –149]. FAT1 gene mutational inactivation is common in many cancer types [148,150], implicating it as a tumor suppressor gene. Diminished Fat1 levels lead to re-localization of β-catenin from the plasma membrane to the nucleus for transcriptional activation of oncogenic targets [148]. However, consistent with the dual nature of β-catenin, overexpression of Fat1 was shown to co-localize β-catenin to the leading edge of an oral squamous cell carcinoma (OSCC) cell line, triggering invasion. This was inhibited by Fat1 knockdown and redistribution of β-catenin [147]. An Oncomine bioinformatics search revealed that Fat1 is more frequently upregulated than deleted in tumors of patients with OSCC [151]. Intratumoral heterogeneity is common, and metastatic lesions often arise from unique founder cells or colonies. Thus, genetic signatures of metastatic colonies may be disparate from clones prominently featuring some of the driver mutations that offer a selective advantage for growth of the primary tumor [152]. As a result, the requirements of tumors at different stages of development can change drastically as the tumor adapts to the constant physiological fluxes in energy demands and environmental changes over time, leading to seemingly conflicting data in isolated model systems that are highly context-dependent.
It is becoming increasingly clear that Wnt signaling is required for homeostatic functions that are regulated in disparate ways in different cell types and organ systems [153 –157]. In cell types requiring β-catenin for expansion and extravasation of progenitor cells, there are often key barriers that must be overcome for β-catenin to exert an oncogenic influence. Compensatory signaling responses may factor into context-dependent Wnt/β-catenin activity. Expression of p53 is critical, as this master tumor suppressor acts in concert with other cell cycle regulators to block the effects of many key oncogenes. Importantly, p53 acts as a negative regulator of canonical Wnt signaling [69,158]. Of note, p53 is also a potent inducer of differentiation in ESCs [159] and iPSC [160]. In HNSCC, Wnt signaling is often associated with inactivation of p53 and/or retinoblastoma protein (pRb) in both chemical-induced [161] and virus-initiated [162] carcinomas. Constitutively active β-catenin increases clonogenicity in p53-competent nasopharyngeal carcinomas but also elicits a compensatory response from p53-dependent signaling [69], further highlighting the importance of p53-mediated feedback mechanisms in preventing pathologic Wnt signaling [158,163]. In cancer progression, several barriers to tumor cell expansion and metastasis are obviated by mutation or inactivation of p53 and/or additional key regulators depending on cell type, and unopposed activation of the canonical Wnt pathway can confer a selective growth advantage in these tumors (Fig. 4B) [152].
Wnt: an incendiary in tumor evolution?
Cancer cell dedifferentiation is not a linear reversion to a pre-differentiated state but rather a disorganized adaptive response in which genomic architecture takes on chimeric characteristics of ESCs, tissue-specific progenitor cells, differentiated tissue-specific cells, and even cells from completely unrelated tissue types [123,124]. This is in stark contrast to ESCs, which maintain a naïve pluripotent state and regulate Wnt/β-catenin signaling in a manner that prevents untimely loss of ground state pluripotency in the absence of additional signaling components that direct cellular response toward a more specific fate. Notably, Tcf3 abundantly occupies super-enhancers of many differentiated cell types [58]. Super-enhancers are large enhancer regions enriched in master transcription factors associated with cell identity [57,58,123]. They are sensitive to perturbations due to cooperative binding of master transcription factors [123], hence they provide different cell types with plasticity in response to external changes to their components. The presence of Tcf3 at these regions in different types of differentiated cells may confer sensitivity to reactivated β-catenin signaling in the presence of permissive chromatin states, allowing for large-scale changes in genomic organization in addition to net genetic changes. It is tempting to speculate that some of the changes observed in dedifferentiation and EMT in response to β-catenin activation may relate to this hidden plasticity that remains in the genomes of differentiated cells. Intriguingly, metastatic cancers consist of very few differences in mutation signatures compared with those of primary cancers, and instead display greater divergence in epigenetics and heterologous signaling from niche factors [164]. Two distinct drivers of metastasis that are secreted by the metastatic niche are tenascin C (TNC) and periostin. Both factors sensitize circulating tumor cells to the effects of Wnt (and Notch in the case of TNC) to allow their survival in hostile microenvironments that would normally lead to the demise of disseminated cancer cells [165].
Tissue regeneration models have shown that Wnt acts in a paracrine, non-cell autonomous manner by stimulating collective expansion of proximal cells following increased Wnt ligand secretion by stable β-catenin mutant-expressing progenitors [166]. It is not difficult to imagine how, in oncogenic scenarios, a similar mechanism may be responsible for co-opting neighboring cells to take on the cancer-initiating or metastasis-promoting characteristics of the founder cell population in cancers where the Wnt/β-catenin pathway is an oncogenic driver. This would allow for rapid spreading of the malignant phenotype in a favorable environment. A major advantage of the overexpression of a secreted oncoprotein such as Wnt during tumor growth is that more than one distinct cell population can be repurposed to benefit the tumor during its evolution. In this regard, tumor heterogeneity is not strictly the competition between specific neoplastic subclones but involves a cooperation that provides a selective advantage for the growing tumor holistically. Proof of concept for cooperative evolution of biclonal tumors was established using mammary tumor cells from a doxycycline-inducible Wnt1 transgenic mouse model to trace the lineages of tumorigenic cells following injection into control and constitutive Wnt1-expressing host recipients [167]. Hras mutations in mammary cells frequently evolve secondary to Wnt1 overexpression in mammary epithelial stem cells [168]. A combination of sorted Wnt1hi, Hras WT luminal cells and Wnt1lo, Hras mut basal cells efficiently reconstituted biclonal tumors originating from donor cells, whereas individual subclones were much less effective [167]. Moreover, reactivation of Wnt signaling by heterologous Wnthi luminal cells or acquisition of downstream Wnt effector mutations in basal cells led to resistance against Wnt depletion by doxycycline withdrawal, causing tumor relapse [167]. In addition to non-cell autonomous effects of Wnt signaling in perpetuating tumor evolution, late activation of canonical Wnt/β-catenin signaling in a cell type harboring prior copy number aberrations and mutations favoring tumorigenesis would be expected to improve tumor cell fitness and transmit more severe phenotypes. Indeed, this is the case in chronic myelogenous leukemia, in which an initial Bcr/Abl translocation is frequently followed by mis-splicing of GSK-3β, causing increased β-catenin signaling [169]. This subsequently leads to blast crisis, which greatly increases drug resistance and mortality. These examples from different tumor types reveal how aberrant Wnt signaling can promote tumor evolution in both hierarchical and multiclonal models of tumorigenesis.
As cancers reactivate several developmental pathways normally silenced in differentiated cells, they provide an ample model for comparison to more primitive developmental stages where coordinated cellular responses drive cell fate specification through dynamic changes in primitive cells exhibiting high degrees of plasticity [170]. A major role of Wnt signaling during dedifferentiation of tumor cells might be to provide a mechanism for reversion to such a progenitor-like state. Wnt overexpression or downstream alterations that enhance Wnt activity likely offer a selective advantage to cancer cells that is accompanied by an overall change in the cellular genetics and tissue microenvironment to favor conditions amiable for tumor development and/or spreading, such as through EMT induction to escape a hostile or hypoxic environment in the region of the primary tumor [171]. The importance of pro-inflammatory signaling in colorectal cancer [98] and HNSCC [172] conform to the hypothesis that chronic inflammation favors selection of colonies capable of enhanced oncogenic or metastatic activity. Increased cytokine and chemokine signaling may aid in inducing the primitive EMT-facilitating microenvironment in adult β-catenin-expressing cells, so the intrinsic invasive capabilities of primordial cells of the PS is regained in an optimal environment for driving processes conserved from those mechanisms that evolved for the initial invasive ingression necessary for coordinating mesoderm and endoderm development [173,174].
Conclusions/Final Perspectives
Wnt signaling is finely tuned to regulate cell fate decisions based on integration of several criteria, including timing of development, parallel metabolic and genetic regulation, localization patterns, concentration, and interaction with other key pathway components. The controversy over functional roles of Wnt in self-renewal or differentiation dissipates in the light of recent findings highlighting the importance of cellular context. The question of whether canonical Wnt signaling is necessary for self-renewal has gradually led to studies that have aided in unraveling the different contexts by which this Wnt/β-catenin gatekeeping switch exerts specific actions that guide the fate of stem or progenitor cells of diverse cell types. The underlying epigenetics, metabolic state, and interactome profile are taking center stage in attempts to harness this powerful signaling pathway for endeavors in disease modeling, cancer pharmacology, and regenerative medicine.
As the genetic, epigenetic, and biochemical characterization of ESCs in various stages of pluripotency and differentiation toward specified lineages become more clearly defined, the context of Wnt/β-catenin signaling in various stages of development will be more closely recapitulated in vitro to uncover mechanistic links not previously anticipated due to the plasticity of stem cells and their tight coordination during developmental processes. Understanding the disparate mechanisms of pleiotropic signaling in various contexts will be crucial, as targeting one particular arm of this pathway can lead to consequences that either prevent the treatment from being effective or potentially enhance the toxic effects of connected pathways. For example, cancer therapies targeting β-catenin transcriptional effects may be beneficial in tumors where destruction complex machinery is inactivated, and Tcf/Lef transcriptional activation leads to expression of oncogenic genes, such as c-Myc [148,175]. However, caution must be undertaken not to enhance cytoplasmic and membrane-associated activities that can lead to EMT and invasion along with disruption of normal cell adhesion. Studies in ESCs have greatly aided in elucidating the seemingly conflicting roles of Wnt/β-catenin activity, and these may provide a cautionary note against broadly targeting specific pathway components without genetic sequencing and detailed analysis of tumor phenotype, including the microenvironmental compartment and invasive potential. Despite these warnings, greater clarification on the canonical Wnt/β-catenin pathway will continue to be gained in the years ahead, which will provide novel insights into how Wnt biology can be controlled and exploited for therapeutic ends.
Footnotes
Acknowledgments
I thank Dr. Ali Brivanlou and Dr. Elaine Fuchs of the Rockefeller University for their helpful comments during the preliminary drafting of this article. I would also like to thank Dr. Lorraine Gudas, Dr. Kwame Osei-Sarfo, and members of the Gudas laboratory for their helpful discussions and insights into Wnt biology.
Author Disclosure Statement
I have no conflicts of interest to report with regard to this article.