
Abstract
Stem cell therapy is a potential method for the treatment of numerous diseases. The most frequent cellular source is bone-marrow-derived mesenchymal stromal cells (BM-MSCs). Human adipose-derived stromal cells (ADSCs) share similar properties with BM-MSCs as they support hematopoiesis, modulate ongoing immune responses, and differentiate into cells of mesodermal origin. On the other hand, ADSCs have higher frequency in situ, higher availability, and very few ethical issues compared with BM-MSCs, giving them an advantage over BM-MSCs for clinical use. Most of the methods used to isolate ADSCs contain a collagenase digestion step, but the type of collagenase and time of sample digestion vary among studies and these differences could have an impact on the cell properties and thus in result comparison. To overcome this obstacle, we propose a new method to isolate ADSCs from lipoaspirate without collagenase digestion step. We compared ADSCs obtained with our method versus classical protocol using collagenase digestion. Cells obtained with our method are equivalent but they have a better long-term hematopoietic support than those obtained with classical method. Moreover, our method has an advantage over the classical one as it is easier, safer, faster, less expensive, and more consistent with good manufacturing practices to obtain large number of ADSCs ex vivo.
Introduction
M
There is currently no specific marker described to characterize MSCs. In 2006, the International Society for Cellular Therapy (ISCT) proposed a standard set of rules to define the identity of these cells. Thus, MSCs must be plastic adherent in standard culture conditions; they must express surface molecules, such as CD105, CD73, and CD90, and they should express neither hematopoietic, nor endothelial markers (CD45, CD34, CD14 or CD11b, CD79a, or CD19) nor MHC class II; and they should be able to differentiate into osteoblasts, adipocytes, and chondroblasts in vitro [5].
The MSCs are considered as good candidates for clinical use due to the following properties. They are able to support hematopoiesis, they have an immunomodulatory capacity, and they are able to differentiate into different cell types [5]. In reconstructive surgery [6,7], cardiology and neurology [2,8], MSCs could be used to repair wounded zones [9 –11]. Nevertheless, the effectiveness of MSCs in reparative medicine seems to be more dependent of their trophic potential than of their capacity to differentiate into the cells of appropriate tissue [12]. MSCs are nonimmunogenic as they express neither costimulatory molecules nor MHC class II and they do not trigger an immune response in an allogeneic setting [13]. The MSC immunomodulatory properties have been quite well documented over the last few years [14]. These cells exhibit capability to suppress the activation and proliferation of different immune cells, such as T-cells [15,16], B-cells [17], NK-cells [18,19], and dendritic cell [20].
Apart from the BM, MSCs have been isolated from various human tissues, such as adipose tissue (AT) [21], skin [22], dental pulp [23], cord blood [24], conjunctive tissue from the umbilical cord (called Wharton's jelly) [25], placenta [26], and others [27]. Adipose-derived stromal cells (ADSCs) share similar properties with BM-MSCs, leading some authors to present them as identical. However, both populations differ in terms of phenotype, proliferation, and functions. These differences could be explained by (a) the different microenvironments where these cells reside in their respective tissues of origin and by (b) the differences in their ex vivo expansion protocols [28]. The advantages of ADSCs over BM-MSCs are their higher frequency in the tissue [29], availability, and presence of very few ethical issues.
Isolation protocols of MSCs from ATs are not standardized and need to be harmonized [10]. Most of the studies report the use of adipose stem/stromal cells isolated by a method based on enzymatic digestion; however, time of digestion with collagenase varies among studies [28]. Enzymatic digestion can induce cell injury and alter cell functions [30]. Multiplying protocol steps and adding xenobiotics increase the risk of contamination and the difficulties to generate cellular product in good manufacturing practice (GMP) conditions [31]. Here, we propose a new method of isolation that is easier, safer, faster, less expensive, and more consistent with GMP to obtain large number of ADSCs ex vivo.
Materials and Methods
Tissue samples
Lipoaspirates (LAs) were obtained from female patients (n=25) undergoing reconstructive lipofilling with informed written consent. Ethic approval was obtained from the institutional ethics committee. Surgeons used Coleman's techniques for removal and lipofilling [32]. Briefly, 20 min prior to collection, a volume of physiological serum with adrenalin (1:400,000) equivalent to the defect to be corrected was infiltrated into the donor site to reduce bleeding (equivalent to the volume to correct). The removal was done with 10-mL syringes and a hollow blunt-tipped cannula. Samples were obtained from supernumerary syringes and processed within a few hours after surgery.
Isolation and ADSC culture
Fresh samples were split into two equal parts to realize the primary culture. The nondigested sample part (collagenase free) was directly plated on the plastic surface in a cell culture flask (Greiner) for 5 days in the culture medium, that is, Dulbecco's modified Eagle's medium (DMEM) with 1.0 g/L glucose, without
When subconfluency was reached for one condition, both with and without collagenase digestion, adherent cells were harvested after detachment with TrypLE Select (Gibco; 10 min, 37°C), washed in PBS, counted, and analyzed for their growth, clonogenicity, and antigen expression. Cells were expanded until passage 3 (P3) by replating at low density (50,000 cells/75 cm2). Differentiation, immunomodulation, and prohematopoietic activity were realized on cells obtained after P2.
Growth and clonogenicity
Viability and cell number were evaluated for each passage by trypan blue (Gibco) exclusion assay. The cumulative cell number was calculated by the cumulative addition of the total cell number obtained at each passage. Cell growth was calculated for each passage using the population doubling formula:
Phenotype
For each passage, the phenotype of ADSCs (n=7) was analyzed. Cells were incubated with monoclonal antibodies (Table 1) for 30 min at room temperature in the dark. After this incubation, cells were washed with MacsQuant Running buffer (Miltenyi Biotec) and fixed with 8% formaldehyde. Data were acquired on a MacsQuant analyzer and analyzed with FCS 3 Express software (DeNovo Software).
In vitro differentiation assays
To assess the multipotential capacity of ADSCs, osteogenic, adipogenic, and chondrogenic differentiation were performed on cells obtained at P2.
Osteogenic differentiation
Two thousand cells per well were plated in a 48-well plate with culture medium. After 5 days, medium was completely discarded and replaced by osteogenic medium (NH OsteoDiff Medium; Miltenyi Biotec). Cells were fed weekly with complete replacement of osteogenic medium. At days 7, 14, and 21, mineralization was demonstrated by Alizarin Red staining after cell fixation (8% formaldehyde); calcium (Ca2+) deposits and alkaline phosphatase (ALP) activity were measured using colorimetric assays [QuantiChrom Calcium Assay Kit (DICA-500), BioAssay Systems and LabAssay ALP, Wako Chemicals, respectively] as described by the manufacturer.
Adipogenic differentiation
Two thousand cells per well were plated in a 48-well plate with culture medium. After 5 days, medium was completely discarded and replaced with adipogenic medium (NH AdipoDiff Medium; Miltenyi Biotec). Cells were fed weekly with complete replacement of adipogenic medium. At days 7, 14, and 21, cells were stained with Oil Red O solution (Sigma) after fixing (8% formaldehyde). Lipid vacuoles were observed and color intensity was measured by spectrophotometry (492 nm; Organon Teknika Cappel Products).
Chondrogenic differentiation
One hundred fifty thousand cells were cultured in the tip of a 15-mL conical tube (Greiner) to enable cell culture in micromass with chondrogenic medium (NH ChondroDiff Medium; Miltenyi Biotec). Cells were suspended carefully and cultured at 37°C with 5% CO2 humidified atmosphere with cap slightly screwed. Half of the chondrogenic medium was replaced weekly. At day 21, aggregates were stained with Alcian blue to highlight cartilage proteoglycans. In some cases, cryosectioned pellets were stained with Alcian blue to confirm chondrogenic differentiation.
Hematopoiesis-supporting activities
MSCs are an essential component of the BM niche for maintenance of stemness of hematopoietic stem cells. They produce important hematopoietic cytokines and support hematopoiesis in long-term cultures [33 –36]. ADSC hematopoiesis-supporting activities were evaluated by blast colony-forming cell (Bl-CFC) assay and long-term culture-initiating cells (LTC-ICs). These assays serve to evaluate the abilities of MSCs to support short-term hematopoiesis and long-term survival of hematopoietic progenitors. In long-term culture, the persistence of primitive progenitors was assessed by transferring cells into a semi-solid medium to enable secondary hematopoietic colonies to develop.
Bl-CFC assay
Bl-CFCs represent a specific subpopulation of relatively primitive hematopoietic progenitors characterized by colony formation only in close contact with a preformed MSC layer.
Five thousand CD34+ cells were isolated from umbilical cord blood (after informed consent of the mothers) with MidiMACS Separation (Miltenyi Biotec) and cultured in 250 μL of semi-solid medium MethoCult H4434 Classic without cytokines (Stem Cell Technologies) for 10 days on a confluent ADSC layer. At days 5 and 10, refringent colonies (>20 cells) were counted using an inverted microscope (Leica).
LTC-IC assay
Fifty thousand purified CD34+ cells (same isolation protocol as described for Bl-CFC assay) were cultured for 5 weeks on a confluent untreated ADSC layer in 1 mL of DMEM, identical to that of ADSC culture (see “Isolation and ADSC Culture” section for details). Five hundred microliters of the medium was replaced weekly. After 5 weeks, cells were harvested with the medium and counted by trypan blue exclusion assay. Distinction was made between ADSCs and LTC-ICs by their difference in size and morphology. Then, 50 μL of the cells was cultured for 21 days in 250 μL of semi-solid medium MethoCult H4434 Classic with cytokines and erythropoietin (EPO; Stem Cell Technologies). At days 7, 14, and 21, the total number of CFUs comprising granulocytic-erythroid-macrophage-megakaryocyte CFU (CFU-GEMM), myeloid granulocyte-macrophage CFU (CFU-GM), and erythroid burst-forming units (BFU-E) was counted with an inverted microscope (×100; Leica).
Immunomodulation assay
Peripheral blood mononuclear cells were obtained by Ficoll-Hypaque gradient centrifugation of peripheral blood from healthy donors with informed consent. Purification of T-lymphocytes (>95% purity) was performed by positive selection using the MACS system (Miltenyi Biotec). Activation of T-lymphocytes was accomplished using mitogenic stimuli [phytohemagglutinin (PHA, 5 μg/mL; Remel) and interleukin-2 (IL-2, 20 U/mL; Biotest AG)]. Activated T-cells and ADSCs obtained through either collagenase-free or collagenase-treated method were plated in cocultures (cell ratio: 1/4) during 5 days. Lymphocyte proliferation was assessed using the CellTrace™ CFSE Cell proliferation kit (Invitrogen, Molecular Probes) and analyzed by flow cytometry as previously described [37].
Statistical analysis
A total of 25 lipoaspirated samples were analyzed. Data are represented as mean±standard error of the mean. Comparison between the two methods was evaluated with the Wilcoxon matched pair test (two-tailed) and differences were significant for P<0.05. All analyses were performed with GraphPad Prism version 5.00 for Windows (GraphPad Software,
Results
Preparation of AT samples
When undergoing reconstructive lipofilling, the average age of 25 female patients was 53±6.7 years with an average body mass index of 24±2.5. The LAs (Fig. 1A) were directly transferred into cell culture flask and the ADSCs were isolated, without any enzymatic treatment, based only on their capacity to adhere to plastic surfaces. A small amount of culture medium was added and the flasks were gently shaken to distribute the aggregates of LAs on the entire surface (Fig. 1B). Adipose aggregates were kept in contact with the plastic surface, requiring the medium volume to be regularly controlled during the first 5 days of culture.

Isolation of ADSCs from LAs.
After 5 days, LAs were discarded by gentle rinsing and fresh culture medium was added. At this time, some colonies of fibroblastic-like cells were observed in collagenase-free samples (Fig. 1C) as well as in collagenase-treated ones. Cell cultures were pursued until reaching subconfluence and then cells were expanded by successive passages. This enzyme-free method for ADSC isolation is easy, fast, and reliable, and reduces the risk of contamination.
Growth and clonogenicity
One of our study's objectives was to quickly obtain a large number of ADSCs from easily available tissue for putative clinical applications. We evaluated the efficiency of our ADSC isolation from AT protocol by determining the cumulative number of cells obtained after three passages and by analyzing their clonogenicity by CFU-F assay. From the 60 LAs obtained from different patients (not all were included in this study due to lack of information or data), our method was successful in 97% of cases. One sample was contaminated and one sample showed no growth event in the case of enzymatic treatment. For the 25 LAs evaluated in this study, after 21±1.52 days of culture from 1 mL of LAs, 4.63±1.49×102 ADSCs were obtained with the collagenase-free method versus 3.95±0.88×102 ADSCs obtained with the collagenase method. After three passages (95.9±5.1 days of culture), 1 mL of LAs gave 4.20±1.13×108 cells using the collagenase-free method versus 3.34±0.79×108 cells from collagenase-digested samples (Fig. 2). The ADSC population doublings observed for the collagenase-free method and for the collagenase digestion method were, respectively, 2.97±0.79 and 2.90±0.53 at P1 (P=0.76), 6.55±0.79 and 6.48±0.54 at P2 (P=0.72), and 9.85±0.95 and 9.81±0.63 at P3 (P=0.81).

Growth and clonogenicity of ADSCs during the different passages.
The number of CFU-F for both cell populations was determined after each passage to evaluate ADSC clonogenic capacities (n=25). CFU-F assay makes it possible to estimate the number of mesenchymal progenitor cells. After three passages, from 1 mL of LAs, no statistical difference was observed; we obtained 6.29±3.31×105 CFU-F with the collagenase-free method versus 4.41±2.45×105 CFU-F with the collagenase method (P=0.29) (Fig. 2).
Phenotype
For each passage, cell phenotype was analyzed by flow cytometry for the expression of mesenchymal, endothelial, and hematopoietic markers (Fig. 3). After primary culture, ADSCs were already positive for the MSC markers CD105, CD73, CD90, CD146, and CD166 and were negative for endothelial marker CD31, hematopoietic marker CD45, and HLA-DR. The percentage of positive cells for those markers was stable over passages. For the hematopoietic CD34 antigen, we observed a significant difference between cells obtained with the collagenase-free method (2.17%±0.93% and 3.16%±1.14% after the first and second passages, respectively) and those obtained with the collagenase method [4.59%±1.17% and 8.06%±2.42%; P<0.05 (P1) and P=0.05 (P2)]. There is also a difference for the percentage of CD90-positive cells after primary culture (82.23%±4.00% vs. 90.75%±2.02%, respectively; P=0.07) and after P2 (88.06%±5.00% vs. 92.97%±4.24%, respectively; P<0.05) (Fig. 4).
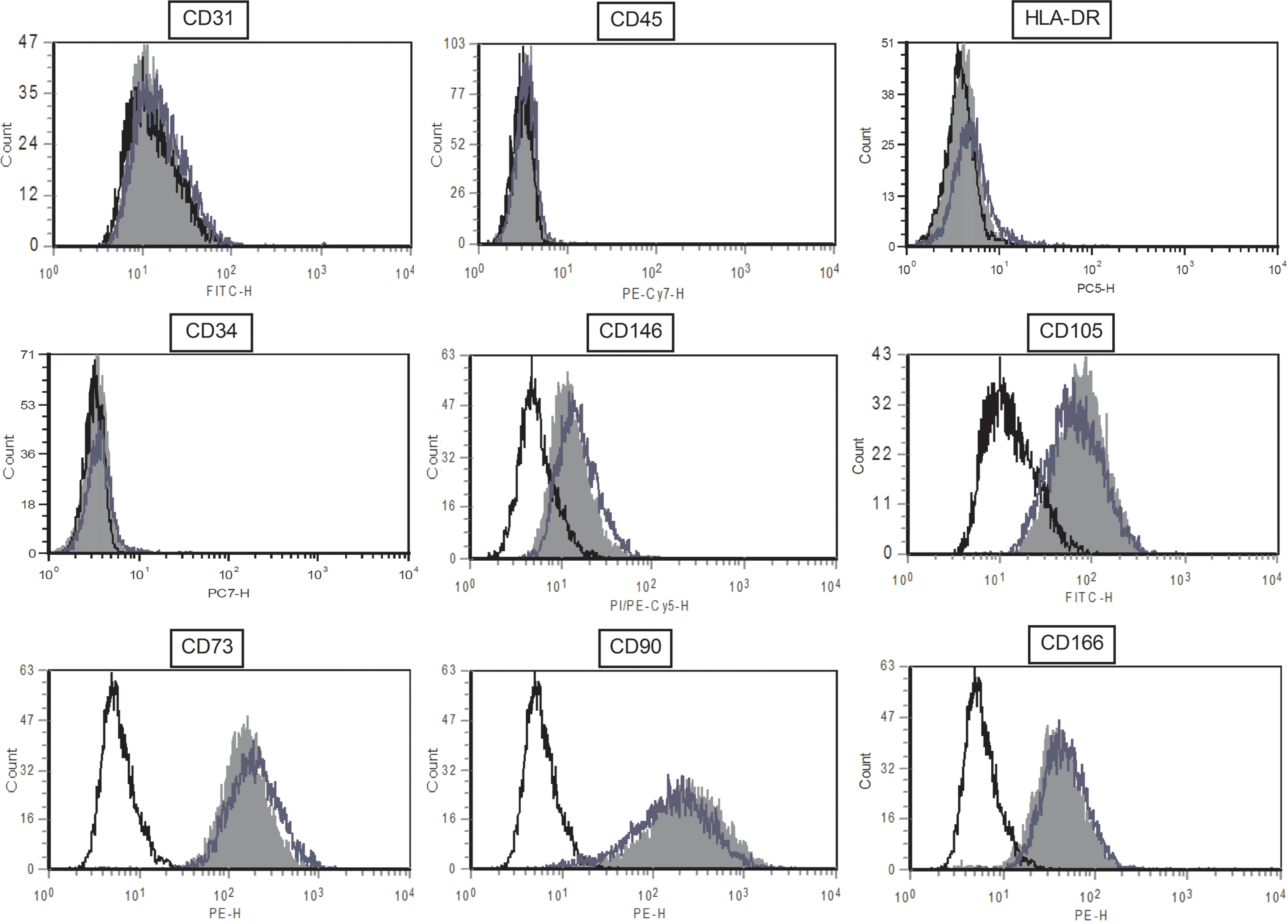
Flow cytometric analysis of ADSC surface marker expression. Data shown are a representative cell phenotype analyzed at P1 (black line: control; blue line: collagenase-free method ADSCs; solid gray histogram: collagenase-treated ADSCs). Color images available online at

Flow cytometry analysis of ADSC surface marker expression during passages. Percentage of positive cells from PM to P3 for the expression of mesenchymal, endothelial, and hematopoietic surface antigens. Results are expressed as mean±SEM (n=7). *P≤0.05; **P≤0.01. SEM, standard error of the mean.
ADSC differentiation potential
To meet the criteria of the ISCT, we evaluated the differentiation potential of ADSCs obtained by our method in comparison with ADSCs obtained after enzymatic treatment.
Osteogenic differentiation
Osteogenic differentiation was demonstrated by Alizarin red staining detecting matrix calcification (Fig. 5A) and confirmed by colorimetric dosage of calcium deposits and ALP activity (n=20). ADSCs obtained with both methods were able to produce a comparable calcificated matrix (Fig. 5B). No difference was observed in terms of calcium level and ALP activity (Fig. 5C).
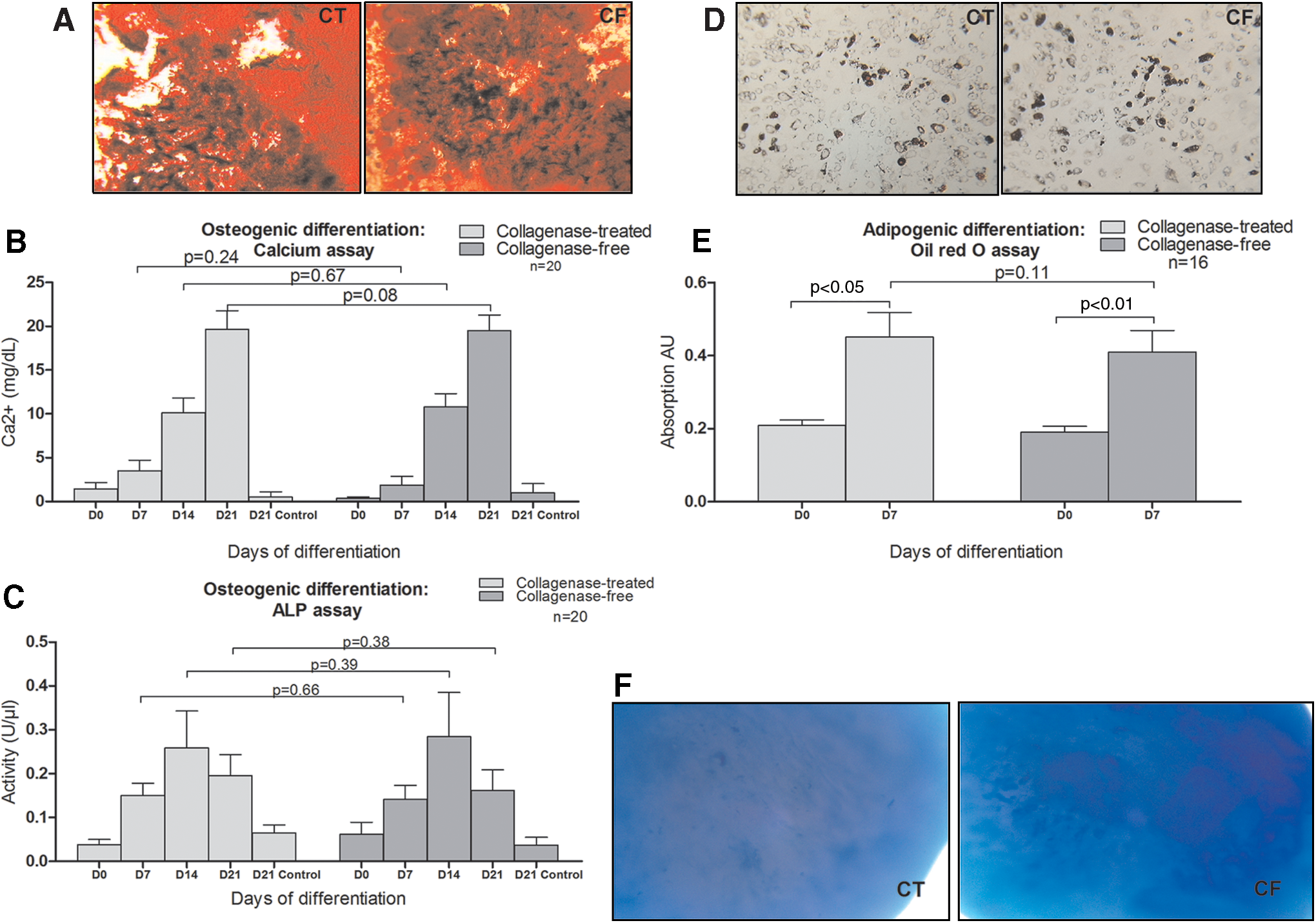
ADSC differentiation into osteoblasts, adipocytes, and chondroblasts.
Adipogenic differentiation
Adipogenic differentiation was observed after 7 days by accumulation of lipids in small vacuoles stained with Oil Red O (Fig. 5D) and lipid accumulation was determined by colorimetry (Fig. 5E). ADSCs from the collagenase-free method and from the collagenase digestion method had a similar potential for adipogenic differentiation.
Chondrogenic differentiation
Chondrogenic differentiation was obtained after 21 days of induction in a pellet culture system and 2-mm-diameter pellets were obtained with ADSCs from both methods. Glycosaminoglycan matrix was shown by Alcian blue staining (Fig. 5F).
Hematopoiesis-supporting activities
To compare the potential of ADSCs isolated by two methods to support hematopoiesis, we evaluated the ADSC support of Bl-CFC proliferation from umbilical cord hematopoietic CD34+ stem cells without the adjunction of cytokines. To assess the hematopoietic-supporting ability of ADSCs, long-term cultures of umbilical cord CD34+ stem cells were established for 5 weeks using ADSCs as feeder layers. Secondary CFU production was used as a measure of primitive progenitor cell activity. Bl-CFCs were obtained within 5 days, with no statistical difference between ADSCs isolated using each method (Fig. 6A). ADSCs from both methods were also able to promote the survival of LTC-ICs with an equivalent efficiency: a mean of 31.2±2.2×104 LTC-ICs with the collagenase-free method versus 30.0±2.7×104 with the collagenase digestion method (P=0.51) was obtained (Fig. 6B). The LTC-ICs obtained were still functional, as they generated different secondary CFU-colonies: CFU-GEMM, CFU-GM, and BFU-E. Figure 6C shows the total number of CFU-Cs counted after 7, 14, and 21 days of culture with adequate cytokines and growth factors. LTC-ICs in contact with ADSCs obtained from the collagenase-free method seemed to be able to yield more progenitor cells after 14 days (61.07±10.41 CFU-Cs) and 21 days (67.88±15.57 CFU-Cs) than those from the collagenase digestion method (50.63±7.75 CFU-Cs at day 14 and 66.54±11.84 at day 21) (P<0.005 and P<0.01, respectively) (Fig. 6C). A 1.32±0.26-fold increase of CFU number between 14 and 21 days for the collagenase-free method can be observed versus a 1.66±0.30-fold change between 14 and 21 days for the collagenase method, which does not represent a significant difference (P=0.92, data not shown).
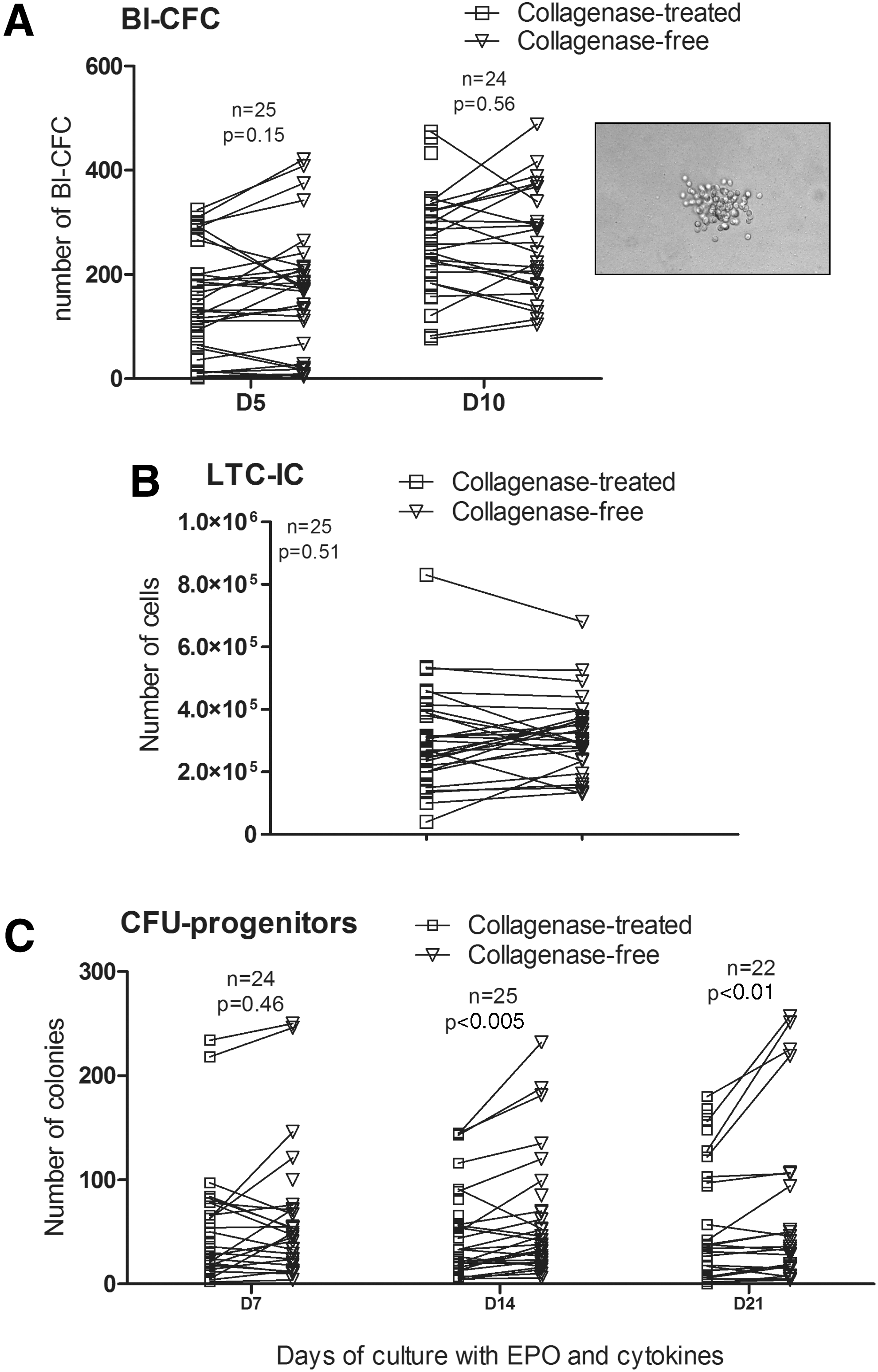
ADSC support of hematopoiesis.
Immunomodulation activities
MSCs have been shown to have immunosuppressive properties. The immunosuppressive effects of ADSCs obtained using both methods were evaluated on activated T-cells by measuring the percentage of mitogen-induced proliferating T-cells through CFSE labeling. When activated T-cells were incubated on ADSCs obtained through both methods, T-cell proliferation decreased significantly (P<0.01). This effect was similar with ADSCs obtained with or without collagenase treatment (28.40%±2.08% vs. 22.33%±1.52% of control T-cell proliferation, respectively; P=0.87) (Fig. 7).
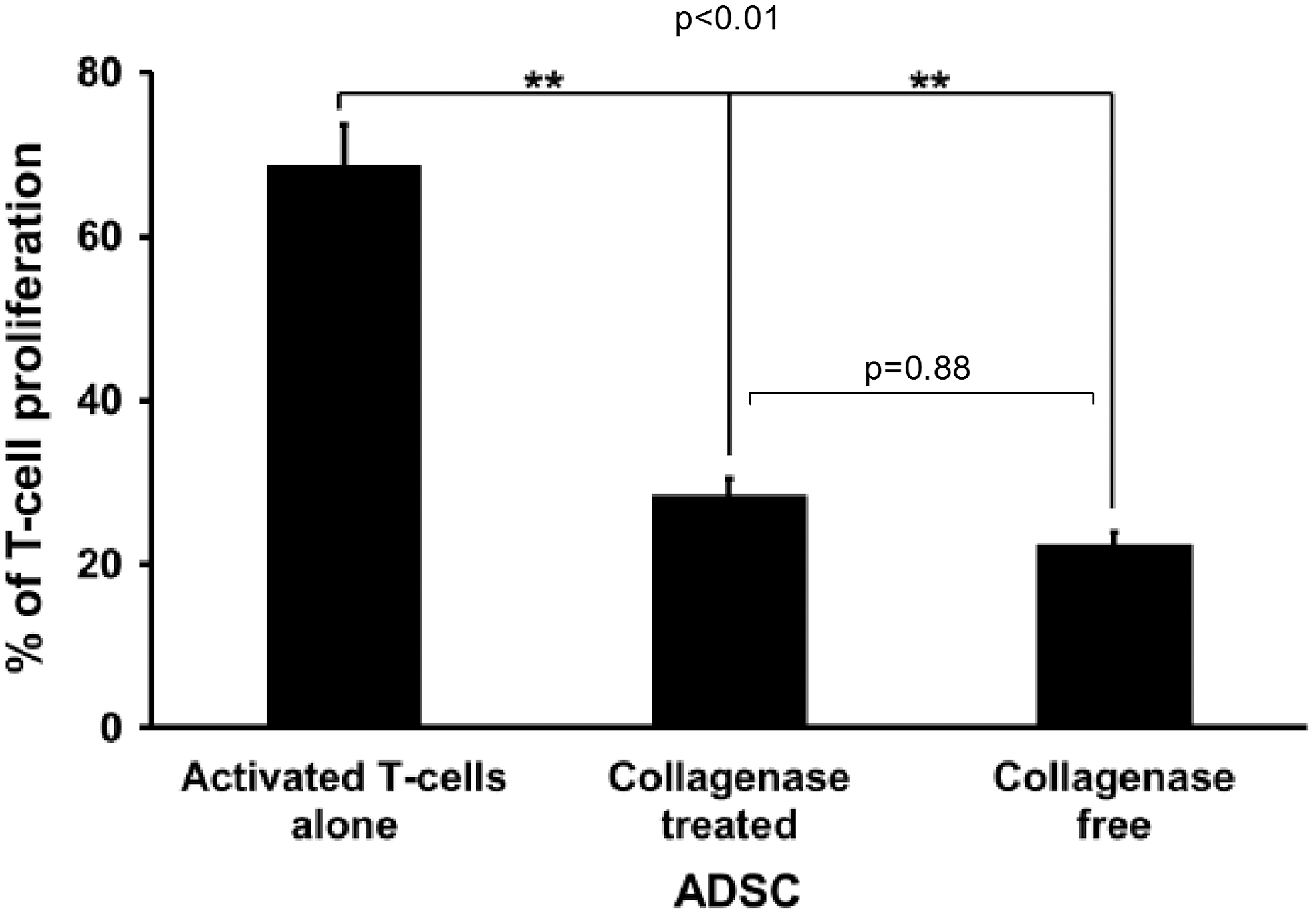
ADSC immunomodulatory capacities. Activated T-cells were labeled with CFSE dye and added to ADSCs (cell ratio: 1/4) isolated from adipose tissue treated or not by collagenase. After 5 days, lymphocyte proliferation was determined by measuring CFSE fluorescence. Data are presented as the mean percentage of lymphocyte proliferation±SEM. **P≤0.01. CFSE, carboxyfluorescein succinimidyl ester.
Discussion
We propose a new, nonenzymatic method to isolate ADSCs from LA samples. This method is easy, fast, and low cost as it is based on MSC’ property of adhering to the plastic surface. Our data showed that ADSCs obtained using this method had comparable growth capacity, clonogenicity, phenotype, multipotentiality, and immunomodulation potential in comparison with ADSCs obtained using the usual method (collagenase digestion). Interestingly, ADSCs obtained without enzymatic treatment had a higher potential to support hematopoiesis than ADSCs obtained using the usual method.
MSCs have been considered as an appropriate source of cells used in the treatment of a number of congenital and degenerative diseases [38]. At present, BM is the main source of cells although it presents some disadvantages, such as the invasiveness of the procedure, low MSC frequency, and aging [39]. AT seems to be an alternative source of MSCs for cell-based therapy. It can be obtained in large quantities through a simple liposuction and is often considered as a surgical waste.
For clinical use, reliable repeatable isolation of an adequate number of MSCs is of great importance. The usual method generally used to extract ADSCs from AT implies the use of collagenase for a digestion step [31]. Commercial collagenases from Clostridium histolyticum are often used in tissue digestion [40]. This bacterial collagenase, also called clostridiopeptidase A (EC. 3.4.24.3) [41,42], is usually not able to efficiently dissociate tissues alone because of the presence of other noncollagen proteins among the extracellular matrix[43]. Crude collagenase is more efficient due to the presence of other proteases in addition to the clostridiopeptidase, particularly clostripain [44], which presents a trypsin-like activity [45]. However, commercial preparations are heterogeneous and present different ratios of different enzymatic activities leading to inconsistent results [31]. Collagenase treatment can affect the quantity and quality of isolated ADSCs. Moreover, the duration of enzymatic treatment is very important as it may be associated with alteration of ADSC properties. Indeed enzymatic digestion may negatively affect cellular properties, mainly through modifications of the natural cell environment. In our study, we used a collagenase preparation presenting a normal-to-high collagenase activity and a very low tryptic activity to minimize unspecific degradation of the surface markers. Recently, Mulder et al. observed reduced expression of some T-cell molecules after collagenase or DNase treatments. Their work was mostly focused on DNase effects on lymphocyte membrane markers. The expression of the CD14 surface marker decreased by more than 50% but not in a significant manner after collagenase digestion. Nevertheless, this nonsignificance may be due to too few samples for this marker. CD19, CD45, and HLA-DR expression was not affected by the collagenase treatment [46]. To our knowledge, no study has been realized on the effects of enzymatic digestion on native MSC surface markers recommended by ISCT (CD105, CD73, CD90, and CD34). For a phenotype study after cell dissociation, it seems important to pay attention to the dissociation enzyme used in the study.
We observed very few differences between ADSCs obtained with our collagenase-free method and that obtained after the collagenase treatment. The percentage of CD34-positive cells was lower for cells obtained with our method. In contrast to BM-MSCs, native ADSCs are contained in the CD34+ cell population and its expression is negatively correlated with cell expansion in vitro [47]. Suga et al. have reported that CD34 expression decreased overtime in culture but this decrease depends on culture conditions such as cell density [48]. As a consequence, ADSCs have been reported to be CD34− in most publications.
In our culture conditions, only a weak percentage of ADSCs express CD34 (<3.5%) but the lower percentage observed for ADSCs obtained by the nonenzymatic treatment could be in relation with more effective cell adhesion and a greater cell density. Indeed treatment degrades the extracellular matrix and can prevent the cells from adhering to the culture substrate.
We also observed a significant difference in the percentage of CD90-positive cells, but ADSCs isolated by both methods remained highly positive for this antigen, as recommended by the ISCT [5].
We also evaluated the capacities of ADSCs to sustain the proliferation of hematopoietic progenitors in two culture systems. We confirmed that ADSCs obtained without enzymatic digestion were able to support hematopoiesis in the same way as ADSCs obtained through the usual method. However, ADSCs from the collagenase-free method were more efficient in supporting LTC-ICs by promoting a greater generation of CFU-Cs. Finally, we confirmed that ADSCs obtained through both methods were able to modulate the immune response in a similar way.
Similar phenotypes and functions between ADSCs obtained by enzymatic method or nonenzymatic method suggest that the properties of ADSCs are not compromised by our method. Since AT is a rich and alternate source of cells for cell-based therapy, one of the major challenges to be faced is to simplify GMP. This can be achieved by avoiding the use of nonhuman products for clinical purposes [49] and to develop a rapid and efficient method for their obtention. Carvalho et al. observed that the use of xenogenic reagents for the cell isolation protocol may be an unsuitable option in terms of patient safety and could present a high risk of infection and/or lead to severe immune reactions [31]. In that article, they compared several alternative enzymatic products for the isolation of ADSCs. An alternative solution to be considered, as we demonstrate here, may be to avoid this digestion step.
In addition, Faustini et al. argue that articles on the optimization of isolation techniques and product characterization are rare [50]. In 2010, our group has established a simple, rapid, and reproducible protocol to isolate abundant MSCs from short segments of umbilical cords [51]. The explant technique seems also simpler and less invasive for isolating MSCs from the synovium [52]. Isolation of ADSCs without tissue destruction has been reported by Ghorbani et al. [53]. However this nonenzymatic method is a two-step protocol requiring 24 h of incubation in fetal bovine serum. A rapid and efficient method described by Zeng et al. [54] enabled the obtaining of ADSCs keeping low level of senescence during long-term culture in vitro. However the functionality of ADSCs was not evaluated.
According to Faustini et al., “the use of minimally manipulated fresh cells may lead to higher safety and efficacy in actual treatments.” [50]. Our method is fully in agreement with those arguments. Moreover, our one-step protocol is reproducible and simple and can be quickly performed.
Footnotes
Acknowledgments
This work was supported by a grant provided by the F.R.S.-FNRS (Fonds National de la Recherche Scientifique) of Belgium (Grant-Télévie 7.4.517.09).
Author Disclosure Statement
The authors have no conflict of interest.