
Abstract
The gene trap method for embryonic stem cells is an efficient method for identifying new genes that are involved in development. Using this method, we identified a novel gene called helicase family gene related to
Introduction
T
The helicase family contains a large number of proteins that regulate many systems, including DNA replication, DNA repair, DNA recombination, DNA transcription, mRNA splicing, and translation, by controlling the polynucleotides that are necessary for cell survival. Some helicases are characterized and classified into DNA and RNA helicase families by virtue of their substrates and structures. The RNA helicase family includes proteins such as eukaryotic translation initiation factor eIF-4A [3,4], Escherichia coli ribosome assembly protein DeaD [5], yeast pre-mRNA splicing factor PRP2 [6,7] and Drosophila X-chromosome dosage compensation factor MLE [8]. However, the analyses of helicases have been limited to cellular functions. Therefore, the developmental functions of helicases, particularly those related to the early stages of mammalian development, are not clear. helG encodes a novel RNA helicase family protein, the motif of which has an especially high homology with a subgroup of RNA helicase proteins that includes human RNA helicase A (RHA) [9], bovine nuclear DNA helicase II [10], and Drosophila MLE protein. This novel helicase protein is also strongly expressed in neural cells during the early stages of mammalian development. This is the first report of a helicase family gene that is strongly expressed in neural cells and directly required for mammalian embryogenesis.
Materials and Methods
General methods
The methods for the construction of the trap vector, gene trapping in ES cells, selection of the trapped ES cells, and X-gal staining, along with other related methods, have been extensively described elsewhere [2].
Construction of the trap vector IG
IG contains the following DNA fragments: (1) an EagI-MscI fragment containing a splicing acceptor site of the mouse growth factor gene fgf4; (2) an oligomer with three different frame termination codons; (3) an internal ribosomal entry site (IRES) derived from encephalomyocarditis virus [11,12]; (4) Geo, which is the fusion gene of E. coli LacZ gene and neomycin resistance gene (neo r ); (5) SV40 early mRNA polyadenylation signal; and (6) pNEB193 vector (New England Bio Labs).
Cloning of the genomic DNA and cDNA
The genomic DNA from the W3 cell line was digested with PstI and ligated to obtain circular molecules, which were then used for polymerase chain reaction (PCR) using a LA PCR Kit (Takara Shuzo) with the oligonucleotide primers (5′-AATTCCGCACCGAGAGACCA-3′ and 5′-ACTGATGGAAACCAGCCATC-3′); these primers were the two reverse complementary sequences to the trap vector IG. The PCR product was cloned within the pGEM-T vector (Promega). A 2.4-kb fragment containing the 5′-flanking region of the trap vector was obtained. Using this fragment as a probe, a 19-kb fragment was cloned from a 129SVJ mouse genomic DNA library (Stratagene).
Poly (A)+ RNA was prepared using the Quick Prep Micro mRNA Purification Kit (Pharmacia Biotech) from the W3 cell. The 3′-RACE method was performed using a Marathon cDNA Amplification Kit (Clontech) with a primer (5′-CTAGAGGGAAACCGTTGCTA′) that was a reverse complementary sequence to the IRES gene in the trap vector IG. A 0.5-kb fragment containing a portion of helG cDNA was obtained. Using this 0.5-kb PCR product as a probe, a portion of the helG cDNA was isolated from an embryonic day 12.5 (E12.5) mouse head cDNA library that had been constructed using a ZAP-cDNA Synthesis Kit and a Zap-cDNA Gigapack III Gold Cloning Kit (Stratagene). One of these cDNA fragments was used as a probe for rescreening of the same cDNA library. DNA sequencing analyses were conducted using the ABI PRISM Cycle Sequencing Kit and ABI PRISM 377 Sequencer (Perkin Elmer).
Reverse transcription polymerase chain reaction
Poly (A)+ RNA was prepared using a Quick Prep Micro mRNA Purification Kit (Pharmacia Biotech) and embryos in different stages. RT-PCR was performed using mRNA from eight blastocysts, five E6.5 and three E7.5 embryos and 10 ng of mRNA from E8.5 to E14.5 embryos and adult brains. The oligonucleotide primers complementary to the helG gene 3′ region (5′-GATTCACGAACTGTGCGGTTGCTA-3′ and 5′-TTTGCACTCTGTACATAGCCCCAG-3′), helG gene 5′ region (5′-CCGCAGCCTAAAAACCTTCTCAAC-3′ and 5′-TTGAAGAGTTGGCAGGCAGCTGCT-3′), neurogenin (5′-CTAGTGGTATGGGATGAAACAGGG-3′ and 5′-GCAGTGTGTCCCCTGTCTGC-3′), neuroD (5′-AACCGCATGCACGGGCTGAA-3′ and 5′-TCCGGGTTCTGCTCAGGCAA-3′), NCAM-180 (5′-GCCAGTGGACAAGCCTCTGA-3′ and 5′-GATGCTCTGGTGAAGCCGCT-3′), Hβ58 (5′-TGCCAATGTCCGCCTGAGGT-3′ and 5′-CAGGAATTGGACCCAGCACC-3′), Wnt-5a (5′-TACCAACTCCTCTGCCCGAG-3′ and 5′-CAAAGCCACTCCCGGGCTTA-3′), nodal (5′-TCACGTTTGCCTCAGGCAGC-3′ and 5′-CTACAGACAGCTGTCCCTCC-3′), brachyury (5′-GCTTGTTCCTGGTGCTGGCA-3′ and 5′-CACTCCGAGGCTAGACCAGT-3′), shh (5′-GTCGAGCAGTGGACATCACC-3′ and 5′-CCAGGAAGGTGAGGAAGTCG-3′), goosecoid (5′-GCCCTACATGAACGTGGGCA-3′ and 5′-TCCGAGGAGGATCGCTTCTG-3′), Fgf-4 (5′-GCGTGGTGAGCATCTTCGGA-3′ and 5′-TAGGGTGTGCTTCCGAGGCT-3′), eed (5′-GCAACACCAGCCACCCTCTA-3′ and 5′-TCTGTGCCCTTCCACACCTC-3′), and amnionless (5′-TTCCAGGTCTCTGCAAGGGC-3′ and 5′-GCACCGTGAAGTGTGGTGCA-3′) were used. PCR experiments were performed under the following temperature cycling conditions: 94°C for 30 s, 64°C for 1 min, and 72°C for 2 min, all for 30 cycles.
Whole-mount in situ hybridization
Whole-mount in situ hybridization (ISH) was performed as previously described [1]. A 1.1-kb fragment (1040–2161) of helG cDNA was used as a probe for the detection of helG transcription. At the end of staining, the embryos were gradually dehydrated with methanol in phosphate-buffered saline and made transparent by incubation with a 2:1 mixture of benzyl benzoate (Nacalai Tesque, Inc.) and benzyl alcohol (Wako Pure Chemical Industries, Ltd.) prior to examination with light microscopy.
Southern and northern analyses
Genomic DNA was prepared from W3 ES cells, yolk sacs of the embryos, and tail tips of the mice for southern analysis. Poly (A)+ RNA was prepared using a Quick Prep Micro mRNA Purification Kit (Pharmacia Biotech). The conditions for the southern and northern analyses have been described previously [13,14]. A 1.2-kb fragment (285–1444) of the helG cDNA was used for detection of the helG transcript.
A Mouse Multiple Tissue Northern (MTN) Blot membrane (Clontech) was used to analyze the helG expression in the various adult mouse tissues. A GAPDH probe was used as a positive control [15].
Construction and expression of midnolin green fluorescent protein fusion proteins
Midnolin green fluorescent protein (GFP) fusion and a series of deletion mutants were introduced into the vector, and the helG cDNA was inserted into the multiple cloning site of the pEGFP-C1 or pEGFP-N1 vector (Clontech). CHO-K1 cells (American Type Culture Collection) were used for the transfection of GFP fusion constructs. The CHO-K1 cells were grown at 37°C with 5% CO2 in F-12 Nutrient Mixture medium (Life Technologies, Inc.) supplemented with 10% fetal bovine serum [1].
Transfection was conducted using the LipofectAMINE PLUS reagent (Life Technologies, Inc.) according to the supplied protocols. Briefly, 1 μg of premixed DNA and PLUS reagent in OPTI-MEM medium (Life Technologies, Inc.) was mixed with LipofectAMINE reagent. Day-old CHO-K1 cells in six-well cluster dishes were incubated with this solution. Each well contained 1 mL of transfection mixture, and the dishes were incubated at 37°C with 5% CO2 for 3–4 h. After changing to serum-containing medium, the cells were further incubated for at least 24 but not more than 48 h to allow for the expression of the GFP fusion genes.
Protein expression, purification, and the HelG activity assay
The plasmid containing helG (pEG32a-helG) was transformed into Bl21 (DE3) competent cells, as per the standard transformation protocol. A single clone was picked from the plate, inoculated in 100 mL luria-bertani (LB) liquid medium and shaken at 37°C to an OD600=0.5. Then, isoproyl-β-
The untwisting HelG experiment was performed in a 50-μL system that contained 25 mM Tris-CH3COOH, 1 mM Mg(CH3COO)2, 1 mM dithiothreitol (DTT), 3 mM adenosine triphosphate (ATP), DNA template (plasmid pBKS digested with BamHI, ∼0.4 nM ends), and SYBR Green I. Each reaction was performed in three independent tubes (Test Group 1) and repeated (Test Group 2). The reaction system was placed on ice immediately after 5, 10, and 15 min of reaction to prevent the unwound single-strand DNA from recovering. The 5-min group test was repeated to validate the untwisting activity. The control involved the same reaction system without the HelG protein.
Accession
The cDNA sequence of helG has been deposited in DDBJ/EMBL/GenBank (accession no. AB047557).
Results
Vector IG functions as a gene trap
A gene-trapped ES cell line was obtained using the gene trap vector IG. A splicing acceptor sequence placed 5′ to the vector components avoided the possibility of Geo being removed by splicing. A termination codon and an IRES were inserted upstream of Geo to allow the frame of the integration position to be expressed independently. After electroporating the vector into CCE ES cells, 23 of 24 (96%) G418-resistant colonies stained positive for β-gal activity, which indicated that IG had trapped an active endogenous gene(s) in the undifferentiated ES cell state. The β-gal staining varied from strong to merely detectable levels. The clone that failed to display β-gal activity might have resulted from the partial integration of the trap vector. We chose to analyze the W3 ES cell line in detail because its β-gal activity was extremely high under the undifferentiated culture conditions [16].
Three chimeric mice were obtained and transmitted the ES cell genotype at rates of 50%–80%. Chimeric mice produced from the W3 cell line were crossed with C57BL/6 mice to obtain heterozygous mice. Genomic Southern blot analyses with several restriction enzymes and a LacZ probe revealed that the heterozygous mice had one copy of the trap vector in their genomic DNA (data not shown). Intercrossing of the heterozygous mice with one another did not result in any homozygous mice (Table 1).
Embryos were isolated at different stages, and genotypes were determined by Southern blot and/or polymerase chain reaction analysis. DNA samples could not be recovered from the resorbed embryos. All embryonic day 8.5 (E8.5) homozygous embryos (*) were abnormal.
From the W3 ES cells, a 2.4-kb fragment of genomic DNA containing the integrated trap vector's 5′-flanking region was cloned. Using this 2.4-kb fragment as a probe, a 35-kb genomic DNA fragment was cloned from a 129SVJ mouse genomic DNA library, and cDNA (encoding HelG) was continuously isolated from a mouse brain cDNA library.
Sequence analysis of helG
The cDNA was 3,903 bp in length (not including the poly A tail, Supplementary Fig. S2) including a polyadenylation signal in the 3′ region. The integration site of the trap vector was found to be in the middle of the 3′-most exon (3705–3723) and contained a 19-base deletion. The sequences at the intron–exon junctions closely matched the consensus splice junction sequences of eukaryotes [17] (data not shown). Northern blot analysis using mRNA samples isolated from wild-type embryos and adult mice revealed a single band with a 4.4-kb transcript (data not shown). The size of the helG cDNA (3.9 kb without the poly-A signal) agreed with the blotting analysis results.
The helG cDNA corresponds to a 31,262-bp region on chromosome 9 of Mus musculus that consisted of 22 exons and contains a polyadenylation signal (AATAAA) at 3878–3883. The longest open-reading frame (ORF) was found to begin with methionine (177–179), and the surrounding ATG (TGGGCATGG) sequence agreed with the consensus translation initiation sequence [18]. Presuming that the ORF begins at this first methionine and terminates at TGA (3828–3830), this region encodes a sequence of 1,217 aa residues (Supplementary Fig. S2). The predicted protein size of HelG was 137 kDa.
A search of the DDBJ, GenBank, and EMBL libraries using the helG cDNA nucleotide sequence data indicated strong homologies to many transcripts, including a 90% identity with the DHX30 transcript originating from the chromosome 3 (3p21.31) of Homo sapiens. This finding led us to believe that a homologue of helG exists in the human genome. BlastP searches revealed 97% identity between HelG and ATP-dependent RNA helicase DHX30 (H. sapiens), and a phylogenetic analysis also showed that HelG was located in the same clade as the human DEAH box helicase (Supplementary Fig. S3).
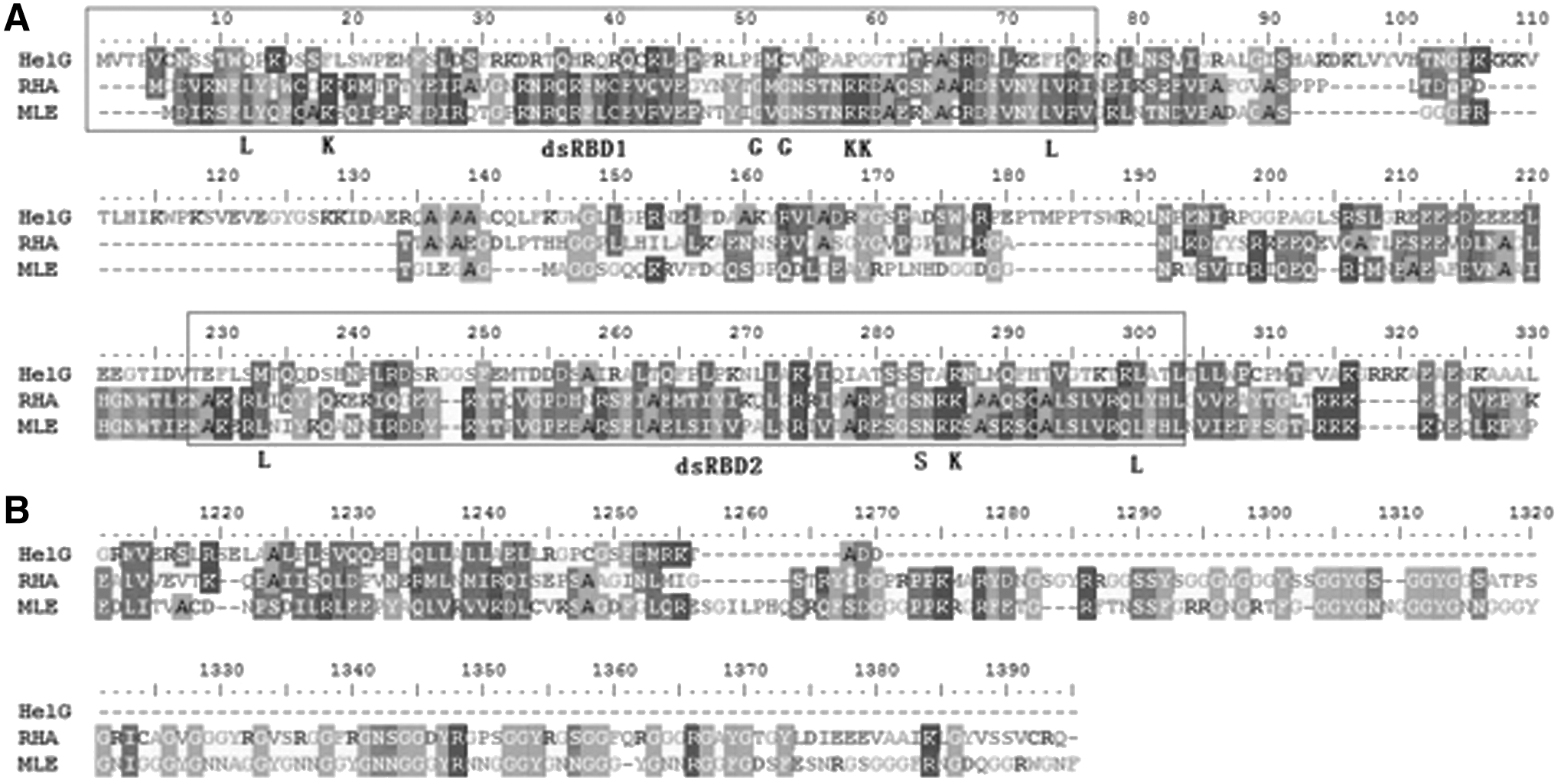
A comparison of the amino acid sequences of the HelG protein to sequences in the Swiss Protein database and the Protein Identification Resource revealed significant homologies with the helicase super family proteins. In addition to homology with mouse helicases and DHX30 helicases, the helG product had shared high levels of high homology with human RHA [9], bovine nuclear DNA helicase II [10], and Drosophila MLE [8], which all belong to the DExH RNA helicase family (Fig. 1).

Comparison of the identical amino acid sequence of the HelG with human RNA helicase A (RHA) and Drosophila MLE. Shaded boxes indicate positions amino acids identical with those in the HelG sequence. Black boxes indicate the I, Ia, II, III, IV, V, and VI helicase domain. Stripe box indicates GYR-rich domains, which are important for function of RHA and MLE.
Structural analysis of HelG
The conserved motifs of HelG were analyzed and compared with RHA and MLE (Fig. 1), which contain DExH motifs belonging to a subset of SF-2 helicases that possess both DNA helicase and RNA helicase activities [19]. As a DExH superfamily helicase, HelG possesses seven conserved motifs in the catalytically active core, and the motifs III, IV, and V were identical among the three helicases.
In motif I (Walker A), the ATP binding site had “GCGKT” (A site), where “K” is known to interact with the phosphates of ATP [20], but the B site “FILDD” was changed into “LLLER” in HelG. Like most other ATP-binding proteins, the “GxxxxGKT” sequence was present in the A-motif, whereas the glycine at the first position is changed into alanine in some helicases [21].
In motif II (Walker B), “DEVH” was present in HelG, whereas “DEIH” is present in RHA and MLE. An “E” is known to act as a catalytic base in ATP hydrolysis, and the fourth residue “H” has been suggested to interact with the first residue “Q” of motif VI [20]. Although residues “E” and “I” are both hydrophobic amino acids, most mammalian ATP-dependent RNA helicases possess “DEVH” rather than “DEIH.”
Motif III of HelG resembled IF4A and contained a “SAT.” It is known that the “S”- and “T”-hydrogen bond the carboxyl of the fourth “D” of the “DEAD” in motif II; thus, the presence of “DEVH” rather than “DEAD” in HelG might affect the interaction between the two motifs.
Motifs Ia and IV are known to interact with oligonucleotides, and the sequences of the two regions are conserved between HelG and RHA. The function of motif V in SF-2 helicase is not clear. In SF-1 DNA helicase, motif V interacts with DNA, motif II, and the ribose of AMPPNP [20].
In motif VI, the nucleic acid-binding region “QRKGRAGR” had one amino acid change; the third amino acid, “K” was an “R” in HelG. As both K and R are strongly basic amino acids, this change might not alter the nucleic acid-binding activity. The “QRRGRAGR” of HelG was similar to the “QRRGRTGR” of HCV helicase.
Additionally, a double-strand RNA-binding domain (dsRBD) was revealed in the amino terminus (Fig. 2A). In both RHA and MLE, two dsRBD domains exist, but the first dsRBD domain in HelG was quite different from those of RHA and MEL; the most conserved residue was changed. This region influences both ATPase and helicase activities, and this difference in the dsRBD1 domain might indicate a unique activity of HelG. Unlike NDH II helicase [19], the 100-amino acid long glycine-rich domain (RGG-box) was absent in the carboxyl terminus of HelG (Fig. 2B). The RGG-box at the COOH terminus enhances nucleic acid binding and might be required for increased unwinding efficiency of the helicase core domain [19].
Alignment of both amino terminus
Through comparison with the PDB database, we found that the N-terminal of HelG displayed homology with the dsRBD of the KIAA0890 protein (Human DHX30); residues 67–175 displayed 97% identity, and residues 262–359 displayed 42% identity. Residues 456–970 (the catalytically active core of HelG) displayed 32% identity with Prp43p (a DEAH helicase) of Saccharomyces cerevisiae [22]. The C-terminus of HelG (residues 878–1100) displayed 23% identity with the C-terminal domain of the human spliceosomal helicase Prp22 [23]. The three dimensional (3D) structure of HelG was constructed based on homology modeling (Supplementary Fig. S4). The two domains (67–175 and 256–354) displayed 3D structures that were similar to those of the dsRBD of human DHX30, and the entire helicase catalytic core formed a 3D structure.
helG is expressed in neural cells during gastrulation
The heterozygous embryos that were crossed with the C57BL/6 strain were analyzed for the expression of the reporter gene LacZ beginning in the fertilized egg stage (E0.5) and continuing through E16.5. Expression was not detected before E5.5, although the gene was expressed in ES cells. Faint expression was first detected at E6.5 (the egg cylinder stage) in the embryonic and extraembryonic ectoderm. At E7.5, expression was detected in the embryonic ectoderm, mesoderm, and ectoplacental cone (Fig. 3A). At E8.5, strong expression appeared in the central nervous system, neural plate, and neural tube. Weak expression was also detected in the somites but not in the heart (Fig. 3B, C). At E9.5, this pattern continued, and expression was primarily detected in the central nervous system and the subset of somite cells that is known to determine the migration paths of trunk neural crest cells and spinal nerve axons. Sections of E9.5 embryos indicated that expression occurred in the proximal cells of the somites and in the epithelial cells of the neural tube (Fig. 3D). The expression became weaker at E10.5, with the exception of the expression in the telencephalon. Given that the LacZ expression in the heterozygous embryos was higher in a part of the brain, the helG gene might have a special function in adult mice because the expression pattern appeared to change in these tissues during embryo development. From E11.5 to E15.5, faint expression was detected. The expression patterns of helG suggested that it is involved in the differentiation of the neural cells. The heterozygous mice of this trap line were not observed to exhibit an obvious phenotype, and the heterozygous embryo results were normal. Wild-type E10.5 was analyzed by whole-mount ISH using a portion of the helG cDNA as the probe. The revealed expression pattern strongly corresponded to the LacZ expression pattern observed in the heterozygous embryos. ISH revealed expression in the central nervous system and the neural plate but not in the heart (Fig. 3F).

Expression in helG during embryogenesis and phenotype of the helG homozygous embryos. Embryos were stained with X-gal (blue signals).
helG expression was analyzed by RT-PCR at E3.5 (blastocysts) and from E6.5 through E14.5 in the wild-type embryos. A faint band of helG mRNA was detected in the E3.5 embryos, and a band was also detected at E6.5. After E7.5 and through E14.5, helG mRNA produced a strong band (data not shown). RT-PCR analysis is highly sensitive. Therefore, we were able to detect helG expression in the mouse embryos from E3.5 in addition to the high levels that occurred after E7.5.
The helG transcription levels in adult tissues (ie, heart, brain, spleen, lung, liver, skeletal muscle, kidney, and testis) were analyzed by northern blot (Fig. 3E). helG expression was detected in all tissues. However, the levels differed between the tissues. Extremely strong signals were detected in the testis and brain, and low levels were detected in the spleen and lung.
The intracellular distribution of HelG was determined using the fusion GFP and full-length HelG proteins. Two expression vectors were constructed: phelGGFP, which encoded a GFP gene that was fused in-frame at the C-terminus of HelG, and pGFPhelG, which encoded a GFP gene fused at the N-terminus. Each construct was transfected into the CHO-K1 cell lines, and the GFP signal localizations were analyzed under fluorescent microscopy. In the cells that expressed both phelGGFP and pGFPhelG, the signals were localized to the cytoplasm and not the nucleus (Fig. 4A, B). Widespread expression throughout the nucleus and cytoplasm of the cell was detected using the pEGFP-C1 vector (positive controls), which encoded only GFP (Fig. 4C). Nuclear Helicase BLM (the causative gene of Bloom syndrome), which is a GFP fusion protein, has the ability to localize to the nucleus [24]. Therefore, we believe the cytoplasmic localizations of the helG-GFP fusion proteins were not artificial. These results suggest that HelG is localized in the cytoplasm and not in the nucleus in vivo.
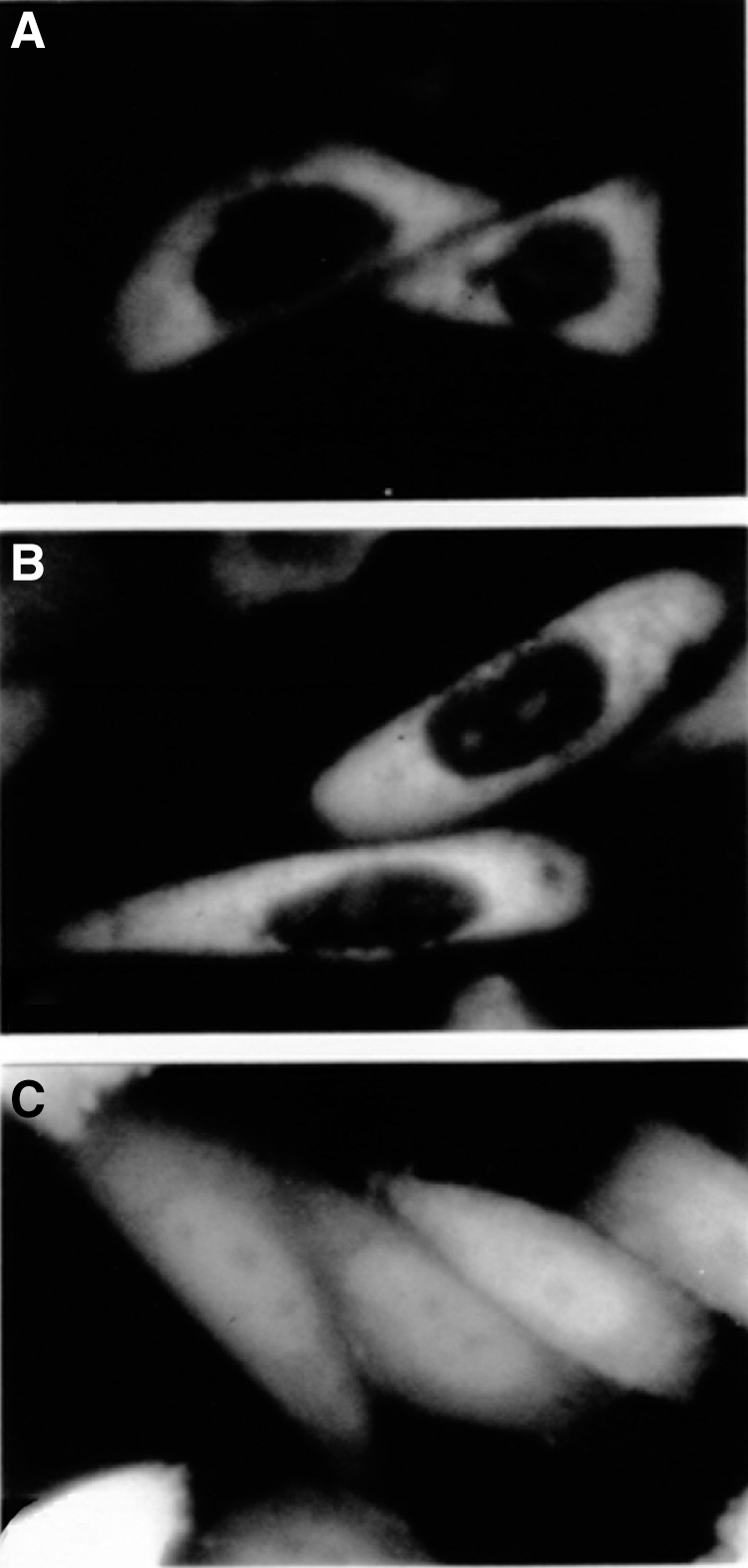
Subcellular localization of HelG and green fluorescent protein (GFP) fusion proteins. CHO cells were transiently transfected with pGFPhelG
helG−/− mice are embryonic lethal
Because no living helG−/− pups could be derived from heterozygous parents as determined by PCR, the genotypes of the embryos derived from heterozygous parents were analyzed. The wild type:heterozygous:homozygotes ratio indicated that the helG−/− mutants died before birth. Homozygous embryos were not found after E9.5. Although some live helG−/− were found at E8.5, their sizes were similar to those of the wild types at E7.5, and some dead helG−/− embryos were also observed at this time (Fig. 3G). The sizes of a number of helG−/− embryos at E7.5 were smaller than those of the wild types. We then performed fixation, dehydration, embedding, and sectioning of both E7.5 and E8.5 helG−/− homozygotes for staining purposes. However, all the sectioning work failed in the fragile E7.5 helG−/− homozygotes. This prevented further analysis of the E7.5 helG−/− homozygotes, and only the whole E7.5 embryo could be stained and used for reverse transcription (RT)-PCR. No bands was detected in the E7.5 or E8.5 helG−/− embryos using a primer set that was constructed to detect the 3′ region of the helG mRNA. These results indicate that the growth of the helG−/− mutant embryos was retarded at E7.5 and that most of these embryos had died by E9.5.
The heterozygous and wild-type embryos had differentiated neural plates, hearts, and head folds. Some of these embryos had also “turned” at E8.5, but the helG−/− embryos only had head folds, hearts, and allantois and never “turned” (Fig. 3H). Many blood cells were observed around the embryos, which indicates that the embryos were on the verge of necrosis. The sections of the E8.5 helG−/− homozygotes revealed neural ectoderm and cephalic mesenchymal cells in the head fold, neural groove, and paraxial mesoderm (Fig. 3I, J) but not in the organized somites of the embryos. Some embryos had cavities in the areas of the somites that were caused by a lack of mesodermal cells. Neural tubes were not observed in the helG−/− homozygotes, with the exception of a single embryo (Fig. 3K, L). The development of the helG−/− embryos was retarded, and their appearances were abnormal. Their phenotype indicated that development stopped at E7.5 prior to the stage of neural plate formation and that the expression of helG affects the development of the neural system.
Expression of the genes related to gastrulation in helG−/− mice
To analyze the phenotype of the helG−/− mice on the molecular level, helG and/or genes with expression related to gastrulation were analyzed using RT-PCR in the E7.5–8.5 helG−/− homozygotes and the E6.5–8.5 wild-type embryos (Table 2).
The helG gene along/or with the genes whose expression is related to gastrulation was analyzed using reverse transcription (RT)-PCR. The signals of the DNA fragment were very strong (++), strong (+), or undetectable (−).
The following genes whose absences cause embryonic lethality were analyzed: Fgf-4 [25], nodal [26], eed [27], Hβ58 [28], amnionless [29], and brachyury [30]. All of these genes were detected in the E8.5 helG−/− homozygotes at the same levels as were detected in the E7.5 wild-type embryos. The absence of any of these genes might not be the trigger that stopped the development of the helG−/− embryos. The expression of genes related to gastrulation was analyzed in the helG−/− mice. The neural cell-specific genes neuroD [31], neurogenin [32,33], and NCAM-180 [34] were not detected at any stage in the helG−/− homozygotes or at E6.5 in the wild-type embryos; however, these genes were detected in the E8.5 wild-type embryos. These results indicate that the neural cells were not differentiated in the helG−/− homozygotes.
The expression of the following cell-type marker genes during gastrulation was also analyzed: nodal for the node [35], brachyury for all mesodermal cells [30,36], sonic hedgehog (shh) for axial mesoderm [37], Wnt-5a for the medial-lateral mesodermal cells [38], and goosecoid for the anterior mesoderm [39]. The expression of all of these genes in the helG−/− embryos were detected at levels similar to those of the E7.5 wild-type embryos with the exception of goosecoid (which was barely detected in the helG−/− homozygotes and strongly detected in the E6.5 wild-type embryos). These findings led us to believe that the expression of goosecoid was suppressed, and the development of the helG−/− embryos passed the E6.5 stage and stopped just before the E7.5 stage during gastrulation. The RT-PCR results agreed well with the histological analyses of the helG−/− embryos.
HelG has DNA-related functions in vitro
In addition to DNA and RNA helicase activities, DEAH box helicases are also involved in the regulation of transcriptional [40] and the initiation of translation initiation [41]. The helicase protein DHX29 has been shown to stimulate cell proliferation, and RHA, another DEAH box helicase, is essential for normal gastrulation [42]. Human RNA helicase dysregulation has been suggested to contribute to cancer [43]. Impaired helicase activity of the ribosome-associated IGHMBP2 is the cause of motoneuron degeneration in DSMA1, which is an autosomal recessive disease [44]. The overexpression of DHX30 even inhibits the human immunodeficiency virus type 1 (HIV-1) RNA packaging [45].
To determine the helicase activity of HelG, we performed untwisting experiments using purified HelG. The plasmid containing helG (pEG32a-helG) was transformed into Bl21 (DE3)-competent cells and cultured in liquid LB. After IPTG induction, sonication lysis, and protein purification (Fig. 5), the purified HelG (contained on His-tagged Beads) was obtained to examine the helicase activity. The untwisting experiment was performed in a 50-μL reaction system using a BamHI-digested pBKS plasmid as the template and SYBR Green I as the dye. This experiment revealed a fluorescent intensity of 18.2 for the single-strand DNA and an intensity of 55 for the double-strand DNA. After 5 min of reaction, the fluorescent intensity displayed a tendency toward a decrease relative to the control with an average value of 51 (Table 3). The results of the other two groups were indeterminate, most likely due to the instability of the entire reaction system. Thus, we repeated the 5-min reaction (Table 4), and the decreased fluorescence intensity validated the untwisting activity of HelG.
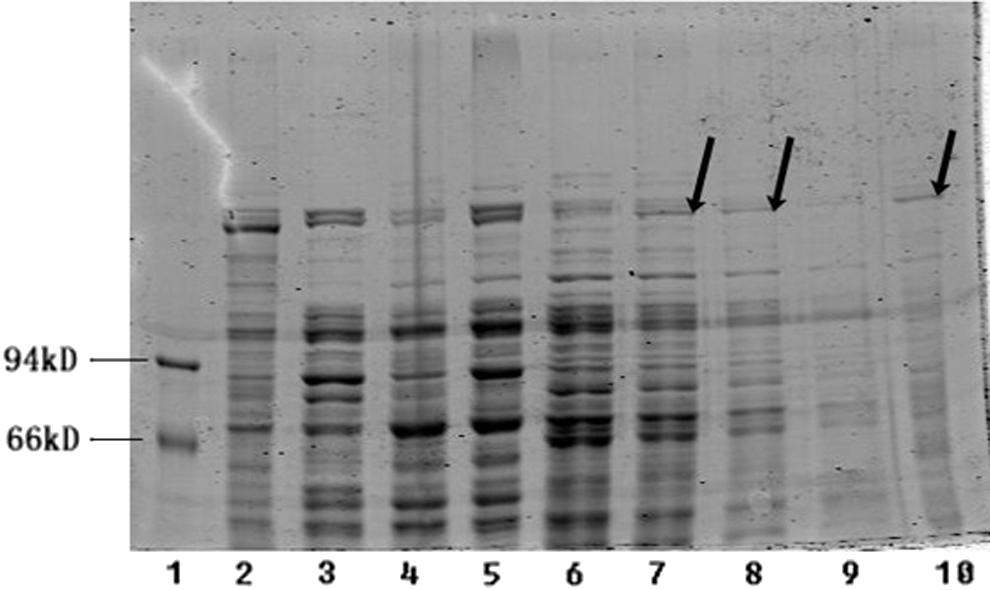
The purification experiment result. From left to right, Lane 1 protein marker, Lane 2 the previous sediment sample contained the induced helicase protein (here as positive control), Lanes 3 and 4 supernatant from the sample untreated with isoproyl-β-
Discussion
Using an improved gene trap method, we “trapped” a helG gene expressed in ES cells. The helG product shared a high level of homology with the DExH RNA helicase family proteins, which include the human RHA, bovine nuclear DNA helicase II, and Drosophila MLE. RHA is a nuclear 3′–5′ double-stranded DNA/RNA helicase and has a role in the regulation of gene transcription via mediating the association of CREB-binding protein and RNA polymerase II [46]. Bovine nuclear DNA helicase II is believed to be a counterpart of human RHA based on their cDNA homology [19]. MLE is a protein that is required for X chromosome dosage compensation in males [8]. RHA, DNA helicase II, and MLE are highly homologous with one another not only in the helicase motif but also in the C- and N-regions outside the helicase motif; these regions include the GYR-rich domains that dictate the functions of these proteins [19,46]. The helG product shared a significantly high homology in the helicase motif and a low homology outside the helicase motif with these proteins. HelG is 24% identical to RHA and MLE. Higher identities were found between HelG and RHA in the helicase motif (45%) and in the helicase domains I–VI (95%) than in the N- and C-terminus regions (13% and 19%, respectively). Therefore, we believe that HelG is a member of the DExH helicase family of proteins. However, with the exception of the helicase motif, HelG does not display significant identity with any of these helicase proteins. The function of HelG might be different from those of these helicases in vivo.
Defective helicase genes have been reported in some mutant mice. The SNF2/SWI2 DNA helicase family gene RAD54 is known to function in DNA repair [47]. RAD54 knockout mice display normal development [48]. Disruption of the mouse xeroderma pigmentosum group D (XPD) gene is lethal at the two-cell stage, which led to the conclusion that XPD is essential for cellular viability [49]. The 3′ region of helG mRNA was not transcribed in the helG−/− homozygotes via integration of the trap vector, and the phenotype of the helG−/− homozygotes is thus believed to have resulted from the absence of wild-type HelG (ie, the knockout of the helG gene) and not from dominant negative effect of a truncated HelG. HelG is the first gene that has been reported to be directly required for embryogenesis.
The development of the helG−/− mutant embryos stopped at E7.5–8.5. At this stage, helG gene expression was detected in the embryonic ectoderm and mesoderm (E7.5), and strong expression appeared in the neural plate and mesoderm at E8.5 (Fig. 3) in the wild-type embryo. This period (E7.5–8.5) corresponds to the most dramatic differentiation that occurs during embryogenesis and specifically during neural development. In this period between the formation of the node and the beginning of neurogenesis, several different types of mesodermal cells are differentiated, including the cells of the axial mesoderm, paraxial mesoderm, lateral mesoderm, and extraembryonic mesoderm.
In the helG−/− mutants, the head fold and neural groove were formed, and the marker gene of the node (nodal) and the genes required for the formation of the node (eed and brachyury) were expressed. However, the beginning of neurogenesis (ie, the head process and head fold formation) was not observed, neural cell-specific genes (ie, neuroD, neurogenin, and NCAM-180) were not expressed in the helG−/− mutants, and the axial mesoderm was not detected. The helG−/− mutants had mesodermal cells, but the somite was not observed. The expression of goosecoid is solely restricted to the mesodermal layer at the anterior end of the primitive streak at this stage in wild-type embryos, and this region later forms the head process. The expression of goosecoid was barely detectable in the helG−/− embryos. These results suggest that the helG−/− embryos lacked axial mesoderm.
The LacZ staining experiment validated the helG expression in the extraembryonic ectoderm from E6.5 to E7.5 in the heterozygous embryo. These results reflect a possible role of HelG in extraembryonic ectoderm formation. Because the RT-PCR analyses revealed that several genes related to gastrulation were expressed in the E8.5 helG −/− homozygotes, we speculate that the extraembryonic ectoderm defect that resulted from the absence of helG might be the cause of the deaths of the homozygous embryos.
A large number of helicase super family genes have been reported, and most of these helicases are related to polynucleotides in the cell. Some helicases that are localized in the nucleus are related to genomic DNA and/or pre-mRNA, and other helicases that are localized in the cytoplasm are related to mRNA. As HelG localized to the cytoplasm, we believed that HelG interacts with mRNA and regulates the gene translation.
Some helicases have been suggested to play roles in embryonic development. The RNA-helicase translation initiation factor eIF4AII is expressed in dramatic variety in the tissues of mice [50] and has been reported to play a role in neural patterning by inducing neural fold genes during Xenopus development via interactions with mRNA during transcription [51]. The similarity of the neural gene regulatory functions of eIF4AII and HelG suggests a relationship between these proteins during mouse development.
To understand neural development, it is necessary to perform detailed analyses of the role of HelG. Specifically, the relation with Sox1 is important for the initial detection of transcripts in the neural fold ectoderm at pregastrulation to early somite stages [52]. The HelG gene likely interacts with Brachyury, Nodal, and Hesx1, which are the key to the formation of nerve tissue and the patterning of the somites. Further analyses of those interactions would deepen our understanding of neural development. It is a novel finding that the HelG gene has the nucleic acid helix cleavage ability that controls one end of mouse neural development. The temporal and spatial localization of helG expression suggested its involvement in neural development.
The predicted role of helG is as a regulator of the translations of genes, including the genes required for embryo development. In the homozygous embryo, the expression levels of several genes whose absence cause embryonic lethality were normal, and an abnormal phenotype was not detected with the exception that the development of the homozygotes embryos was retarded. These results suggest that HelG regulates many genes or a gene that is related to the development of the entire body. Because we were able to separate the amnion from the homozygous (helG −/−) embryos at E8.5 for use in genotyping, the E8.5 homozygous (helG −/−) embryos could be detected and analyzed. However, the homozygous (helG −/−) embryos at E7.5 had no amnions, and we had to use the whole embryos for genotyping, which prevented further analyses. We attempted to divide the bodies of E7.5 homozygous (helG −/−) embryos to obtain both genotyping and section samples, but the sectioning work failed due to the fragile bodies and small sizes; thus, we were unable to obtain clear data. We confirmed that all E7.5 embryos were normal in morphology, but some of the embryos were smaller than the wild types. We then detected some helG −/−embryos (the samples were grouped). Further analysis of the helG homozygous embryos and the determination of the target of HelG will aid the elucidation of the function of helG in mouse development.
One defect in the HelG untwisting assay was the lack of single-stranded DNA-binding protein (SSB) in the system. Additionally, to generate enough helicase to test the unwinding activity, we will need to change the expression system. As the molecular weight of the helicase protein was above 137 kDa, a eukaryotic expression system might not be appropriate. Moreover, the use of fluorescence to test helicase activity is not a well-developed method. Indeed, the wavelength of the dye and DNA combination was different from that of the dye and RNA combination. Finally, to date, no reference exists that indicates the fluorescence intensity difference between single-strand RNA and double-strand RNA. Further, an isotope-labeling method has been used for all RNA unwinding analyses in recently published articles.
Conclusions
We “trapped” a helicase family gene (helG) that was expressed in ES cells using a modified gene trap method for ES cells and analyzed its sequence and function. helG is strongly expressed in neural cells during the early stages of mouse development, and the helG−/− mice were confirmed to be embryonic lethal. Moreover, we performed an untwisting assay and validated the RNA-related activity of HelG in vitro. This is the first report of a helicase family gene that is directly required for embryogenesis.
Footnotes
Acknowledgments
We thank Dr. E.J. Robertson for kindly providing us with CCE ES cells and Dr. H. Suemori, Dr. T. Masaki, and K. Tsukahara for helpful technical advice. Authors did part of this work when they belonged to the Bio Signal Pathway Project, Kanagawa Academy of Science and Technology in Japan.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.