
Abstract
Mesenchymal stem cells (MSCs) are somatic, multipotent stromal cells with potent immunomodulatory and regenerative properties. Although MSCs have pattern recognition receptors and are modulated by Toll-like receptor ligands, MSC-microbial interactions are poorly defined. The objectives of this study were to determine the effect of bacterial association on MSC function. We hypothesized that gastrointestinal bacteria associate with MSCs and alter their immunomodulatory properties. The effect of MSC-microbial interactions on MSC morphology, viability, proliferation, migration, and immunomodulatory functions was investigated. MSCs associated with a remarkable array of enteric pathogens and commensal bacteria. MSC interactions with two model organisms, the pathogen Salmonella typhimurium and the probiotic Lactobacillus acidophilus, were further investigated. While ST readily invaded MSCs, LB adhered to the MSC plasma membrane. Neither microbe induced MSC death, degeneration, or diminished proliferation. Microbial association did not upregulate MHC-II, CD80/86, or CD1 expression. MSC-microbial interaction significantly increased transcription of key immunomodulatory genes, including COX2, IL6, and IL8, coupled with significantly increased prostaglandin E2 (PGE2), interleukin (IL)6, and IL8 secretion. MSC-ST coincubation resulted in increased MSC expression of CD54, and significant augmentation of MSC inhibition of mitogen-induced T-cell proliferation. T-cell proliferation was partially restored when PGE2 secretion was blocked from ST-primed MSCs. MSC-microbe interactions have a profound effect on MSC function and may be pivotal in a variety of clinical settings where MSCs are being explored as potential therapeutics in the context of microbial communities, such as Crohn's disease, chronic nonhealing wounds, and sepsis.
Introduction
M
MSCs are in phase I–III clinical trials for the treatment of immune-mediated and inflammatory intestinal diseases, such as graft-versus-host disease and inflammatory bowel disease in people [5 –8]. The therapeutic rationale is based on the ability of MSCs to home to and engraft in the lamina propria of the gastrointestinal (GI) tract during intestinal inflammation and exert potent immunomodulatory functions. As the gut mucosal barrier is compromised in patients with intestinal injury, engrafted MSCs may be exposed to bacteria and bacterial products [9].
The mammalian host and its enteric microbiome form a complex ecosystem in which dynamic reciprocal events dictate various host cellular and metabolic pathways, including the active shaping of the mucosal and systemic immune system [10,11]. The gut microbiome communicates with cells of the gut, including epithelial cells, M cells, dendritic cells, and macrophages, and is required for normal development and maturation of the gut-associated lymphoid tissue [12]. The microbiome confers resistance to enteric pathogens by competing for nutrients, enhancing mucosal barrier function, and improving innate immunity [12,13]. Relevant investigation of the GI environment and the associated microbiome requires an intact animal model whose GI system mimics the human system. The dog is increasingly recognized as a valuable preclinical, large animal model for a variety of human diseases, including cancer, respiratory disease, and inflammatory disease [14].
In multiple animal models of sepsis, MSCs have an indirect beneficial effect [15 –17]. MSCs express functional pattern recognition receptors [ie, Toll-like receptors (TLRs) and NOD-like receptors (NLRs)] that may allow them to sense and react to a variety of microbes and their associated molecules [18 –24]. These reports suggest that MSC-microbe interactions occur, but specific and direct evidence of these interactions and their influence on the immunomodulatory capacity of MSCs remains elusive. Recently, Fiedler et al. determined that MSCs can interact with common skin-associated bacteria, but their exact interaction and their effect on immunomodulatory properties were not determined [25]. In individuals with intestinal inflammation who receive MSC therapy, microbes may contribute to the engrafted MSC's niche and, along with other mediators, participate in determining whether administered MSCs will adopt an anti-inflammatory or a proinflammatory phenotype [19,26 –29].
Considering the interplay between chronic GI inflammation and the gut microbiome in modulating the immune system along with the use of MSC therapy to regulate GI inflammation and immune cell subsets, we hypothesized that during intestinal inflammation or injury, GI bacteria will associate with MSCs to alter their immunomodulatory properties. To that end, the study found that many common GI pathogens and probiotic bacteria adhere to and invade MSCs. Bacterial adherence and invasion did not alter MSC viability or proliferation. Interestingly, Salmonella association inhibited MSC migration, but it did not induce an antigen-presenting phenotype. Rather, Salmonella augmented the capacity of MSCs to inhibit mitogen-induced T-cell proliferation, which was coupled with a significant induction of peroxisome proliferator activator receptor gamma (PPARγ), interleukin (IL)6, IL8, HGF, and cyclooxygenase 2 (COX2) transcription; increased cell surface expression of CD54; and increased secretion of IL6, IL8, and PGE2. We further showed that increased secretion of PGE2 by bacterial-primed MSCs played a significant, though partial, role in the inhibition of T-cell proliferation.
Materials and Methods
Animal use
Canine fat harvest was approved by the Institutional Animal Care and Use Committee and the Clinical Trials Review Board at the University of California, Davis (UCD). Approximately 10–15 g of falciform fat was collected from healthy dogs that presented to the William R. Pritchard Veterinary Medical Teaching Hospital at UCD for routine abdominal surgery unrelated to fat collection. All dog owners signed an informed consent form.
MSC culture
MSCs were isolated from fat and cultured exactly as previously described [8,30]. MSCs were cultured in low-glucose Dulbecco's modified Eagle's medium (DMEM; Mediatech, Manassas, VA), 10% fetal bovine serum (FBS; HyClone, Inc., Logan, UT), and 1% penicillin/streptomycin (P/S; Invitrogen, Carlsbad, CA) in tissue culture flasks (Nunc, Roskilde, Denmark) and incubated at 37°C with 5% CO2. Cells were passaged once they reached ∼70% confluency and they were replated (at ∼5×104 cells/cm2) 12 h prior to incubation with bacteria. All experiments were conducted using MSCs at passages 3–6.
Bacteria
Salmonella enterica ssp enterica serotype Typhimurium 14028S, LT2, and a Type III secretion system mutant (ΔInvA); Salmonella enterica ssp enterica serotype Enteritidis (BCW_4673 and BCW_1342); Salmonella enterica ssp enterica serotype Heidelberg (ATCC 8326 and BCW_89); Salmonella enterica ssp enterica serotype Newport (BCW_1378); and Salmonella enterica ssp enterica serotype Saint Paul (BCW_88) were thawed and grown in Luria broth (Teknova, Holister, CA) and incubated with shaking (200 rpm) at 37°C. Escherichia coli [O157:H7 (ATCC 35150) and K12] and Listeria monocytogenes EGDe were thawed and grown in Brain Heart Infusion broth (Sigma-Aldrich 53286, St. Louis, MO), and rocked at 37°C. Lactococcus lactis ssp lactis SK11 was thawed and grown in Elliker's broth (Difco) overnight at 28°C; bacteria were then diluted 1:100 in fresh broth before use. Bifidobacterium infantis (ATCC 15697) and Bifidobacterium longum (BCW_0855) were grown in an anaerobic hood with a gas mix of 20% CO2 and 6% H2, balanced with N2 at 37°C in Reinforced Clostridial Medium (Sigma-Aldrich 91365) for 16 h, after which the bacteria were transferred 1:100 into fresh broth and incubated at 37°C. Lactobacillus acidophilus NCFM was thawed and grown in Elliker's broth and incubated at 37°C. Prior to use, all bacterial cultures were centrifuged (2,000 g for 10 min), resuspended in DMEM, and adjusted with a spectrophotometer to the appropriate optical density. Bacteria were plated at each step to confirm culture purity.
MSC/bacteria association
Adherence and invasion assay
Total microbe association with canine MSCs was determined by the gentamicin protection assay as described by Elsinghorst [31], with modifications described in the next paragraph and quantified by quantitative polymerase chain reaction (qPCR). MSCs were plated (5×104/well) in a 96-well plate and incubated overnight at 37°C with 5% CO2. Bacteria were collected at second-transfer mid-log phase. Bacteria were centrifuged (6,500 rpm for 10 min), growth media were aspirated off, and pellets were suspended in DMEM. Salmonella, E. coli, Listeria, Lactococcus, and Bifidobacterium were resuspended to a concentration of 108 CFU/mL; Lactobacillus was adjusted to a concentration of 109 CFU/mL before adding 50 μL of each microbe suspension independently [multiplicity of infection (MOI) 1:100 and 1:1,000, respectively] to the MSCs. Plates were immediately centrifuged at 500 rpm for 30 s and incubated for 60 min at 37°C in 5% CO2. After incubation the bacteria were aspirated from the well and the MSC monolayer was washed twice with 200 μL of 1×Dulbecco's phosphate-buffered saline (DPBS) to remove nonadhered bacteria. Invaded bacteria were determined by incubating the coculture with 200 μL gentamicin, minimum inhibitory concentration established for each strain (data not shown), for 2 h at 37°C with 5% CO2. Total associated bacteria wells were incubated with DMEM. Following the incubation, cells were washed twice with 200 μL of DPBS buffer and lysed with 50 μL of Warnex lysis buffer (EX1/EX2 buffer, EX20501A; AES Chemunex Canada, Inc., Laval-Des-Rapides, Canada). Invasion and total cell association samples were run in biological triplicate. Microbe association was quantified using real-time PCR with a Bio-Rad CFX96 platform (Bio-Rad real-time system C1000 thermocycler) (Table 1). The difference in total associated versus invaded microbe was used to determine microbe adherence. For long-term culture experiments, MSCs were washed three times with DPBS after the gentamicin treatment and were supplemented with complete culture media (DMEM, 10% FBS, and 1% P/S).
Light microscopy
For cytologic examination, MSCs were dissociated with trypsin (0.05% Trypsin-EDTA; Gibco, Grand Island, NY) and cytofuge preparations were made (Cytospin 4; Thermo Shandon, Wilmington, DE). Slides were stained with Hema 3® (Fisher Scientific, Kalamazoo, MI) per manufacturer's instructions and examined with a light microscope (Olympus BX161; Olympus, Inc., Center Valley, PA). Digital images were captured with a digital camera (Penguin 600CL; Pixera Corporation, Santa Clara, CA) and compatible computer software (View Finder 3.0.1; Pixera Corporation).
Transmission electron microscopy
MSCs were plated (8×104/chamber) on multichamber glass slides (Nalge Nunc International, Naperville, IL) and bacterial-MSC coincubation was completed exactly as described previously. The supernatant was aspirated at various time points up to 8 h post-bacterial–MSC coincubation. The cells were washed with DPBS and fixed with 150 μL of Karnovsky's fixative solution. Slides were submitted to the Electron Microscopy Laboratory, Department of Pathology and Laboratory Medicine, School of Medicine, UCD. Samples were processed routinely, stained with uranyl acetate and lead citrate, and scanned with a transmission electron microscopy (TEM; Philips EM400 with Goniometer; FEI Company, Hillsboro, OR, made in Eindhoven, the Netherlands).
MSC migration
Matrigel inserts (BD Biosciences, Franklin Lakes, NJ) were thawed and rehydrated per manufacturer's instructions. Seven hundred fifty microliters of complete culture media was placed in the bottom wells (10% FBS served as migration stimulus). MSCs were coincubated with bacteria as described previously, after which MSCs were trypsinized and 4×105 MSCs were resuspended in media with low FBS (DMEM, 1% FBS, and 1% P/S), placed in a transwell insert, and incubated at 37°C with 5% CO2. After 22 h, cells from the upper surface of the matrigel membrane were removed. Membranes were stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) solution and mounted on a glass slide. Slides were examined via fluorescent microscopy (Olympus BX161; Olympus, Inc.) and six images per slide were captured with a digital camera (Penguin 600CL with View Finder 3.0.1 software). Cell nuclei were automatically counted using a computer software program (Image J,
MSC viability
MSC viability was determined up to 24 h after bacterial coincubation using 7-amino-actinomycin D (7-AAD; BD Biosciences) incorporation. At each time point, both the media and adherent cells were collected, washed, resuspended in DPBS, and incubated with 7-AAD (10 min in a 37°C water bath). MSC viability was determined via flow cytometry (Cytomics FC500; Beckman Coulter, Pasadena, CA). Data were analyzed using FlowJo flow cytometry software (Tree Star, Inc., Ashland, OR).
MSC proliferation
MSCs were plated in 12-well plates (2.5×104 cells/cm2) and coincubated with bacteria exactly as described previously. MSCs were incubated for 72 h (37°C, 5% CO2) during which the cell number was determined prior to bacterial coincubation and after 72 h to estimate proliferation. Additionally, MSCs were enumerated using Trucount tubes (BD Biosciences) according to manufacturer's instructions using flow cytometry to accurately determine the cell count. Briefly, cells were harvested with 200 μL of trypsin and neutralized with 800 μL of complete media, and 520 μL of the cell suspension was placed in a Trucount tube and vigorously vortexed. The cell-to-fluorescent bead ratio was determined via flow cytometry (Cytomics FC500). Data were analyzed using FlowJo flow cytometry software (Tree Star, Inc.).
MSC phenotype
MSC surface phenotype was determined at various time points after MSC-bacteria coincubation. All antibodies were purchased from the Leukocyte Antigen Biology Laboratory, UCD, unless otherwise indicated. Antibodies included MHC-II (clone CA2.1C12), CD1 (clone CA13.9H11), CD80 (clone CA24.5D4), CD86 (CA24.3E4), and CD54 (clone CL18.1D8, a kind gift from C. Smith, Houston, TX). Surface protein expression was determined using flow cytometry (Cytomics FC500). Flow cytometry data were analyzed using FlowJo flow cytometry software (Tree Star, Inc.).
MSC gene expression
MSCs were plated in six-well plates (4×105/well) and coincubated with bacteria as described previously. Lipopolysaccharide (LPS; L3012 Sigma-Aldrich) treatment was administered at 10 ng/mL. At 60 or 120 min, MSCs were washed twice with DPBS and lysed with RLT buffer, and RNA was extracted (RNAeasy mini kit; Qiagen, Gaithersburg, MD) per manufacturer's instructions. Genomic DNA was digested (Turbo DNase Ambion, Grand Island, NY) and cDNA was synthesized (First-Strand cDNA synthesis; Origene, Rockville, MD) per manufacturer's instructions. qPCR was performed on a 7300 Real Time PCR System (Applied BioSystems, Foster City, CA). Primers were designed using Integrated DNA technology website (
MSC–peripheral blood mononuclear cell cocultures
Peripheral blood mononuclear cell (PBMC) isolation and mixed leukocyte reactions (MLRs) were carried out as described by Wunderli and Felsburg [42] and Carrade et al. [2], with modifications described in the next paragraph. About 9 mL of Ficoll-Paque (GE Healthcare, Piscataway, NJ) was diluted with 1.5 mL of tissue culture water for a final specific gravity of 1.066. Whole blood (10 mL) was mixed with 20 mL of modified Tyrode's/HEPES buffer containing EDTA (12 mM NaHCO3, 138 mM NaCl, 2.9 mM KCl, 10 mM HEPES, and 1 M EDTA), and layered on top of the diluted Ficoll-Paque layer. The blood was centrifuged and PBMCs were collected and resuspended in activation medium (DMEM+10% heat-inactivated FBS+1% P/S), and stored on ice until plating. PBMCs were activated with 5 μg/mL concanavalin A (Con-A; Sigma-Aldrich). Cells were collected and processed per manufacturer's instructions (BrdU Flow Kit; BD Biosciences), stained with a viability dye (Fixable Viability Dye eFlour®780; eBioscience, San Diego, CA), anti-canine-CD3 conjugated to Alexa Flour®488 (clone CA17.2A12; Leukocyte Antigen Biology Lab, UCD) and anti-BrdU conjugated to Alexa Flour®647 (clone MoBU-1; Invitrogen), and analyzed on a flow cytometer (Cytomics FC500). Flow cytometry data were analyzed using FlowJo flow cytometry software (Tree Star, Inc.).
Statistical analysis
Data were examined for normal distribution using the Kolmogorov–Smirnov method. Paired Student's t-tests and analysis of variance (ANOVA) with post hoc Tukey tests were performed using GraphPad InStat 3 (GraphPad Software, Inc., La Jolla, CA) or JMP version 10 (SAS Institute, Triangle Park, NC). P<0.05 was considered to be statistically significant.
Results
Bacteria associate with canine adipose-derived MSCs in microbe-dependent patterns
Canine MSCs were susceptible to microbial association in vitro (Fig. 1A). As expected, microbe-specific adherence and invasion differences were observed. Salmonella serotypes demonstrated differential ability to associate and localize within MSCs. Surprisingly, the ΔInvA mutant, which lacks the type III secretion system, retained the ability to invade MSCs, as opposed to complete inhibition of invasion in epithelial cells [43]. Among Salmonella serotypes, a 400-fold difference in MSC association was observed. Within the assay time, a number of Salmonella serotypes were exclusively invasive with no MSC adherence noted. S.H BCW_8326, S.H BCW_89, S. Newport, and S. Saint Paul displayed different association patterns with 75.5%, 40.9%, 26.7%, and 29.4%, respectively, for invasion. L. monocytogenes EDGe behaved similarly to Salmonella, but the two E. coli strains exhibited opposite MSC association patterns. Of the total associated E. coli K12, 78.5% of the bacteria invaded MSCs while E. coli O157:H7 exclusively adhered to and did not invade MSCs, which was also observed by He et al. [44].
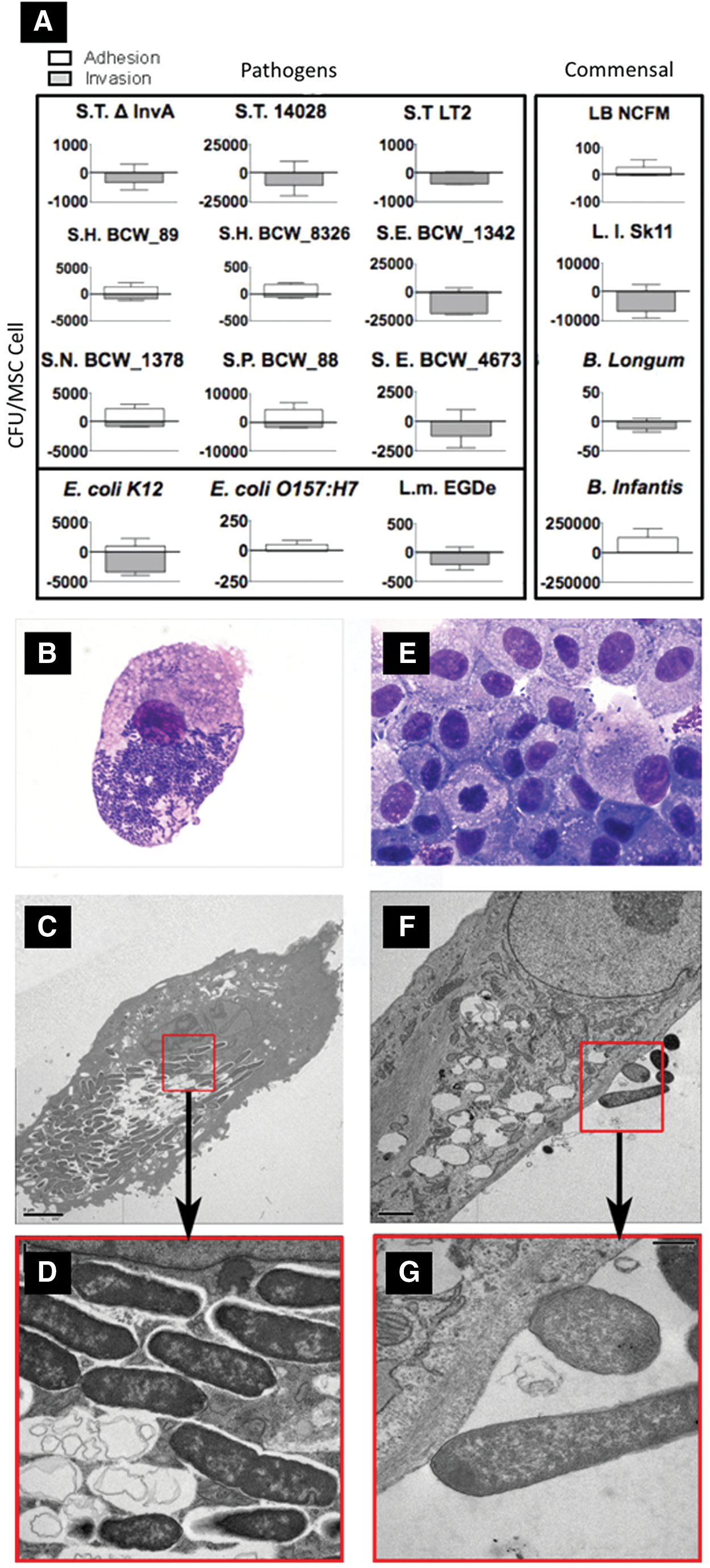
Canine mesenchymal stem cells (MSCs) are susceptible to microbial association in vitro. Adipose-derived MSCs were coincubated with intestinal bacteria and adhesion/invasion was quantified using the gentamicin protection assay. Bacteria used in
MSC association was not exclusive to pathogens. All selected commensal microbes also associated with MSCs to similar amounts as pathogens. B. longum and L. lactis SK11 exclusively invaded MSCs, while B. infantis and L. acidophilus NCFM (LB) were mostly adherent to MSCs (99.9% and 92.7% adherence, respectively).
Salmonella enterica spp enterica serotype typhimurium LT2 (ST) was found within MSCs within 30 min post-coincubation when viewed with light microscopy (Fig. 1B). The MSC membrane developed numerous variably sized (0.1–5 μm) irregular projections during microbe association. Surprisingly, most bacteria were not noted within distinct intracellular vacuoles. Using TEM, at 2 h post-coincubation, low numbers of ST were present intracellularly, either within membrane-bound vacuoles, or, occasionally, free within the cytoplasmic matrix. After 7 h of association, most MSCs contained increasing concentrations of intracellular bacteria, presumably within membrane-bound vesicles (Fig. 1C, D). MSCs did not show any morphologic changes indicative of cellular toxicity or degeneration (ie, mitochondrial or endoplasmic reticulum swelling, chromatin modification, etc.).
In contrast to ST, LB was found primarily extracellularly and appeared to adhere or associate with the MSC membrane rather than invade the cell (Fig. 1E). Small and irregular membrane projections were often noted at sites of bacterial contact. There was no evidence of host cell degeneration. TEM confirmed that the majority of LB were found in the extracellular space (Fig. 1F, G). Bacteria were often closely associated with the MSC membrane and, in some instances, the MSC membrane and bacterial cell wall were indistinguishable. Occasionally, the MSC membrane formed dome-shaped to elongated (up to 1 μm) processes at the site of bacterial adherence. Rare intracellular LB bacteria were seen. Intracellular bacteria were not membrane bound within the cytoplasmic matrix and appeared degenerated. No ultrastructural evidence of cellular degeneration was noted by 7 h post-coincubation. Adherence/invasion assays and microscopy confirmed that (1) a wide array of pathogenic and probiotic microbes can adhere to or invade into canine MSCs and (2) there is a disconnect between microbe pathogenicity and its capacity to invade MSCs. Based on the interactions and the additional literature surrounding the microbes used in this study, ST and LB were selected for further investigation. We next set out to determine how these model microbes, a pathogen capable of invasion and proliferation (ie, ST), and a probiotic that was rarely invasive (ie, LB), alter MSC viability, proliferation, and migration as well as critical immunomodulatory functions.
MSC migration is inhibited, though viability and proliferation are not altered by microbial adherence or invasion
Coincubation of MSCs with ST or LB did not result in MSC death at 24 h even with marked ST uptake and microbe proliferation (n=3 MSC lines, Fig. 2A). Similarly, MSCs continued to proliferate normally for up to 3 days after microbial challenge (Fig. 2B). These observations confirm those found in Figure 1, indicating that the MSC/microbe association is very different to that of epithelial cells, which motivated further exploration of the modulation mechanisms of these organisms when associated with MSCs. Further, ST significantly (P<0.01) inhibited MSC transmigration (Fig. 2C); however, MSCs treated with LB were not significantly different to control cells.
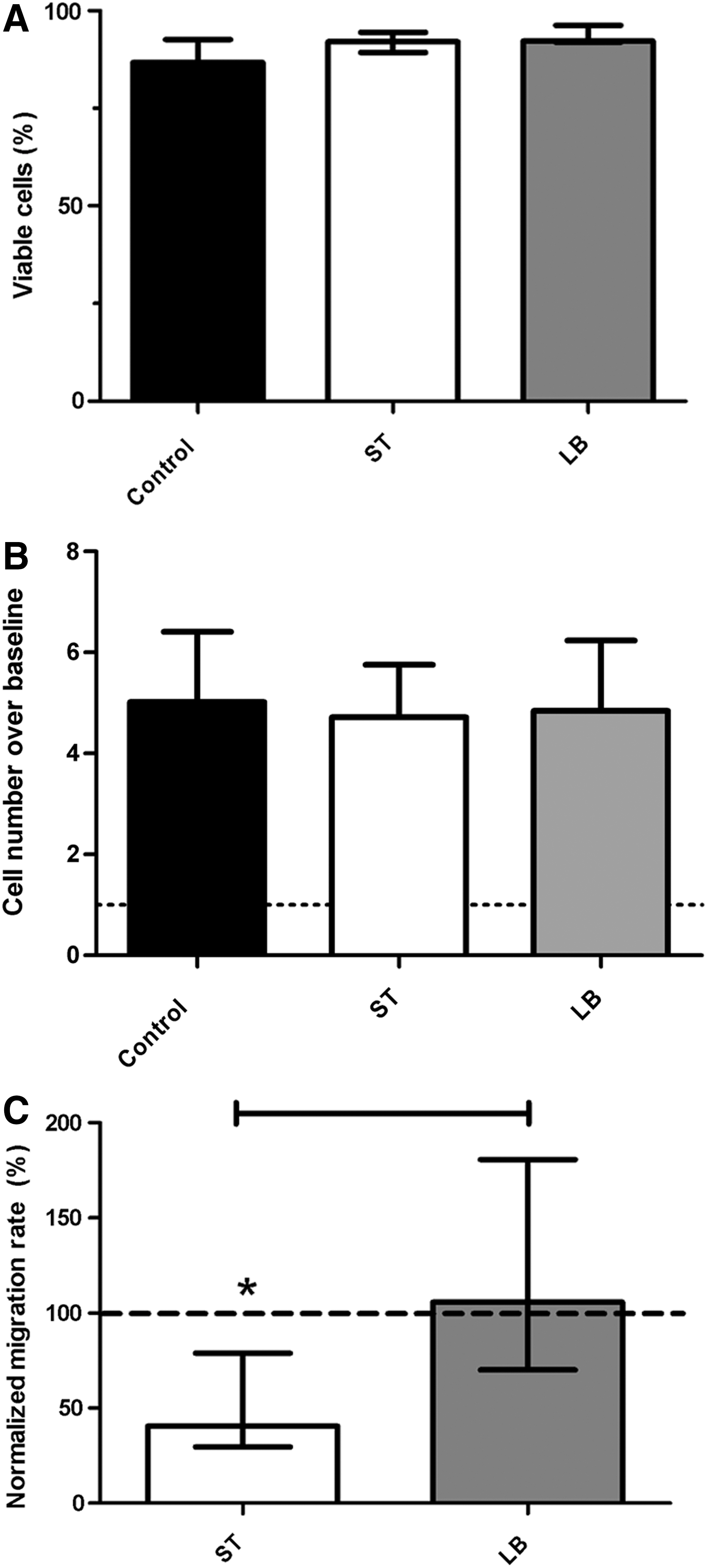
Microbe-MSC interaction effects on basic MSC functions. Coincubation of canine MSCs with ST or LB did not affect MSC viability
MSCs are activated by ST but do not take on an antigen-presenting cell phenotype
Like MSCs from other species, canine MSCs do not express CD80, CD86, CD1, or MHC-II when unstimulated [8]. Stimulation of MSCs with inflammatory mediators, such as interferon gamma (IFNγ) and/or tumor necrosis factor alpha (TNFα), may induce MSCs to express MHC-II, costimulatory molecules, and function as antigen presenting cells (APCs) [45,46]. Since association with bacteria modulates MSC behavior and may have the capacity to activate proinflammatory pathways, we sought to determine whether interaction with viable bacteria would induce the switch to a proinflammatory phenotype. Incubation of MSCs with ST or LB did not induce MSC expression of MHC-II, CD80, CD86, or CD1 expression (data not shown).
GI bacteria induce cytokine gene transcription, immunomodulatory mediator secretion, and surface protein expression in canine MSCs
We next wanted to determine whether microbial association elicited transcription and expression of immunomodulatory or proinflammatory mediators in canine MSCs (Fig. 3). Initially, the effect of ST, LB, and LPS (a TLR4 agonist) treatment on the expression of a wide battery of genes was examined. Microbial association with canine MSCs induced expression of cell cycle, stress response, and immunomodulatory regulators (Fig. 3A) [47 –51]. Microbial association with MSCs induced an increase in the expression of COX2, IL6, and IL8. A less marked increase was noted in the expression of HGF and PPARγ. MSC treatment with LPS mostly mirrored the effect of ST. Notably, while COX2 was markedly induced by ST it was only borderline increased by LPS treatment. MSC treatment with LB mostly resulted in a less pronounced increase in gene expression (compared with ST treatment) other than PPARγ and IL6. The proinflammatory cytokine IL1β was undetected in any of the treatments. Experiments were then repeated with four additional MSC lines to establish more robust results with the genes that were most markedly altered (Fig. 3B). IL6, IL8, and COX2 gene transcripts were significantly (P<0.01) induced in MSCs by both bacterial species. IL6 transcription was equivalently induced in canine MSCs by both bacterial species, but IL8 and COX2 gene expression was significantly (P<0.01) higher in MSCs treated with ST as compared with treatment with LB. Gene expression changes were confirmed by measuring the secretion of IL6, IL8, and PGE2 that mirrored the gene expression data (Fig. 3C). CD54 [aka intercellular adhesion molecule-1 (ICAM-1)] is a cell surface receptor that binds the β2-integrins on leukocyte membranes and is implicated in immune modulation by MSCs [2 –4,29]. Incubation of MSCs with ST significantly (P<0.01) increased CD54 expression compared with baseline and LB (P<0.05) treatments (Fig. 3D). Induction of CD54 in activated MSCs is important in MSC-T-cell adherence and inhibition of Th17 differentiation [4]. These findings suggest that microbial interaction with and invasion of MSCs may promote their immune-regulatory properties.
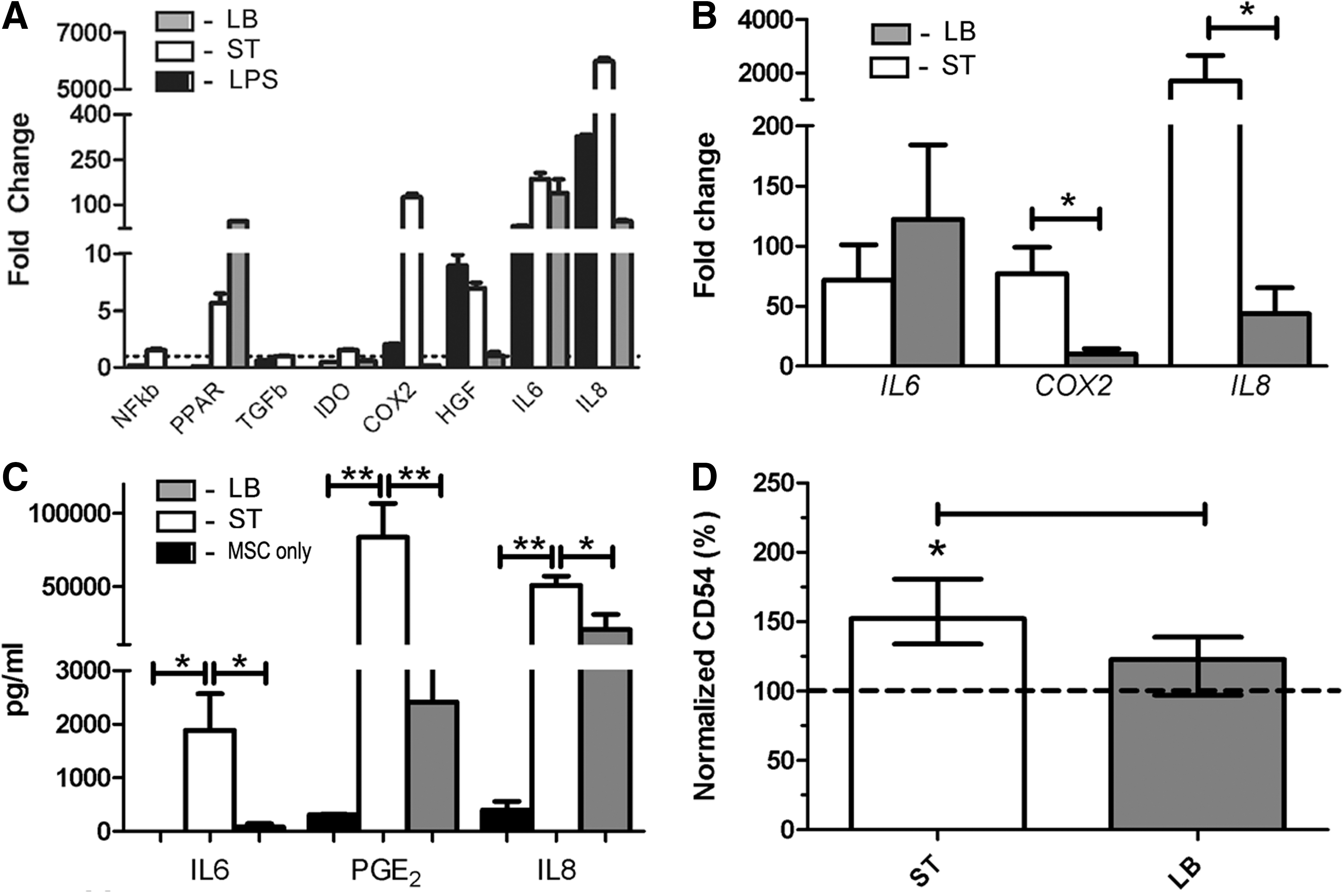
MSCs are activated by intestinal bacteria and upregulate transcription, translation, and cell surface expression of key immunomodulatory genes and mediators, respectively. MSCs were coincubated with ST and LB per protocol prior to cell lysis, RNA extraction, and qPCR. For inflammatory mediator determination, MSCs were further cultured for 4 days, media were harvested, and specific cytokine was determined via ELISA. For cell immunophenotyping experiments, MSCs were cultured for 24 h post-microbial coincubation protocol prior to imunophenotype determination via flow cytometry. Initial gene expression screening experiments
ST augment MSC capacity to inhibit T-cell proliferation via PGE2 secretion
To further define how these organisms modulated MSCs, we used an MLR system to investigate MSC capacity to inhibit lymphocyte proliferation [2]. Pre-exposure of MSCs to ST significantly (P<0.05) increased the inhibition of mitogen-induced T-cell proliferation compared with untreated MSCs (Fig. 4), but this was not observed with LB. Moreover, when PGE2 synthesis was blocked using indomethacin, T-cell proliferation was significantly (P<0.05), though partially, restored in the ST-treated MSCs. These findings support the hypothesis that GI pathogens interact with MSCs and activate the MSCs to become potent anti-inflammatory and immunomodulatory cells in a process that is partially dependent on PGE2. Interestingly, commensal microbes did not have this effect. These findings shed new light on the role that specific GI microbes may have in modulating MSC-host immune cell interactions in the GI, local, or systemic immune system.
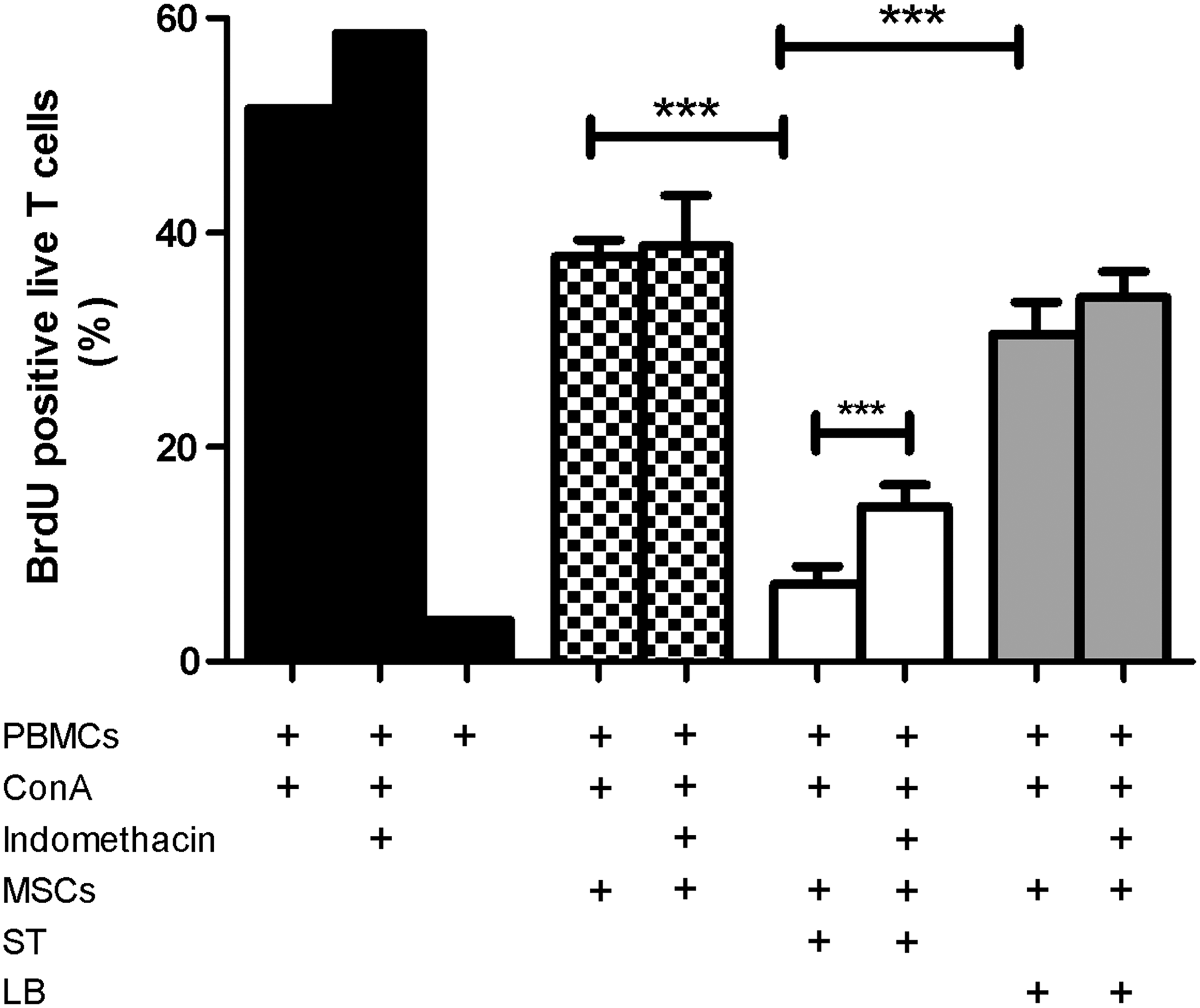
Bacterial interactions augment MSC capacity to inhibit T-cell proliferation. Peripheral blood mononuclear cells (PBMCs) were activated with the T-cell mitogen concanavalin A (Con-A), in the presence or absence of MSCs and microbial-primed MSCs. Cocultures were incubated for 3 days prior to spiking with BrdU. Cells were harvested 24 h post-BrdU spiking; fixed and stained with a viability dye, a T-cell marker (ie, CD3), and anti-BrdU antibody; and read on a flow cytometer. All experiments were repeated with five different MSC lines. Stimulation of PBMCs with Con-A resulted in the maximal BrdU incorporation by viable T-cells. Coincubation of stimulated PBMCs with native MSCs resulted in a marked decrease in T-cell incorporation of BrdU. While ST augmented MSC capacity to inhibit T-cell proliferation, compared with untreated MSCs and LB-treated MSCs, inhibition of T-cell proliferation by LB-treated MSCs was not different from untreated MSCs. When COX function was blocked with indomethacin, T-cell proliferation was significantly, though partially, restored in ST-treated MSCs. ***P<0.001.
Table 2 summarizes the effects that MSC interactions with the two model organisms (ie, ST and LB) exert on MSC biology. A proposed model for MSC activation by GI microbes in the context of intestinal inflammation is depicted in Figure 5. MSCs migrate to inflamed and compromised intestinal lamina propria. Within this injured niche, MSCs may interact with microbes/microbial products that translocate from the gut lumen, leading to stem cell activation, induction of immunomodulatory pathways and gene transcription, and ultimately dictating MSC phenotype. Different microbes and microbial communities may induce specific activation patterns of engrafted MSCs with subsequent modified immunomodulatory and regenerative capacity.
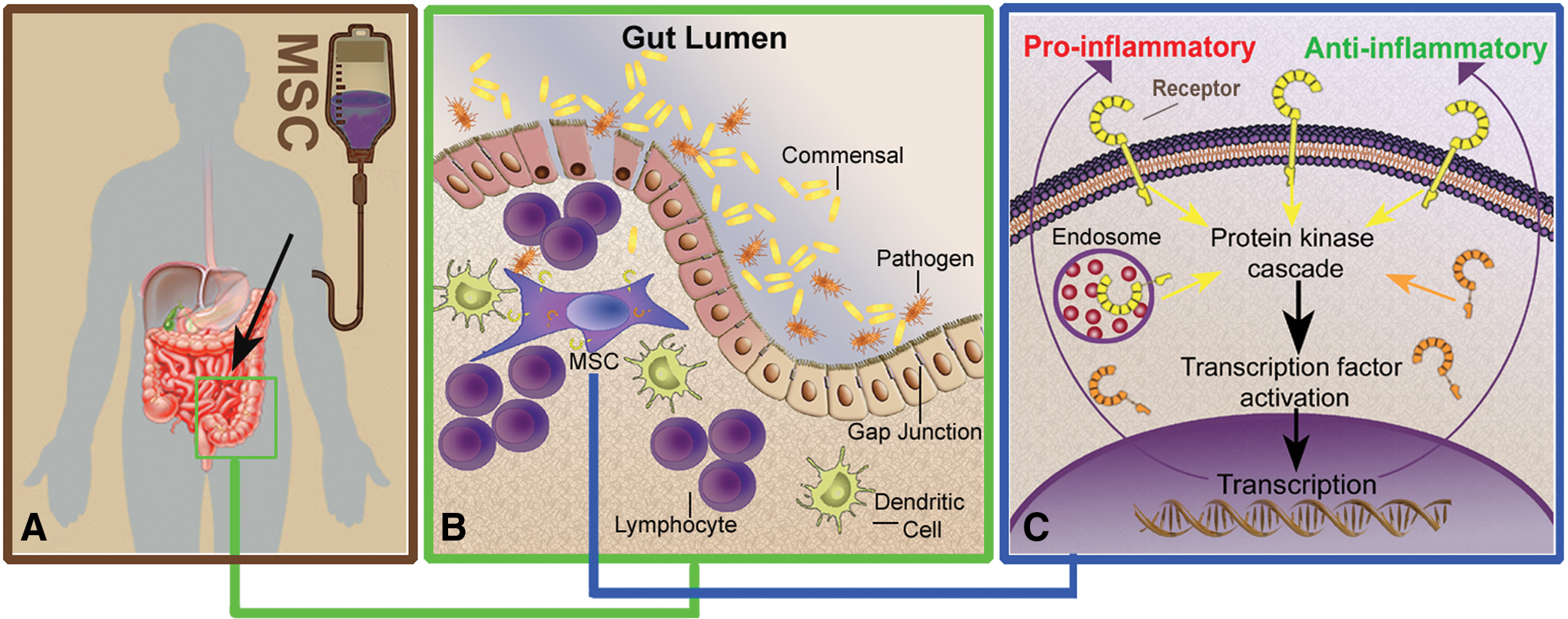
A proposed model for MSC interaction with intestinal bacteria and subsequent MSC activation in patients with intestinal injury. Exogenously administered MSCs home to the injured gut where they engraft in the lamina propria
APC, antigen presenting cell; MSC, mesenchymal stem cell.
Discussion
This is the first study to examine the interaction of viable intestinal microbes with MSCs and their effect on immunomodulatory properties. We found significant changes in MSC behavior that was microbe dependent. These data have profound implications for GI health and disease management for immune function and stem cell therapies. This study provides a solid infrastructure for future studies that will elucidate further the mechanisms of MSC-microbial interaction and their capacity to affect the immunomodulatory properties of MSCs.
Bacterial interactions with MSCs were not limited to a few bacterial species (Fig. 1A); it was observed across many types of bacteria from pathogens to commensal organisms that are common in the gut. All of the microbes tested in this study associated with MSCs with markedly different localization and amount of association, yet it remains unclear as to the impact of the differences in the total amount of association. The 400-fold difference between the bacteria-MSC associations has profound implications for microbiome membership and ratios. Equally important and surprising, none of the bacteria induced MSC death or apoptosis, as seen with epithelial association where inflammation and subsequent apoptosis result in clinical infections [52]. With this in mind, the study progressed to examine the possible changes that a pathogen and commensal microbe may have on MSC physiology.
While ST invasion into epithelial cells [52], macrophages [43], and dendritic cells [53] induces marked cell death within 24 h, invasion of ST into MSCs was not associated with increased cell death nor did it affect cellular proliferation. These findings are compatible with a previous report that describes prolonged culture of synovial fibroblasts after ST infection [54]. These findings suggest that while ST may invade into stromal-type cells without causing significant cell death, invasion does elicit marked and distinct cellular responses.
MSCs are often administered intravenously and then home to the site of injury [55]. MSC-directed migration is crucial for optimal clinical efficacy. While canine as well as murine fat and bone-marrow-derived MSCs migrate to the gut lamina propria in health, gut migration is increased in the presence of intestinal inflammation [8,56]. We demonstrated that while the interaction of LB with MSCs did not affect their migratory properties, MSCs that were coincubated with ST had marked and significant decreased migration toward a chemotactic gradient. These findings suggest that the migratory capacity of MSCs that have interacted with bacteria in the intestinal lamina propria will be determined by the nature of the bacterial-MSC interaction. Previous studies that investigated the role of TLR ligation on MSC migration found that ligation of TLR3 by synthetic agonists induced a mild increase in MSC migration, whereas LPS (TLR4 agonist) and flagellin (TLR5 agonist) did not influence MSC migration [22]. Another study reported that TLR2 ligation inhibited MSC migration in “wound healing” assays [24]. Although ST carry TLR4 and TLR5 agonists (ie, LPS and flagellin, respectively), the live bacteria in our study may operate via additional pathogen-associated molecular patterns that may activate specific pattern recognition receptors using undefined mechanisms that impact MSC migration. Similarly, while LB organisms carry TLR2 ligands (ie, lipoteichoic acid), they also express additional pathogen-associated molecular patterns with specific pattern recognition receptor targets [57]. Further studies are needed to elucidate migration-related cellular pathways and cytoskeleton modifications after MSC-microbial interactions.
Previous studies have reported that under specific conditions, MSCs may undergo a phenotype switch and function as APCs [45,46,58,59]. Moreover, several other studies have reported that ligation of TLR agonists by MSCs may also induce a proinflammatory phenotype switch [18,20,22,23,60,61]. In our study, direct bacteria-MSC coincubation did not induce cell surface expression of antigen presentation molecules (ie, MHC-II and CD1) or costimulatory molecules (ie, CD80/86). These findings suggest that while specific in vitro manipulations may induce an APC phenotype shift in MSCs, the interaction with live and physiologically relevant bacteria, including pathogenic bacteria (ie, ST), did not induce this potentially harmful phenotype shift. We further noticed that intestinal bacteria induced the pleiotropic cytokine IL6 and the enzymatic precursor for PGE2 (ie, COX2). While the chemotactic factor IL8 was markedly induced by ST, the proinflammatory mediator IL1β was not detected. PGE2 [62] and IL6 [63] are well-characterized cytokines with both pro- and anti-inflammatory mediators that are known to be secreted by MSCs and to inhibit lymphocyte proliferation, Th17 differentiation, and M1 differentiation of monocytes [64,65]. The final biologic effect of these cytokines is dependent upon the overall cytokine milieu and the cellular components within the niche. CD54 is a ligand for the β2 integrin (CD11/CD18 complex) which is present on the cell surface of leukocytes and facilitates their extravasation [66]. CD54 expression is increased on the cell surface of activated MSCs, and β2 integrin ligation was shown to mediate MSC-lymphocyte interaction, cell signaling, and immunomodulation [4,67]. Specifically, β2 integrin ligation by CD54 on the cell surface of activated MSCs was shown to be important in MSC inhibition of the differentiation of the proinflammatory Th17 subset from naive Th cells [4,67].
We further investigated bacteria-primed MSC capacity to inhibit T-cell proliferation. Compatible with multiple previous reports, native, untreated MSCs inhibited mitogen induced T-cell proliferation [1,2]. In this study, MSC interactions with the pathogenic bacteria ST augmented the inhibitory effect that the MSCs had on mitogen-induced T-cell proliferation compared with untreated MSCs or LB-treated MSCs. Moreover, we showed that PGE2 was a key inhibitory mediator that is delivered by microbe-primed MSCs, as microbe-primed MSCs that were treated with a COX inhibitor (ie, indomethacin) had less inhibitory effect on mitogen-treated T-cells.
Conclusions
MSCs interact with a broad range of intestinal bacteria; each of which has the capacity to activate the host cell with distinctively different phenotypes. MSC-microbe interactions have a marked effect on MSC function, including MSC migration and immunomodulation, which may be pivotal in vivo and impact a variety of clinical settings where MSCs are being explored as potential therapeutics. MSCs from different tissue sources (ie, cord tissue, bone marrow, etc.) and different animal species may have variable specific responses to microbial interaction, and as such, our findings should be further explored in other model organisms and in MSCs derived from various tissue sources. Nevertheless, MSCs from many tissue types and animal species share a common core of biologic behavior and activation pathways [68,69]. The proposed model ties the clinically relevant points together with the mechanistic changes that were observed in this study. This study definitively found that MCS-microbe interactions change the fate of MSC biology.
Footnotes
Acknowledgments
The authors gratefully acknowledge the staff in the blood bank and the Regenerative Medicine Lab at the UC Davis Veterinary Medical Teaching Hospital for technical support. The authors further wish to thank the dog owners who have generously agreed to donate blood for this study. The authors gratefully acknowledge Dr. Geraldine Hunt and the staff in the Veterinary Medical Teaching Hospital soft tissue surgery service for fat tissue collection. This project was supported by a generous gift from Mr. Dick and Carolyn Randall to D.L.B. and funding to B.C.W. from National Institutes of Health (NIH-1R01HD065122-01A1 and NIH-U24-DK097154), the U.S. Food and Drug Administration (FDA: FD003572-03), and Agilent Technologies Thought Leader Award program.
Author Disclosure Statement
No competing financial interests exist.