
Abstract
Differentiation of pluripotent cells into endoderm-related cell types initially requires in vitro gastrulation into the definitive endoderm (DE). Most differentiation protocols are initiated from colonies of pluripotent cells complicating their adaption due to insufficiently defined starting conditions. The protocol described here was initiated from a defined cell number of dispersed single cells and tested on three different human embryonic stem cell lines and one human induced pluripotent stem cell line. Combined activation of ActivinA/Nodal signaling and GSK3 inhibition for the first 24 h, followed by ActivinA/Nodal signaling efficiently induced the DE state. Activation of ActivinA/Nodal signaling alone was not effective. Efficient GSK3 inhibition allowed the reduction of the ActivinA concentration during the entire protocol. A feeder-independent cultivation of pluripotent cells was preferred to achieve the high efficiency and robustness since feeder cells hindered the differentiation process. Additionally, inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway was not required, nonetheless yielding high cell numbers efficiently committed toward the DE. Finally, the endoderm generated could be differentiated further into PDX1-positive pan-pancreatic cells and NGN3-positive endocrine progenitors. Thus, this efficient and robust DE differentiation protocol is a step forward toward better reproducibility due to the well-defined conditions based on dispersed single cells from feeder-free-cultivated human pluripotent cells.
Introduction
P
For the differentiation of human and mouse ESCs it is generally accepted that high concentrations of ActivinA (ActA) direct this process by activating the transforming growth factor-beta (TGF-β) signaling pathway [9,10]. However, it is not likely that this well-concerted process is solely controlled by one pathway because it includes the differentiation of the epiblast-like hESCs [11], via the primitive streak to mesendodermal progenitor cells and finally their division into either mesoderm or endoderm. Consequently, most groups combine ActA with other molecules like Wnt3a [12,13], phosphatidylinositol 3-kinase (PI3K) inhibitors [14,15], or HDAC inhibitors [16,17] to differentiate hESCs into the DE. More recently, it has been shown that inhibition of the PI3K signaling is beneficial for the DE differentiation with ActA [18].
Nearly all protocols initiate differentiation from human pluripotent cell colonies because it is the most popular cultivation technique either feeder dependent or feeder free. This complicates the adaption of published protocols by other laboratories due to the undefined starting conditions, such as colony size, colony number per cavity, passaging procedure, and a potential crosstalk between feeder cells and pluripotent cells during differentiation. Therefore, this study used dispersed single cells in defined cell numbers to differentiate human pluripotent stem cells into the DE lineage. These dispersed single cells can be differentiated efficiently into the DE by a combined treatment with CHIR-99021 (Chir), activating canonical Wnt signaling [19], and ActA for the first 24 h followed by ActA alone. Further, an efficient inhibition of the GSK3 [19] allowed a reduction of the usual ActA concentration. An inhibition of PI3K was not necessary under these conditions, which resulted in a high proliferation rate during the differentiation process. Interestingly, an additional chemical inhibition of the PI3K decreased the proliferation rate without increasing efficiency. ESCs differentiated with this well-defined protocol were able to differentiate into the pancreatic lineage under appropriate conditions reflecting the functionality of the differentiated DE cells.
Materials and Methods
Human ESC culture
Human ESC work was performed in accordance with the German Stem Cell Act. High priority of the research objectives in this study was confirmed by the Central Ethical Committee for Stem Cell Research (ZES) in Berlin, Germany. Approval of the experiments was granted by the Robert Koch-Institute (Berlin), which is the responsible authority for hESC research in Germany.
Routine culturing of the hESC lines HUES4 and HUES8 was performed with minor changes as described earlier [17]. Briefly, hESCs were cultured as colonies on cell culture plates coated with hESC-qualified Matrigel (Corning, Amsterdam, The Netherlands) according to the manufacturer's instructions in mTeSR1 medium (StemCell Technologies, Köln, Germany) and passaged every 5–7 days. For passaging, the cells were incubated for 5–10 min with Dispase (1 U/mL; StemCell Technologies), washed twice with Knockout Dulbecco's modified Eagle's medium (DMEM)/F12 (Life Technologies, Darmstadt, Germany), and scraped with a 1-mL tip. The cells were reseeded as clumps in a ratio of 1:2–6 in mTeSR1 medium on plates coated with Matrigel. The supernatant of this Matrigel coating, for routine feeder-free ESC cultivation, was collected and stored at 4°C for a maximum of 2 weeks. Such “reused” Matrigel was employed for the coating of differentiation experiments equal to the normal Matrigel coating procedure.
A third human ESC line, HES3, and the human iPSC (hiPSC) line hCBiPS2 [20] were routinely cultivated on feeder cells as described elsewhere [21,22] with a split ratio of 1:8–12. To adapt these cell lines to the aforementioned feeder-free culture conditions, they were passaged as clumps on Matrigel-coated plates in mTeSR1. After the first passage most of the feeder cells were removed and the cells were cultivated as described previously with a split ratio of 1:6–12.
Differentiation experiments from dispersed single cells
ESC colonies were dissociated by washing once with Knockout-DMEM/F12 or phosphate-buffered saline (PBS), incubated for 5–8 min with gentle cell-dissociation reagent (StemCell Technologies) or trypsin/EDTA, collected with Knockout-DMEM/F12, and centrifuged for 3 min at 300 g. The cell pellet was resuspended in mTeSR1 containing 10 μM Y-27632 (Selleck Chemicals, Munich, Germany). Cells were counted with the Cellometer Auto T4 counter (Nexcelom Bioscience, Lawrence, MA) and seeded on dishes coated with “reused” Matrigel (see second paragraph in “Human ESC culture” section) at densities of 95,000–100,000 cells/cm2 (HUES4 and HUES8) or 65,000–72,000 cells/cm2 (HES3) in mTeSR1 with 10 μM Y-27632 to allow reattachment of the cells overnight.
Advanced RPMI 1640 (Life Technologies) supplemented with penicillin/streptomycin, Glutamax (Life Technologies), and 0.2% fetal bovine serum (FBS; PAA, Vienna, Austria) was used as base medium for all differentiation protocols. For randomized differentiation no substances were added. A classical DE induction protocol [12] was applied as positive control using a sequential treatment with 25 ng/mL Wnt3a (Peprotech, Hamburg, Germany) and 100 ng/mL ActivinA (Peprotech) for the first 24 h without FBS, followed by a 72-h treatment with 100 ng/mL ActivinA plus 0.2% FBS. Differentiation exclusively via TGF-β signaling was conducted with 50–100 ng/mL ActivinA. Inhibition of the GSK3 was performed with 5 μM CHIR-99021 (Biozol, Eching, Germany) and 50 or 100 ng/mL ActivinA for the first 24 h followed by 72-h cultivation in base medium supplemented with 50 or 100 ng/mL ActivinA. Inhibition of the PI3K was performed by supplementation of 10 μM LY294002 to the respective medium. Media were changed daily.
Differentiation after DE induction was performed with minor modifications according to a published protocol [23]. Briefly, to induce a randomized differentiation, the medium was changed to DMEM (Life Technologies) supplemented with penicillin/streptomycin, Glutamax, 1% B27 (Life Technologies), and 50 ng/mL recombinant human FGF-10 (ReliaTech, Wolfenbüttel, Germany) for the following days. Directed differentiation into the pancreatic lineage was conducted with 2 μM retinoic acid (RA; Sigma-Aldrich, Taufkirchen, Germany), 1 μM Dorsomorphin (DM; Tocris Bioscience, Bristol, United Kingdom), and 10 μM SB-431542 (SB; Tocris Bioscience) supplemented to this medium. The medium was changed daily until day 10 of differentiation and thereafter every second day.
Flow cytometry
For flow cytometric analysis the in-vitro-differentiated cells were washed with PBS, dissociated using trypsin/EDTA, and resuspended in PBS plus 2% FBS. Antibody staining was performed following standard protocols. About 2.5×105 cells were washed, incubated for 45–60 min with primary conjugated antibodies, washed three times, and measured with a CyFlow ML flow cytometer (Partec, Münster, Germany). At least 2.0×104 events of each sample were analyzed with FlowJo (Ashland, OR, USA). The conjugated antibodies anti-human CD49e-FITC (Biolegend, London, United Kingdom) and anti-human CXCR4-PE (Neuromics, Minneapolis, MN) were used.
Gene expression analysis
Isolation of total RNA was carried out with the RNeasy Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Synthesis of cDNA was performed from 500 to 2,000 ng of total RNA per reaction using the RevertAid™ H Minus M-MuLV Reverse Transcriptase (Thermo Fisher Scientific, Braunschweig, Germany) with random hexamer primers (Life Technologies). The cDNA samples were diluted and 5–15 ng (of the total, initial RNA) was used per reaction. All reactions were performed on a ViiA7 real-time PCR system (Life Technologies) with the following protocol: 50°C for 2 min, 95°C for 10 min, and 40 cycles comprising a melting step at 95°C for 15 s and an annealing/extension step at 60°C for 60 s. To verify the correct amplification for SybrGreen-based PCRs, a melting curve was performed. Each sample was amplified as triplicate using specific primer pairs or TaqMan assays (Supplementary Table S1; Supplementary Data are available online at
Immunocytochemistry
Immunocytochemistry was performed according to standard procedures. Briefly, hESCs were seeded on Matrigel-coated glass slides (Zellkontakt, Nörten-Hardenberg, Germany) and subjected to differentiation. Alternatively the cells were passaged on Matrigel-coated glass slides 1 day prior to fixation in the respective medium containing 5 μM Y-27632. Cells were fixed in 4% (w/v) paraformaldehyde for 20–30 min at 4°C and subsequently blocked for 20 min in PBS plus 0.2% Triton X-100 with either 6% bovine serum albumin (BSA) plus 1 mg/mL NaBH4 or 5% donkey serum (Dianova, Hamburg Germany). Primary and secondary antibodies were diluted in PBS with 0.1% Triton X-100 plus 0.1% BSA. Primary antibodies were incubated on the slides for 1–3 h at room temperature or overnight at 4°C. Secondary antibodies were diluted 1:250–500 and incubated for 1 h at room temperature. The following primary antibodies were used: anti-SOX17 (AF1924; R&D Systems, Minneapolis, MN), anti-FOXA2 (07-633; MerckMillipore, Schwalbach, Germany), anti-Oct3/4 (sc-5279; SantaCruz, Heidelberg, Germany), anti-PDX1 (AF2419; R&D Systems), and anti-NGN3 (AF3444; R&D Systems). Secondary antibodies were obtained from Dianova conjugated with AlexaFluor or Cy fluorophores. Finally, staining of the nuclei was performed with 4′,6-diamidino-2-phenylindole (DAPI) and the slides were mounted with Mowiol/DABCO (Sigma-Aldrich) anti-photo-bleaching mounting media. Stained cells were examined using an Olympus IX81 microscope (Olympus, Hamburg, Germany) and multiple representative pictures of each slide were taken.
Quantification of SOX17/FOXA2 double-positive cells per picture was carried out by manual counting. Initially, double-positive cells were counted, followed by single-positive cells and finally the nuclei to calculate the percentages of double-positive cells. The single-positive cells were counted as negative for the subsequent quantification of double-positive cells. More than 4,000 nuclei were counted per condition from three independent experiments and representative pictures. Quantification of PDX1 and NGN3 single-positive cells was performed with the BZ Analyzer Software (Keyence, Neu-Isenburg, Germany). More than 13,000 nuclei for PDX1 and more than 9,000 nuclei for NGN3 were automatically counted from different representative pictures for small molecule (SM)–treated cells. Without SM treatment more than 3,000 nuclei (PDX1) or 4,000 nuclei (NGN3) were counted by the software, respectively.
Statistics
Unless stated otherwise the data values were expressed as mean±standard error of the mean. Statistical analyses were performed using the GraphPad Prism analysis software (Graphpad, San Diego, CA) applying Student's t-test or ANOVA followed by Bonferroni's or Dunnett's post hoc test for multiple comparisons.
Results
Flow cytometric quantification of the definitive endodermal differentiation potential under different conditions
Human ESC colonies were separated into single cells to obtain a defined starting population and were allowed to reattach overnight on dishes coated with “reused” Matrigel. Subsequent differentiation was performed under the conditions depicted in Figure 1A. The two surface markers CXCR4 and CD49e were measured by flow cytometry for quantification of DE-committed cells (Fig. 1B). Random differentiation (7.5%±0.8%) and the treatment with ActA alone (A) (7.6%±0.5%) did not yield high numbers of CXCR4-positive cells (Fig. 1C). The reference protocol (RP), published by D'Amour et al. [12], yielded significantly increased higher quantities (39.6%±5.3%). However, the combined treatment with Chir and ActA for the first 24 h followed by ActA alone (CA-A) resulted in an additional significant (>2-fold) increase of CXCR4-positive cells (82.9%±1.4%) compared with the RP (Fig. 1C). To exclude that the CA-A protocol was cell line dependent, a second hESC line (HUES4) was subjected to these conditions with similar findings after 4 days of differentiation (Fig. 1D). This cell line yielded with the CA-A protocol 73.2%±1.6% of CXCR4-positive cells, a >2-fold increase compared with the RP.
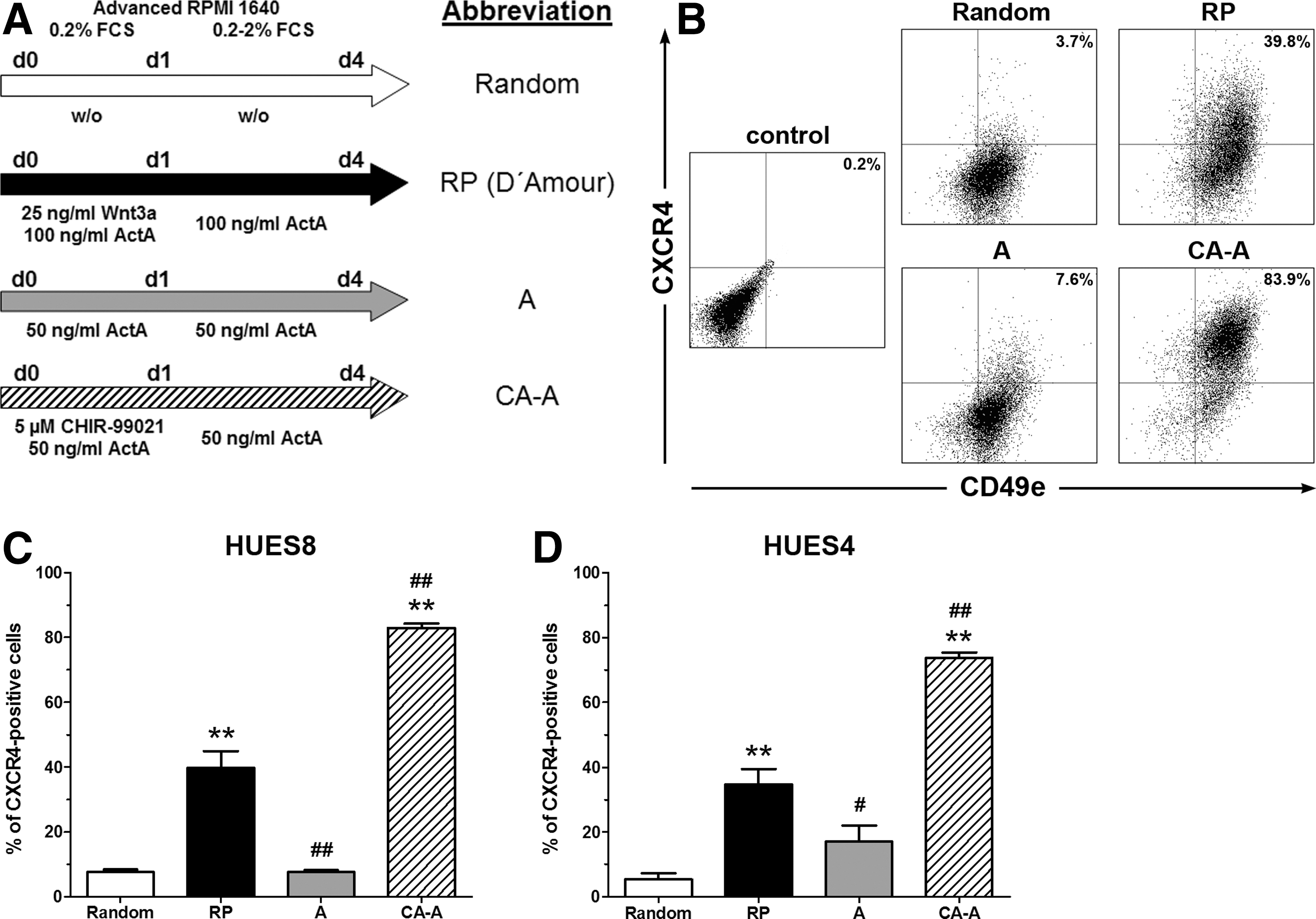
Comparative flow cytometric analysis of definitive endoderm (DE) development for the human embryonic stem cell (hESC) lines HUES4 and HUES8 initiated from dispersed single cells. Human ESCs were dissociated into single cells, cultivated for 24 h (overnight) in the presence of Y-27632 in mTeSR1, and thereafter differentiated following the protocols depicted in
In a recent publication [24] we could show that replacement of Wnt3a by Chir allowed a reduction of the typically required ActA concentration. Thus, 50 ng/mL of ActA was used throughout the study but data for 100 ng/mL ActA were gathered additionally to exclude growth factor concentration effects (Supplementary Fig. S1A–D). The CA-A protocol revealed no differences between 50 and 100 ng/mL ActA (Supplementary Fig. S1A, B). This was also consistently observed for protocol A. Neither the flow cytometry data nor the gene expression data revealed any significant differences between 50 and 100 ng/mL ActA for HUES8 and HES3 cells (Supplementary Fig. S1A, C, D).
Differentiation potential toward the DE of HUES4 and HUES8 cells under these conditions
Differentiated populations were analyzed upon the expression of GSC, SOX17, and FOXA2 by quantitative real-time PCR (qPCR) (Fig. 2A, B). GSC was significantly increased for CA-A-treated cells compared with randomly differentiated samples after 4 days of differentiation. Only the CA-A protocol resulted in significant induced gene expression levels for the two DE marker genes SOX17 and FOXA2 compared with the RP (Fig. 2A). Overall these three genes showed the highest expression levels after CA-A treatment, demonstrating the DE commitment. Embryonic transcription factors [POU5f1 (OCT3/4), NANOG, and SOX2] were expressed at very low levels after 4 days of differentiation with the CA-A protocol. The expression was similar or even lower compared with the Random protocol and significantly reduced compared with the RP (Fig. 2A and Supplementary Fig. S1E). The extraembryonic marker gene SOX7 was equally induced in the RP and the CA-A condition and low in the Random protocol (Fig. 2A). Thus, both conditions induced some extraembryonic endodermal cells without a correlation to the DE differentiation efficiency, indicating no favored extraembryonic endodermal differentiation with the CA-A protocol compared with the RP. These expression patterns were in accordance with the flow cytometric data (Fig. 1C), thereby validating the two surface markers. The expression pattern of the second hESC line HUES4 was closely matching the data obtained with the HUES8 line (Fig. 2B).

Gene and protein expression analysis of DE generated from HUES8 and HUES4 cells.
Nuclear colocalization of SOX17 and FOXA2 was regarded as a hallmark of DE commitment and double-positive cells were quantified. Representative pictures of the HUES8 line are depicted in Figure 2C and the quantification is shown in Figure 2D. A nearly confluent double-positive layer for the CA-A protocol (82.9%±1.4% SOX17/FOXA2 double-positive cells) was observed. The RP protocol (42.3%±2.2%) showed clusters of double-positive cells, while the Random (5.3%±1.0%) and A protocols (13.6%±3.1%) showed only scattered double-positive cells (Fig. 2C, D). Single-positive cells were seldom detected under all conditions (Fig. 2C, white arrowhead) and defined as negative for the quantification of double-positive cells. These results are in line with the flow cytometric quantification (Fig. 1C) and the gene expression analysis (Fig. 2A) substantiated the commitment of these cells into the DE lineage. Quantification of the second hESC line HUES4 (Fig. 2E) yielded similar results to those measured by flow cytometry (Fig. 1D) with significantly increased numbers of 23.7%±3.0% (RP) and 71.7%±2.9% (CA-A) of double-positive cells compared with randomly differentiated cells (Random). In summary, these results clearly demonstrate that the CA-A protocol was able to robustly and efficiently differentiate the dispersed single cells from both hESC lines into the DE lineage.
A third hESC line differentiated as dispersed single cells into the DE
The HES3 cell line was routinely cultivated on feeder cells, which allowed the analysis of (a) the effect of a feeder-dependent versus feeder-free cultivation and (b) the effect of mTeSR1 versus mouse embryonic feed cell-conditioned medium (MEF-CM) prior to the differentiation. HES3 colonies were directly dispersed together with the feeder cells and cultivated for one passage in mTeSR1 or MEF-CM, or under feeder-free conditions for at least two passages prior to differentiation. For the 24-h reattachment phase either mTeSR1 or MEF-CM was used and differentiation was performed according to the specified protocols. No significant differences for all applied protocols could be detected under feeder-dependent versus feeder-reduced conditions prior to differentiation. Only the CA-A protocol yielded for these conditions >40% of CXCR4-positive cells (Fig. 3A). Expression analysis of SOX17, FOXA2, and GSC confirmed this finding with significantly increased expression only for the CA-A protocol (Supplementary Fig. S2A). In contrast, feeder-free-cultivated HES3 cells yielded significantly increased numbers of CXCR4-positive cells (68.2%±4.9%) compared with all other conditions when differentiated with the CA-A protocol (Fig. 3A). Hence, the CA-A protocol works ideally with cells cultivated under feeder-free conditions and two to three passages without feeder cells were sufficient to ensure the efficient DE generation.
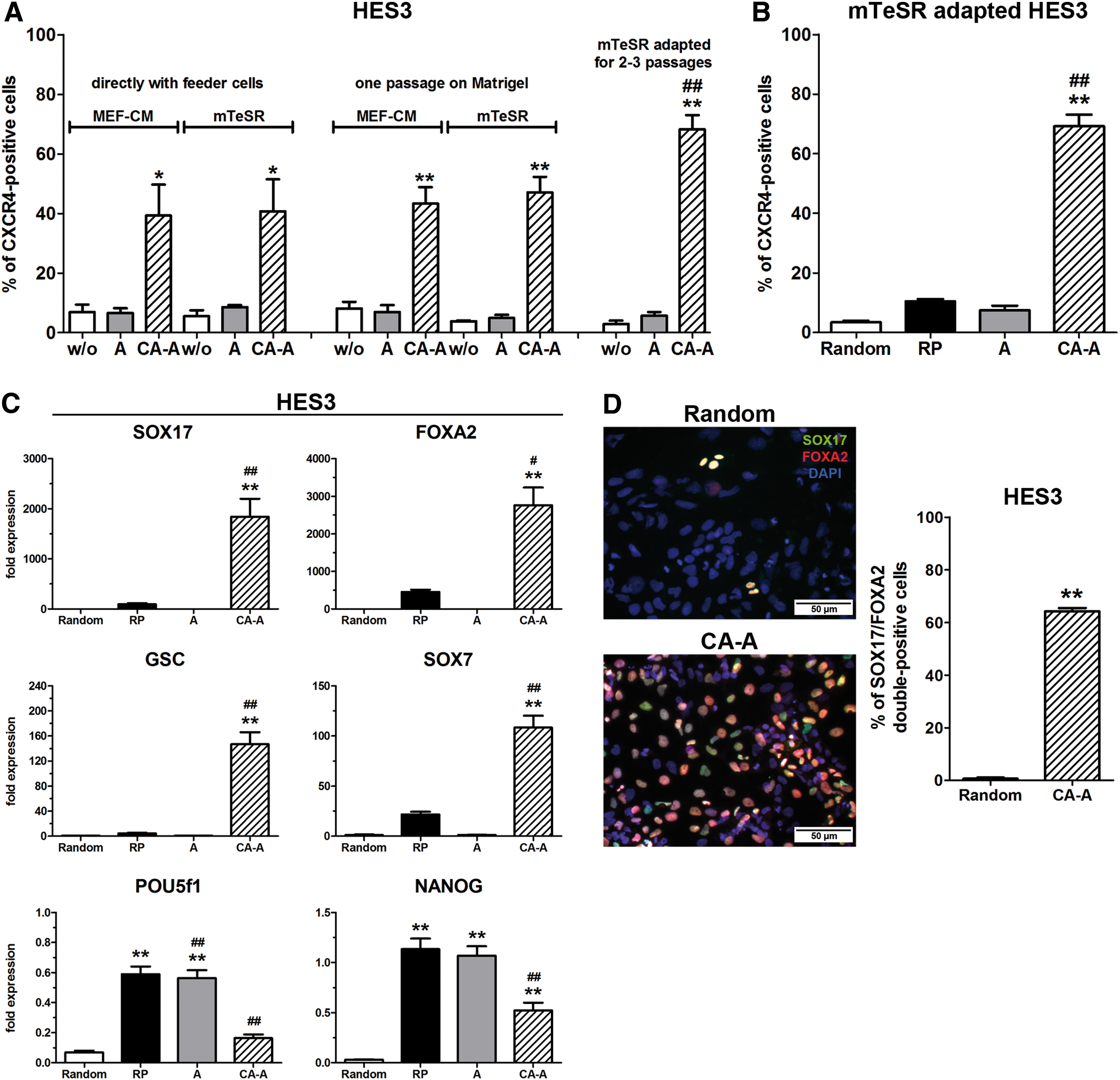
DE differentiation from HES3 cells. Depicted in
In comparison to HUES4 and HUES8, HES3 cells reached 69.3%±3.9% of CXCR4-positive cells after 4 days of differentiation with the CA-A protocol, significantly increased numbers compared with all other protocols (Fig. 3B). As well for this hESC line, a treatment only with ActA was not efficient to obtain sufficient numbers of DE-committed cells (Fig. 3B). The expression of the DE marker genes GSC, SOX17, and FOXA2 was significantly induced upon differentiation with the CA-A protocol, whereas the expression of the embryonic transcription factors POU5f1 (OCT3/4), NANOG, and SOX2 exhibited low levels compared with undifferentiated cells as well as with the RP and A conditions (Fig. 3C and Supplementary Fig. S1E). Interestingly, the HES3 line exhibited a lower SOX7 expression in the RP compared with the CA-A condition (Fig. 3C) but also a lower DE efficiency compared with the HUES lines. Quantification of SOX17/FOXA2 double-positive cells revealed 64.3%±2.9% double-positive cells for the CA-A protocol (Fig. 3D), matching the flow cytometry results (Fig. 3B) and the gene expression analysis (Fig. 3C). Thus, the CA-A protocol also differentiated the third hESC line HES3 with high efficiencies into the DE lineage.
Importance of cell number, concentration dependency, and extracellular matrix for the DE differentiation from dispersed single cells
To further define the conditions for an efficient DE generation, the optimal cell densities (cells/cm2) before initiation of the differentiation were assessed. HUES8 cells were passaged as single cells in the presence of Y-27632 and reseeded with cell densities from 5.3×104 to 1.3×105 cells/cm2. Differentiation was initiated the next day by applying the CA-A protocol and DE-committed cells were quantified 4 days later by flow cytometric measurement of CXCR4-positive cells (Fig. 4A). Below 9.2×105 cells/cm2 the DE differentiation efficiency significantly decreased, whereas above this number no significant increase was detectable (Fig. 4A). This demonstrates the requirement of a minimal cell density to ensure a highly efficient DE differentiation with this protocol. For HES3 cells lower cell amounts were required to ensure the efficient DE differentiation compared with both HUES lines. This can be explained by the higher reattachment rate of HES3 cells (∼85%–95%) compared with the HUES lines (∼65%–75%). Hence, the specific reattachment rate of a particular cell line has to be considered to calculate the ideal cell density to ensure efficient and robust DE differentiation with the CA-A protocol.
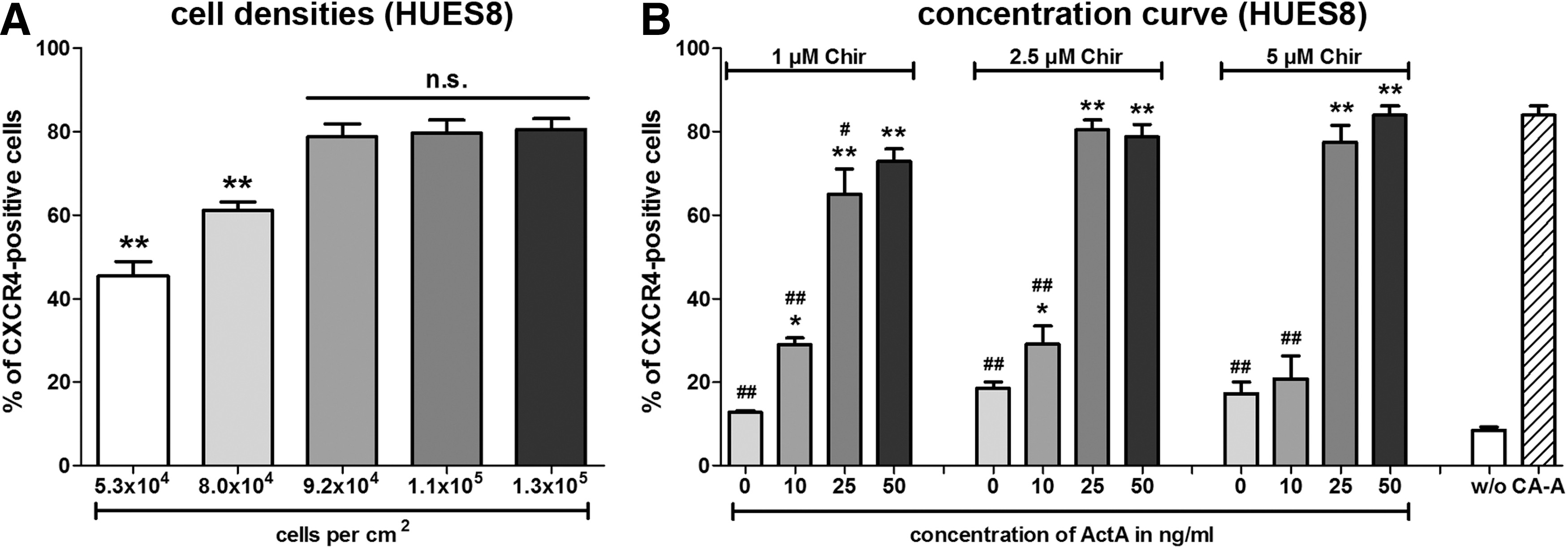
Minimally required cell densities and concentration dependencies of Chir and ActivinA (ActA) for the DE differentiation potential of HUES8 cells.
Further, different concentrations of ActA and Chir were tested to define the required signaling intensities. HUES8 cells were subjected to the indicated concentrations after overnight reattachment of the dispersed single cells in the presence of Y-27632 (Fig. 4B). Chir was first applied together with the indicated ActA concentration (first 24 h) and subsequently ActA was supplemented alone. HUES8 cells showed a minimally required concentration of 10 ng/mL ActA to ensure differentiation above background levels (zero ActA) (Fig. 4B). However, 25 ng/mL of ActA resulted for all three tested Chir concentrations in significantly increased differentiation efficiencies (Fig. 4B). An initial concentration of 2.5 μM Chir was ideal and lower concentrations (1 μM Chir) yielded slightly decreased CXCR4-positive cell numbers, whereas 5 μM of Chir had no beneficial effect. This result indicates that an activation of the Wnt/β-catenin signaling pathway above a particular threshold may reduce the requirement of high ActA concentrations during endoderm differentiation. A comparison of these results with HUES4 cells (Supplementary Fig. S2B) revealed stronger dependency on the activation of the Wnt/β-catenin signaling pathway than HUES8 cells. Only high concentrations of Chir (5 μM) permitted a decrease of the ActA concentration to 25 ng/mL (Supplementary Fig. S2B) without reducing the efficiency.
Additionally, the effect of a “reused” or fresh Matrigel surface was analyzed for HUES8 (Fig. 5A, B and Supplementary Fig. S2C) and HES3 cells (Fig. 5C, D and Supplementary Fig. S2D). No difference could be detected for the Random, RP, and CA-A protocols for both cell lines in the flow cytometry analysis (Fig. 5A, C). The expression of different marker genes (POU5F1, SOX2, NANOG, SOX17, FOXA2, GSC, and SOX7) was not significantly changed under the tested conditions between “reused” and fresh Matrigel coating under the respective differentiation condition (Fig. 5B, D and Supplementary Fig. S2C, D).
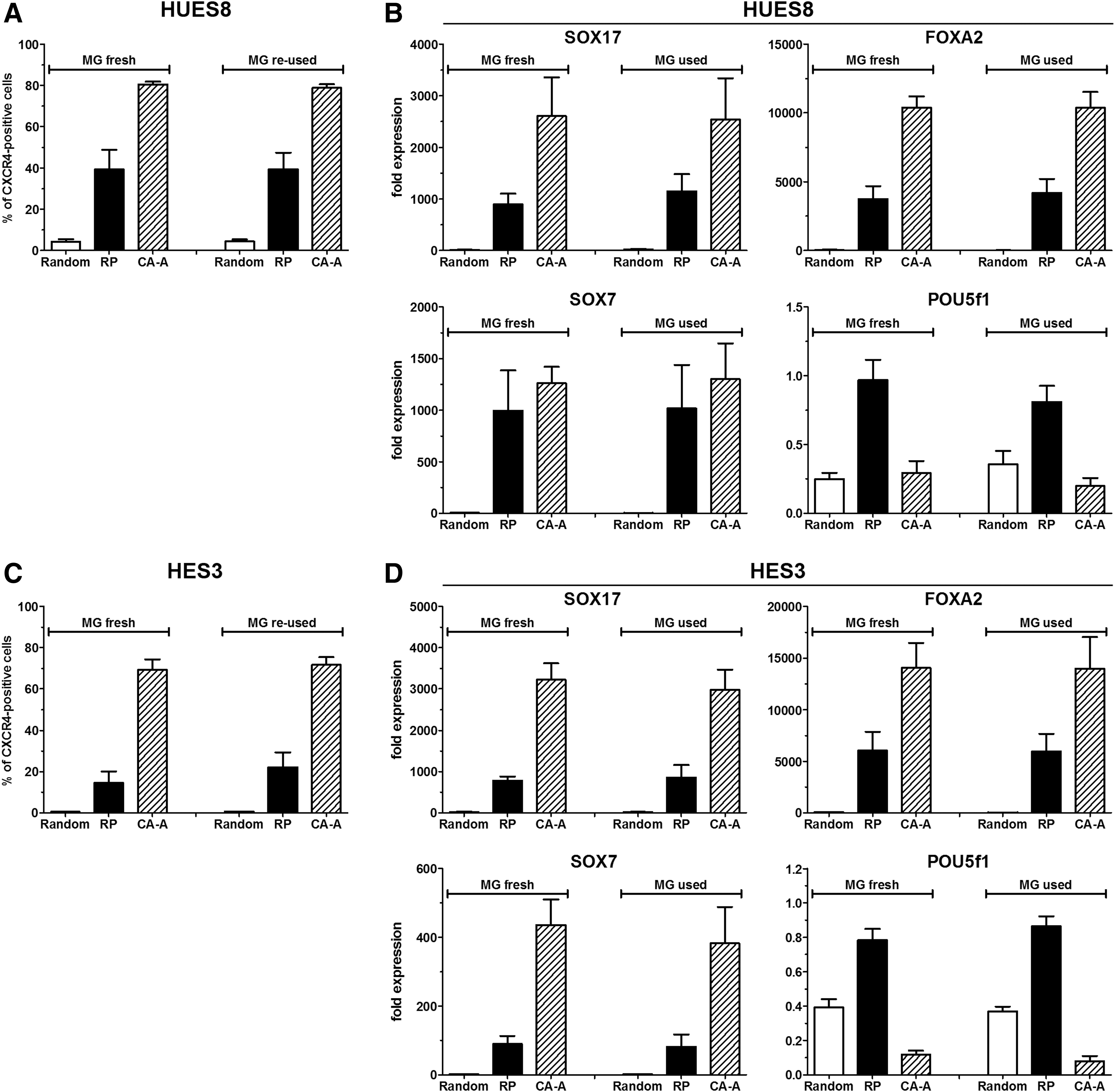
Effect of the fresh and “reused” Matrigel for DE differentiation. The flow cytometric quantification of CXCR4-positive cells after 4 days of differentiation is shown for the HUES8 line
PI3K inhibition and proliferation during differentiation
McLean et al. [18] postulated that PI3K inhibition is a necessary prerequisite for the induction of the DE by ActA. Hence, four conditions were tested: PI3K activation within the advanced RPMI 1640 (containing 10 mg/L insulin) and regular RPMI 1640 media, and the effect of the PI3K inhibitor LY294002 in both base media (Fig. 6 and Supplementary Fig. S3).
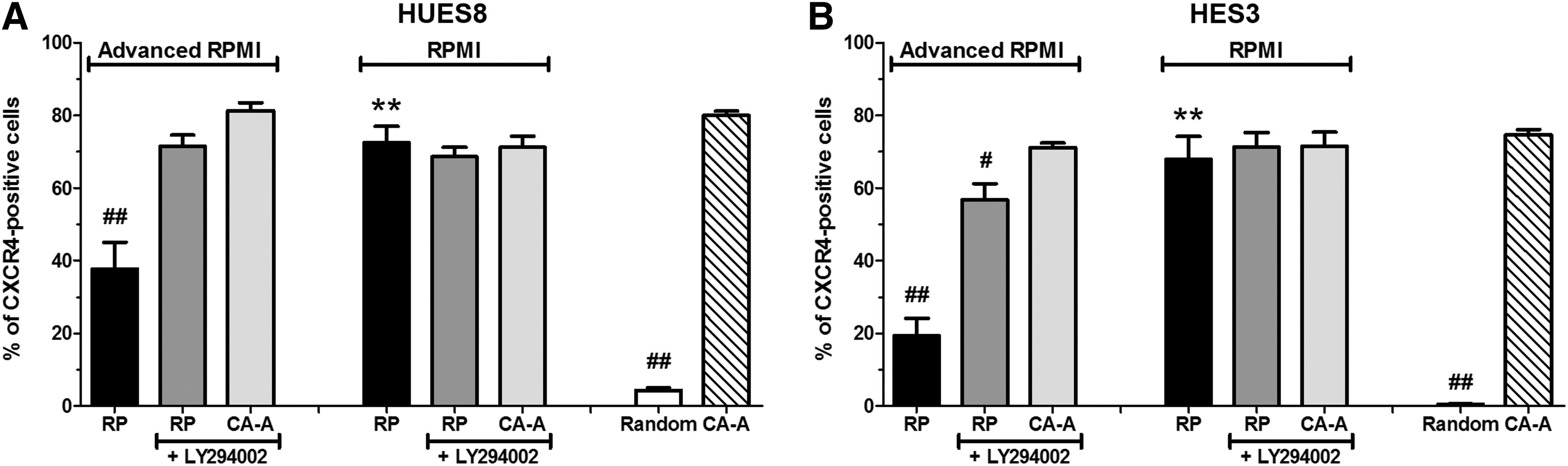
Effects of phosphatidylinositol 3-kinase (PI3K) signaling for the DE formation and endodermal differentiation potential of human pluripotent stem cells. Shown in
In RPMI 1640 medium, HUES8 cells yielded ∼70% CXCR4-positive cells but only ∼40% when advanced RPMI 1640 was used for the RP (Fig. 6A). Addition of the PI3K inhibitor did not significantly change the outcome in RPMI 1640 medium but increased the quantity of CXCR4-positive cells in advanced RPMI 1640. However, the proliferation rate during differentiation in RPMI 1640 medium, especially in the presence of LY294002, was significantly reduced compared with the CA-A protocol, which revealed the highest increase in cell quantity (Table 1). Thus, an additional inhibition of the PI3K in advanced RPMI 1640 medium was only beneficial for the RP, indicating that this protocol is highly dependent on PI3K inhibition. In contrast, no increased efficiencies were detectable for the CA-A protocol, neither in advanced RPMI 1640 or RPMI 1640 supplemented with LY294002 (Fig. 6A). A similar pattern of the changed efficiencies was detected for HES3 cells (Fig. 6B).
The significant differences between the analyzed conditions were calculated by ANOVA followed by Dunnett's post hoc test for multiple comparisons to the CA-A condition.
P≤0.01, b P≤0.05 compared with the CA-A protocol in advanced RPMI 1640 of the respective cell line.
LY, LY294002; RP, reference protocol.
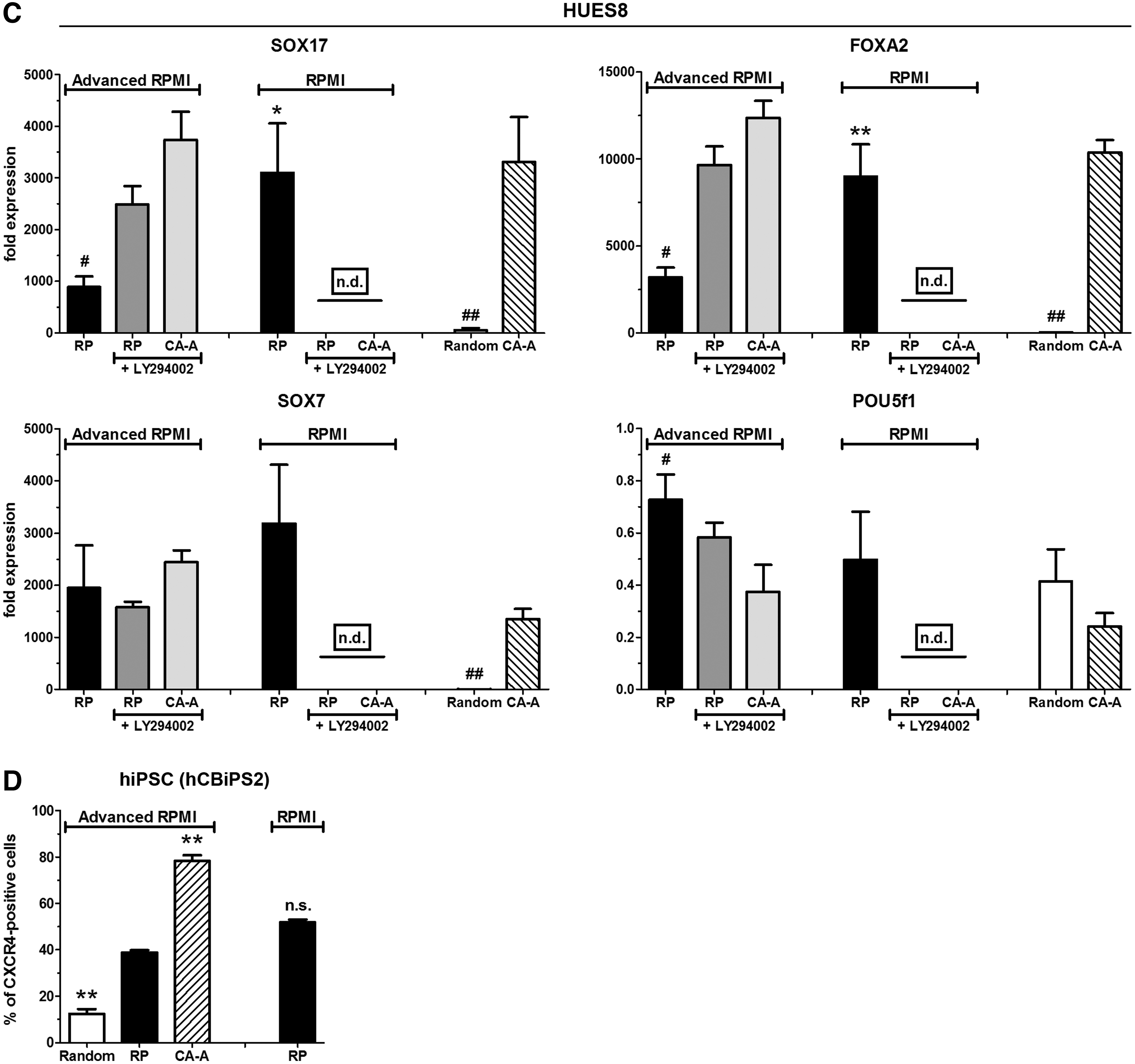
The presence of LY294002 in RPMI 1640 resulted in a loss of cells (fold increase below one) and the RNA quantity of these cells was low, too, which excluded a reliable qPCR gene expression analysis for HUES8 cells (Fig. 6C and Supplementary Fig. S3A). Otherwise the gene expression profiles of the depicted marker genes (Fig. 6C and Supplementary Fig. S3A) were in accordance with the respective DE differentiation efficiency (Fig. 6A). Further, the CA-A protocol exhibited the lowest expression values of the embryonic marker genes (POU5f1, SOX2, and NANOG) compared with the other conditions (Fig. 6C and Supplementary Fig. S3A). A nearly identical expression profile for these genes was measurable for HES3 cells (Supplementary Fig. S3B). Of particular interest is the observation that all protocols in RPMI 1640 exhibited much higher SOX7 expression levels for both hESC lines than in advanced RPMI 1640 (Fig. 6C and Supplementary Fig. S3B).
The cell proliferation rate during differentiation is another important point to obtain large numbers of DE-committed cells. This was assessed by counting cells prior and after 4 days of differentiation to calculate the fold increase of cell quantity (Table 1). Compared with all other conditions, the CA-A protocol exhibited for the three hESC lines significantly increased proliferation rates with values around∼5-fold (HUES8) or ∼8-fold (HUES4 and HES3). PI3K inhibition decreased the proliferation and the change from advanced RPMI 1640 to RPMI 1640 massively reduced the proliferation, which resulted in a lower cell number than initially seeded (Table 1). A possible strong proliferation of undifferentiated cells under the CA-A condition could be excluded by the very low expression of embryonic markers (POU5f1, SOX2, and NANOG), the distinctness of CXCR4 staining, and immunofluorescent costaining of OCT3/4 and SOX17 (Supplementary Fig. S3C). The differentiated population contained mainly SOX17-positive cells and a minority of OCT3/4-positive cells, which had obviously resisted the differentiation procedure. Seldom double-positive cells were detected (arrowhead, Supplementary Fig. S3C). A third, rare population of cells was negative for both transcription factors, indicating a differentiation into a different germ layer (arrow, Supplementary Fig. S3C).
To demonstrate that the CA-A protocol is also able to differentiate hiPSCs into the DE, the hCBiPS2 line [20] was subjected to the differentiation conditions. As controls the RP (in advanced RPMI 1640 and RPMI 1640) and the Random protocol were used. The CA-A protocol generated with the hiPSC line significantly increased numbers of CXCR4-positive cells (>75%) compared with the RP in both media. Thus, the CA-A protocol is suited also for efficient hiPSC differentiation into the DE lineage.
Further developmental potential of the differentiated DE
Next, the developmental potential of the differentiated DE was further characterized by in vitro differentiation toward the pancreatic lineage. HUES8 and HUES4 cells were initially differentiated into the DE with the CA-A protocol. Further differentiation toward the pancreatic lineage was performed with an adapted protocol published by Kunisada et al. [23]. For this purpose a combined treatment with SB-431542 (TGF-β inhibition), DM (BMP inhibition), and RA (with SM; Fig. 7A, orange arrow) was applied. As a control condition, the small molecules were excluded from the medium (without SM; Figure 7A, gray arrow) to permit a randomized differentiation.
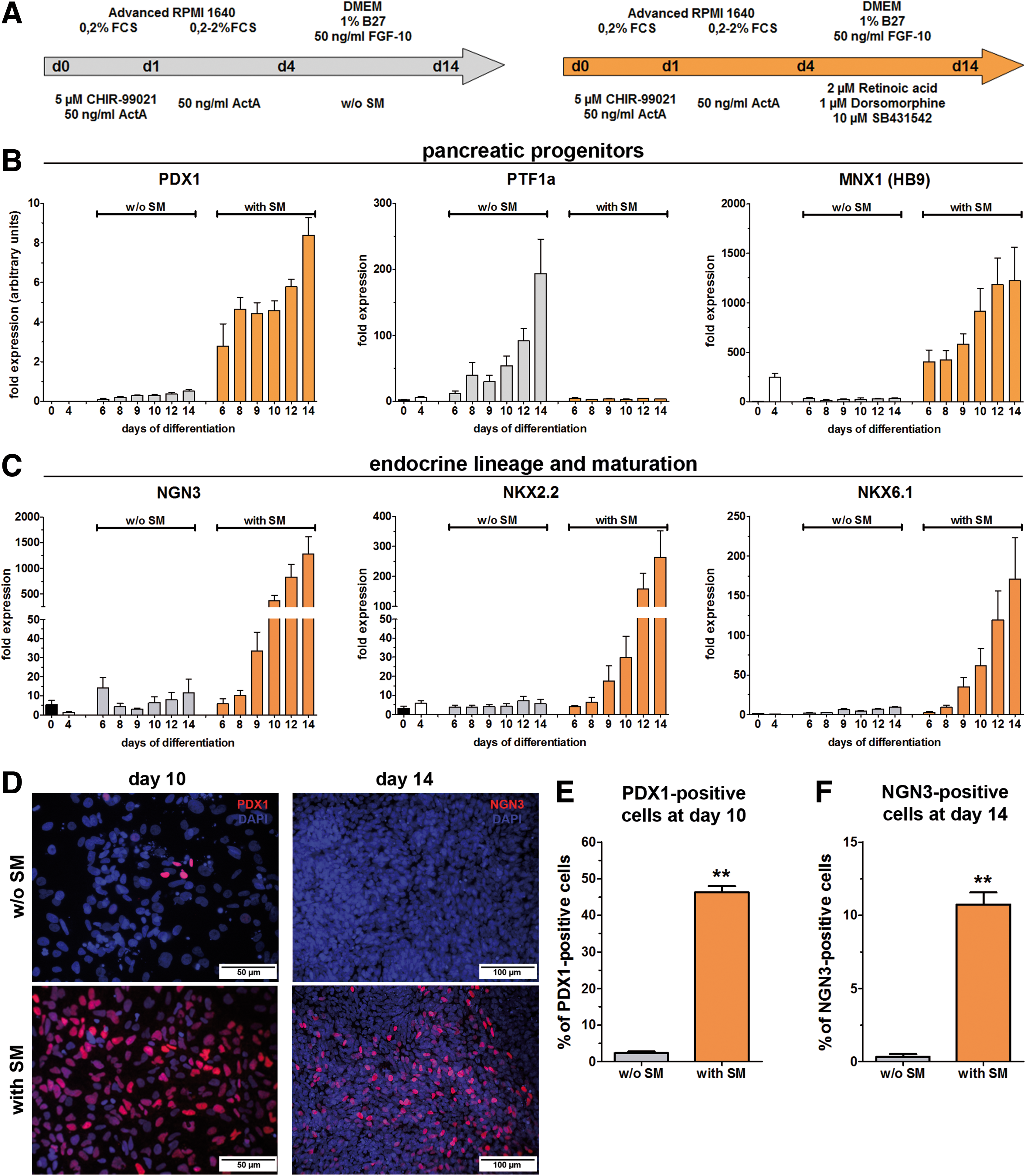
The pancreatic differentiation potential of HUES8 cells upon DE induction with the CA-A protocol from dispersed single cells. The two applied differentiation protocols for further differentiation toward the pancreatic lineage are shown in
After 4 days of differentiation the embryonic transcription factors POU5f1, NANOG, and SOX2 showed only low residual expression levels. Longer differentiation further decreased the expression of POU5f1 and NANOG. In contrast, SOX2 expression increased after 4 days in the medium with SM (Supplementary Fig. S4A). GSC was highly induced at day 4 of differentiation and its expression decreased thereafter to low levels (Supplementary Fig. S4B). The SOX17 and FOXA2 gene expression peaked after 4 days but especially the FOXA2 expression remained high in the medium with SM (Supplementary Fig. S4C). This is necessary because the expression of FOXA2 is passed from the DE toward the gut tube and the developing derivatives [25]. The early gut tube marker HNF1b showed the highest gene expression after 6 days and slightly decreased thereafter (Supplementary Fig. S4D). Expression levels of HNF6, a gut tube marker and required to specify different pancreatic lineages [26], were increased after 6 days of differentiation only in the medium with SM and remained at this level until day 14 (Supplementary Fig. S4D). As well PDX1, a pan-pancreatic marker, showed increased expression only in the medium with SM. This expression was first detectable after 6 days of differentiation and increased until day 14. A marker for the dorsal pancreatic bud, MNX1 (HB9), exhibited an expression profile similar to that of PDX1 (Fig. 7B). PTF1a showed an inverse expression profile with a remarkable increase in randomly differentiated cells after 8 days of differentiation (Fig. 7B). Maturation of these pancreatic progenitor cells toward the endocrine lineage is characterized by NGN3 expression, which increased dramatically after 10 days in the medium with SM (Fig. 7C). The same pattern, with a 2-day delay, was observed for NKX2.2 and NKX6.1 (Fig. 7C). Together with NGN3 these genes are considered as a hallmark of endocrine progenitors and their maturation.
The expression pattern of the differentiated hESC line HUES4 (Supplementary Figs S5 and S6) showed nearly identical results compared with HUES8 cells. Expression levels of the embryonic transcription factors (Supplementary Fig. S5A) decreased rapidly. In contrast to this, the expression of PS (Supplementary Fig. S5B) as well as DE marker genes (Supplementary Fig. S5C) increased to their maximum after 4 days of differentiation. Only the high FOXA2 expression was passed in the medium with SM to the next differentiation stages. The gene expression of the early gut tube markers showed strong inductions after 6 days of differentiation and remained highly expressed in the medium with SM during further differentiation (Supplementary Fig. S5D). PDX1 and MNX1 (HB9) showed increased expression profiles only after cultivation with SM (Supplementary Fig. S6A). The expression of NGN3 increased distinctly after 10 days of differentiation, whereas NKX2.2 and NKX6.1 were strongly induced after 12 days of differentiation with SM (Supplementary Fig. S6B).
Protein expression of PDX1 and NGN3 was analyzed by immunofluorescence staining after 10 or 14 days, respectively. Only very rare PDX1-positive cells were detectable after 10 days in the medium without SM. In contrast, the medium with SM frequently yielded PDX1-expressing cells with different signal intensities (Fig. 7D). Quantification of the PDX1-positive cells after 10 days (Fig. 7E) revealed significantly increased numbers in the medium with SM (46.3%±1.7%) compared with the medium without SM (2.4%±0.4%). Only differentiation with small molecules produced significant numbers of NGN3-positive cells (10.7%±0.8%, Fig. 5F) compared with without SM (0.3%±0.2%), which is in accordance with the gene expression analysis. These results clearly demonstrate that the DE-committed cells from dispersed single cells were able to differentiate further into the gut tube stage and subsequently into the pancreatic lineage.
Discussion
The in vitro differentiation of ESCs into adult cell types is a potential approach to generate surrogate cells for replacement therapies of degenerative diseases. Somatic cells derived from organs of the endoderm germ layer, such as liver, lung, or pancreas, are of particular interest [1]. Endoderm-derived cells require as a first step the differentiation toward the DE as a key stage [8] for further patterning toward the desired organ lineage [27].
Most protocols for DE differentiation are based on hESCs grown as colonies. Generally, hESC colonies are suboptimal for differentiation attempts due to the poorly defined starting conditions, such as differences in colony sizes or colony numbers per cavity. Growth factors, cytokines, or small chemical molecules added to the medium as directive cues may not uniformly activate all cells in a given colony. Finally, cell-to-cell contacts may alter extrinsic cell signaling. In addition, ESCs grown on a feeder layer are hampered by cross-contamination with these mesenchymal cells. The contamination may influence the function and differentiation potential of hESCs. Therefore, a DE differentiation protocol based on dispersed single cells from feeder-free-cultivated pluripotent cells with a defined cell number as starting population is a step toward better reproducibility. In the present study we described a very efficient and robust protocol fulfilling these criteria of feeder-free-cultivated human pluripotent cells.
The DE differentiation has earlier been considered as mainly ActA/Nodal dependent [9,12]. However, the results shown here and by others [24,28 –30] demonstrate that ActA/Nodal signaling alone, independent from the strength, was not able to induce sufficient numbers of cells committed to the DE lineage. A combined activation of ActA/Nodal signaling and GSK3 inhibition by Chir for the first 24 h followed by ActA/Nodal signaling alone (CA-A protocol) was able to yield high numbers of cells truly committed to the DE from dispersed human pluripotent cells. HUES8 cells required lower Chir concentrations nevertheless ensuring an efficient DE induction compared with HUES4 and HES3 cells. This confirms results of Jiang et al., who observed that HES3 cells expressed lower WNT3 levels resulting in a lower differentiation efficiency when compared with HUES8 cells [28]. In line with earlier studies [24,28 –30] this provided proof that the Wnt/β-catenin pathway plays an important role during the early differentiation steps of the DE differentiation process. The different response to GSK3 inhibition demonstrates lineage propensities of pluripotent cells [31]. As in our recent publication [24], the required ActA concentration could be reduced upon replacement of Wnt3a by Chir. The differentiation efficiencies for the hESC lines HUES8 and HES3 in the study from Jiang et al. [28] were lower compared with the described CA-A protocol. Further, sufficient numbers of DE-committed cells could be differentiated with the CA-A protocol from HUES4 cells (>72%) and the hiPSC line hCBiPSC2 [20] (>76%), demonstrating the robustness of this protocol using dispersed single cells as starting population.
Inhibition of the PI3K was proposed to be required for the Nodal/TGF-β-mediated differentiation of hESCs into DE [18]. According to our findings the RP highly depends on low or inhibited PI3K signaling because high DE efficiencies could only be obtained in RPMI 1640 medium. An additional PI3K inhibition resulted also in advanced RPMI 1640 in higher efficiencies. The advanced RPMI 1640 contains a high insulin concentration, which activates the PI3K signaling pathway. An activation of the PI3K signaling pathway is essential for growth and viability of cells in culture [32] and its inhibition in mESCs as well as their differentiated progeny decreases proliferation by accumulation of cells in the G1-phase [33]. This explains the higher proliferation rates in advanced RPMI 1640 or generally in media without PI3K inhibition and the cytotoxicity observed in media with low or inhibited PI3K activity. The CA-A protocol is independent from PI3K inhibition, which is beneficial and superior to other reports [18,34]. An interesting aspect is that upon insulin stimulation GSK3β can be phosphorylated by AKT, a downstream kinase of the insulin receptor/PI3K pathway, resulting in its inactivation [35]. Therefore, an efficient GSK3 inhibition by Chir is probably supported by PI3K signaling.
The supernatant of the Matrigel coating for routine ESC cultivation could be used as coating matrix for differentiation experiments, which further reduces the costs and shows that this “reused” Matrigel is an adequate substrate for dispersed single cells. Other protocols applied an additional Matrigel layer on top of the cell monolayer to increase the robustness of the protocol by promoting the epithelial-to-mesenchymal transition during cardiomyocyte differentiation [36]. However, such a “Matrigel-sandwich” would complicate the procedure, since it is not necessary for the DE induction. Feeder cells, routinely used for long-term cultivation of ESCs, harbor the problem of a potential contaminating cell source during differentiation. Here, we demonstrated that feeder-free-cultivated hESCs are generally preferable even though the cells could be differentiated directly with the feeder cells, although with lower efficiencies and higher variability between individual experiments. The beneficial feeder-free cultivation in this protocol is advantageous for potential clinical applications because feeder-dependent cultures are inappropriate for a therapeutic use of hESCs.
The ability of the DE to differentiate into endoderm-derived organs was analyzed by differentiation toward the pancreatic lineage (adapted from Kunisada et al. [23]). The expression of the pluripotency-associated genes quickly decreased but SOX2 was re-expressed after 6 days, when RA signaling combined with TGF-β and BMP inhibition was applied. This, in combination with the expression profiles of HNF1b and HNF6, showed that the protocol with SM induced the foregut state because SOX2 is, next to its role during pluripotency, expressed mainly in the foregut of the developing embryo with functions for its later specification [37,38]. Interestingly, only the treatment with SM was able to induce a significant expression of the pan-pancreatic marker PDX1 that together with MNX1 (HB9) and HNF6 pattern the foregut/gut tube endoderm toward the pancreatic lineage. In particular the high MNX1 expression is interesting as this gene is a marker for the dorsal pancreatic bud [39,40]. High gene expression of the transcription factors NKX2.2 and NKX6.1 noted upon cultivation with SM points to a further maturation of these pan-pancreatic progenitors. NKX2.2 is normally detected in the early pancreatic endoderm; it induces beta cell differentiation from NGN3-positive cells and has essential functions in adult beta cells [41]. NKX6.1 plays a similar role during development [41] and has recently been shown to be a crucial biomarker for the in vivo maturation potential of ESC-derived pancreatic progenitors [42]. The proendocrine marker gene NGN3 is a target of HNF6 [25,26,43 –46] and marks the transition toward a potential further endocrine differentiation. Therefore, the detectable high expression of NGN3 upon SM addition showed that the pancreatic progenitor cells matured and differentiated toward endocrine progenitors. Thus, the DE-committed cells, differentiated with the protocol described here, were capable for pancreatic differentiation up to endocrine progenitor cells.
In summary, we present an efficient endoderm differentiation protocol, which is robust and reproducible using three different hESC lines and one hiPSC line. Further, the starting conditions are well defined and allow differentiation in a medium with high PI3K signaling, yielding high cell numbers after endoderm commitment. Finally the differentiated endoderm is physiologically relevant and these DE cells could be directed into multipotent PDX1-positive pan-pancreatic cells and NGN3-positive endocrine progenitors.
Footnotes
Acknowledgments
This work has been supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the framework of the Cluster of Excellence REBIRTH (From Regenerative Biology to Reconstructive Therapy). The skilful technical assistance of Rebecca Strauß is gratefully acknowledged. The authors thank Prof. F. Buettner, Dr. R. Zweigerdt, and Prof. U. Martin for providing the HES3 and hCBiPS2 cell lines.
Author Disclosure Statement
No conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.