
Abstract
The developmental fate of the multipotent neural crest (NC) is determined along with the neural axis in which NC cells are generated. Only the cranial NC can differentiate into mesectodermal derivatives such as osteoblasts, chondrocytes, and adipocytes in vivo. Here, we attempted to selectively differentiate mouse embryonic stem (ES) cells into cranial NC stem cells and propagate them to explore their developmental potential to differentiate into mesectodermal derivatives. Using aggregation cultures in feeder- and serum-free neural induction medium (NIM) without serum replacement and
Introduction
I
NC cells at a given axial level follow distinct migratory routes and differentiate into a predictable array of progeny at their final destination [4 –6]. Studies investigating the mechanisms of lineage progression and disease pathogenesis have shown that the majority of cranial NC cells contribute to the morphogenesis of the craniofacial region, including the peripheral nervous system and connective tissue of the cranial muscles, adipocytes, chondrocytes, osteoblasts, and odontoblasts [7,8]. A subpopulation of posterior cranial NC cells, called the cardiac NC cells, contributes to the cardiac outflow tract [9]. The NC cells in the trunk give rise to limited cell types, including peripheral nerve cells, melanocytes, and chromaffin cells.
Transplantation experiments in the amniotic vertebrate have suggested that the trunk NC could be scarcely differentiated into cranial NC derivatives [10], although avian trunk NC cells were differentiated into chondrocytes by long-term in vitro cultures [11,12]. Moreover, when mouse trunk NC cells were recombined with branchial arch 1 epithelium, they differentiated into dentin and bone [13]. Trunk NC cells were differentiated into mesenchymal derivatives in cultures with specific factors [14 –16]. These data suggest that differences in developmental potency between the cranial and trunk NC cells depend on the environmental signals.
To clarify when and how the differences of developmental potency are established between two segments of the NC, analyzing their undifferentiated NC precursor or stem cells, respectively, is necessary. Several studies have reported that mammalian multipotent migratory trunk NC cells were isolated from the neural tube in the rat embryo [17]. Recently, a cranial NC cell line was isolated from the mouse embryo [18]. In mammals, some NC cells having characteristics that resemble those in embryonic NC cells are retained after a period of organogenesis and in the adult tissues [19]. Therefore, a part of the stem cells isolated from dental pulp and the periodontal ligament may be derived from cranial NC stem cells [20,21]. However, whether these stem cells are necessarily equivalent to embryonic NC precursor or stem cells has not been confirmed.
Therefore, the derivation of precursor/stem cells of the cranial and trunk NC from embryonic stem (ES) cells might be effective in elucidating the determination of NC fate. ES cells can be used to analyze early events in mammalian development because of their pluripotency and ability to recapitulate the developmental process in vitro. In addition, ES cells are capable of proliferating indefinitely in vitro, and that ability can facilitate biochemical analyses. Moreover, ES cells are useful in a variety of gene analyses, because they can easily undergo gene transfection.
To differentiate NC cells from ES cells, one should first differentiate the neuroepithelium. Since embryoid bodies (EBs) formed in culture medium with serum tend to differentiate into three germ layers consisting of non-neural cells, fluorescence-activated cell sorter (FACS) is necessary to obtain a small number of NC cells in EBs [22]. A single-step method for the generation of NC-like stem cells from human ES and induced pluripotent stem (iPS) cells in chemically defined media was reported [23]. This approach is efficient for NC differentiation via neuroepithelium, but region specificity of the NC cells was not elucidated. In addition, several methodologies for deriving neuroepithelium from ES cells with a high efficiency have been developed. These protocols are based on stromal-derived inducing activity and serum-free suspension cultures, which can selectively differentiate into a nervous system that alters the rostral/caudal or dorsoventral axes by the addition of a variety of inducing factors [24 –27]. In particular, neurosphere culture [28] is suited for analyzing the process of NC differentiation from the neuroepithelium of a specific region because of the serum-free culture withdrawal of stromal feeder cells.
In this study, we showed that NC cells were differentiated from mouse ES cells via neurospheres that had rostral characteristics under feeder- and serum-free conditions, without serum replacement and
Materials and Methods
Cell culture
Mouse ES (mES) cells (E14; ATCC) were maintained without feeders in ES cell medium consisting of Grasgow's modified Eagle's medium (GMEM) (Sigma-Aldrich) supplemented with 15% fetal bovine serum (FBS), 2 mM
For sphere formation to neuroepithelial induction, mES cells were dissociated using 0.25% trypsin-EDTA to single cells and seeded in 100-μL of serum-free neural induction medium [NIM; 1:1 ratio of Dulbecco's MEM (DMEM)-F12 and neurobasal medium supplemented with 0.5×N2, 0.5×B27, 20 ng/mL insulin, 20 ng/mL basic fibroblast growth factor (bFGF), 20 ng/mL epidermal growth factor (EGF), and 100 U/mL penicilin and 100 μg/mL streptomycin; all from Invitrogen] in low cell-adhesion wells of 96-well plates (Sumilon Spheroid plates; Sumitomo).
For NC differentiation, NIM neurospheres were transferred to a 24-well plate that was coated with 50 μg/mL fibronectin in serum-free medium consisting of αMEM supplemented with 15% Serum Replacement 3 (Sigma), 2 mM
For NC cell expansion, NC cells were cultured on fibronectin-coated plates in the expansion medium consisting of DMEM/F12 supplemented with Serum Replacement 3, 10 μg/mL bFGF, 50 μg/mL EGF, 1 mg/mL Fibronectin, and 100 ng/mL Heparin.
For differentiation to NC derivatives, NC cells were cultured on fibronectin-coated plates in the respective differentiation media.
For osteogenic differentiation, NC cells were cultured in osteogenic medium consisting of αMEM with 15% FBS, 2 mM
For adipocyte differentiation, NC cells were cultured in adipogenic medium consisting of αMEM with 10% FBS, 1 μM dexamethasone, 0.5 mM isobutylmethylxanthine, and 10 μg/mL insulin for 2 weeks. For chondrocyte differentiation, NC cells were densely cultured in chondrogenic medium consisting of 200 μM ascorbic acid, 10 ng/mL TGFβ3 in αMEM containing 10% FBS for 4 weeks.
For myocyte differentiation, NC cells were cultured in myogenic medium consisting of a 1:1 ratio of DMEM/F12 and neurobasal medium supplemented with 1 mM
For neural differentiation, NC cells were cultured in neural differentiation medium No. 1 consisting of 10% chicken embryo extract, 20 ng/mL bFGF, 1× N2, 1× B27 (-vitamine A), 50 μM 2-mercaptomethanol, 35 ng/mL retinoic acid, 25 U/mL penicillin, and 25 μg/mL streptomycin in DMEM-low glucose. After 4 days of culture, the medium was changed to the neural differentiation medium No. 2, consisting of 1% chicken embryo extract and 10 ng/mL bFGF in neural differentiation medium No. 1.
Immunostaining
Cells were fixed with 3.5% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, permeabilized using 0.2% TritonX-100 for 5 min, and blocked with Image-iT™ FX Signal Enhancer (Invitrogen) for 30 min. Spheres were fixed with 3.5% paraformaldehyde and embedded in paraffin. The 6-μm sections were deparaffinized in xylene, and rehydrated in alcohol and deionized water. Primary antibodies (Supplementary Table S1; Supplementary Data are available online at
Histochemistry
To assess the osteogenic, chondrogenic, and adipogenic differentiation, we stained the cells with Alizarin Red S, Alcian Blue, and Oil Red O, respectively. For Alizarin Red S staining, cells were fixed in 3.5% paraformaldehyde for 10 min at room temperature, washed thrice with PBS and H2O, and incubated for 15 min with a 2% Alizarin Red S (Sigma-Aldrich). For Alcian Blue staining, specimens were fixed in 3.5% paraformaldehyde for 10 min at room temperature, washed thrice with PBS and H2O, incubated with an Alcian Blue stain solution (pH 2.5; Nacalai Tesque) for 24 h, and, finally, washed with dH2O several times. For Oil Red O staining, cells were fixed with 3.5% paraformaldehyde for 10 min at room temperature, equilibrated with 60% isopropyl alcohol (IPA), incubated with 0.18% Oil Red O solution in 60% IPA, and washed in 60% IPA to eliminate excess staining.
Reverse transcription-polymerase chain reaction assay
Total RNA was extracted from the cultured cells using TRIzoL (Invitrogen), and 500 ng of total RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen) following the manufacturer's instructions. PCR analyses were performed using a standard protocol. Primer sequences and annealing temperatures are provided (Supplementary Table S2). The amplification fragments were electrophoresed on a 1.5% agarose gel and stained with ethidium bromide. The expression level of each sample was normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA expression.
Results
Neuroepithelium differentiation from undifferentiated mouse ES cells
The NC arises on the dorsal neural tube during the early stage of vertebrate embryonic development [29]. Thus, we first differentiated mouse ES cells into neuroepithelial cells before carrying out NC differentiation.
ES cell aggregates in serum- and feeder-free cultures have been found to be susceptible to differentiation into neuroepithelium; however, large-sized aggregates tend to develop cavities, resulting in increased cell death and generation of non-neural lineages [30]. We examined the appropriate aggregate size for efficient neural differentiation; however, controlling the sphere size in suspension culture on low cell-adhesion culture dishes coated with polyhydroxyl-ethyl-methacrylate (Poly-Hema) or EZ-BindShut dishes (Asahi Glass Co. Ltd.) was difficult. We attempted to control the size of cell aggregates depending on the number of seeding cells using low cell-adhesion U-shape-bottomed 96-well plates following the method of Eiraku et al. [31]; 1×103, 2×103, and 5×103 of dissociated ES cells were seeded and cultured in NIM on the plates. Aggregate size could be reproductively controlled by the number of cells used (Fig. 1A).
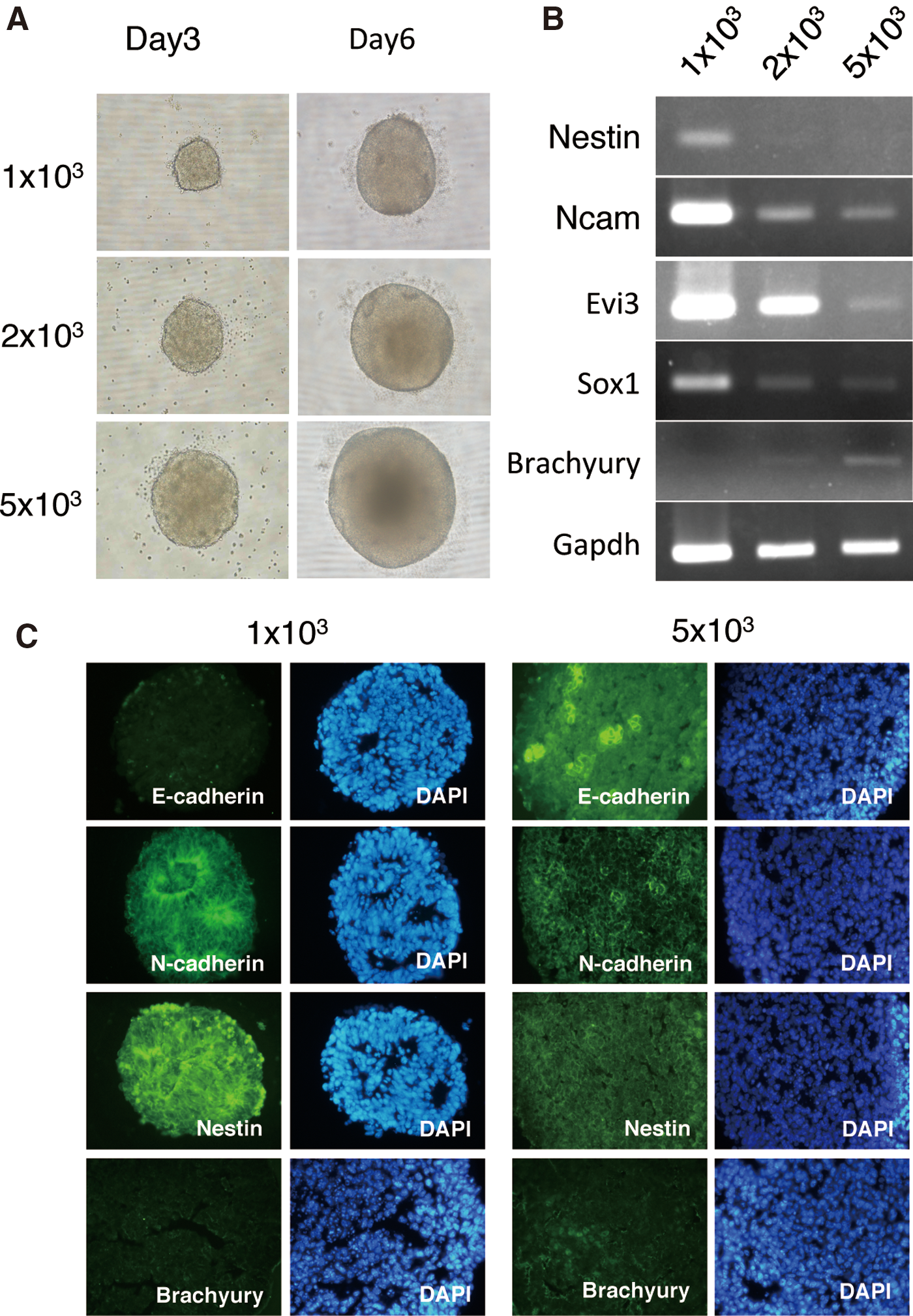
Differentiation of aggregates from various numbers of cells (1×103, 2×103, and 5×103) of mouse embryonic stem (ES) cells.
These aggregates were collected, and the RNA was extracted and examined for neuroepithelial differentiation using reverse transcription-polymerase chain reaction (RT-PCR) (Fig. 1B). On day 6 of culture, expression levels of Nestin, neural cell adhesion molecule (Ncam), Evi3/Zfp521, and Sox1 in aggregates generated from 1×103 of ES cells were higher than those from 2×103 and 5×103 of ES cells. In addition, the expression of the mesoderm marker Brachyury was detected in larger aggregates from 2×103 and 5×103 cells, but not in smaller aggregates from 1×103 cells.
The localization of marker proteins in 1×103 and 5×103 aggregates was examined by immunohistochemical staining (Fig. 1C). The neuroepithelial marker Nestin was detected in most cells in both the 1×103 and 5×103 aggregates. Colonization of the undifferentiated cell marker E-cadherin was detected in 5×103 cell aggregates but not in 1×103 aggregates. Rosette-like structures were observed in culture cells in which the apical side was densely stained by N-cadherin in 1×103 aggregates. However, such rosette-like structures were not observed in 5×103 aggregates. A small number of cells in which Brachyury was detected was observed in 5×103 aggregates but not in 1×103 aggregates.
These results (summarized in Table 1) suggest that ES cells were induced to differentiate into neuroepithelium by cell aggregation using low-cell-adhesion U-shaped 96-well plates. Furthermore, it is critical to make small aggregates of 1,000 cells for efficient neuralization, because larger aggregates tended to be induced to multiple cell types and undifferentiated cells remained.
−, no expression;±, weak expression; +, moderate expression; ++, strong expression; +++, very strong expression, ND; not done; ++*, colonize positive cells; ++**, strong expression was localized at the apical side of rosette-like structure.
During the growth of 1×103 aggregates in NIM, we analyzed the expression of undifferentiated ES cells, neuroepithelium, mesoderm, and endoderm markers (NIM in Fig. 2A). The undifferentiated ES cell markers Nanog and Oct3/4 were expressed in day 3 aggregates, but not in day 6 aggregates. The neuroectodermal markers Evi3/Zfp521, Nestin, Sox1, and Ncam were detected at low levels in day 3 aggregates, which were upregulated in day 6 aggregates. Furthermore, the dorsal marker Pax3 was clearly detected in day 6 aggregates, whereas the ventral marker Nkx2.2 was not. These data indicate that 1×103 ES cell aggregates cultured in NIM converted neurospheres containing dominant dorsal neuroepithelium. In this article, the aggregates cultured in NIM are referred to as NIM-cultured neurospheres.
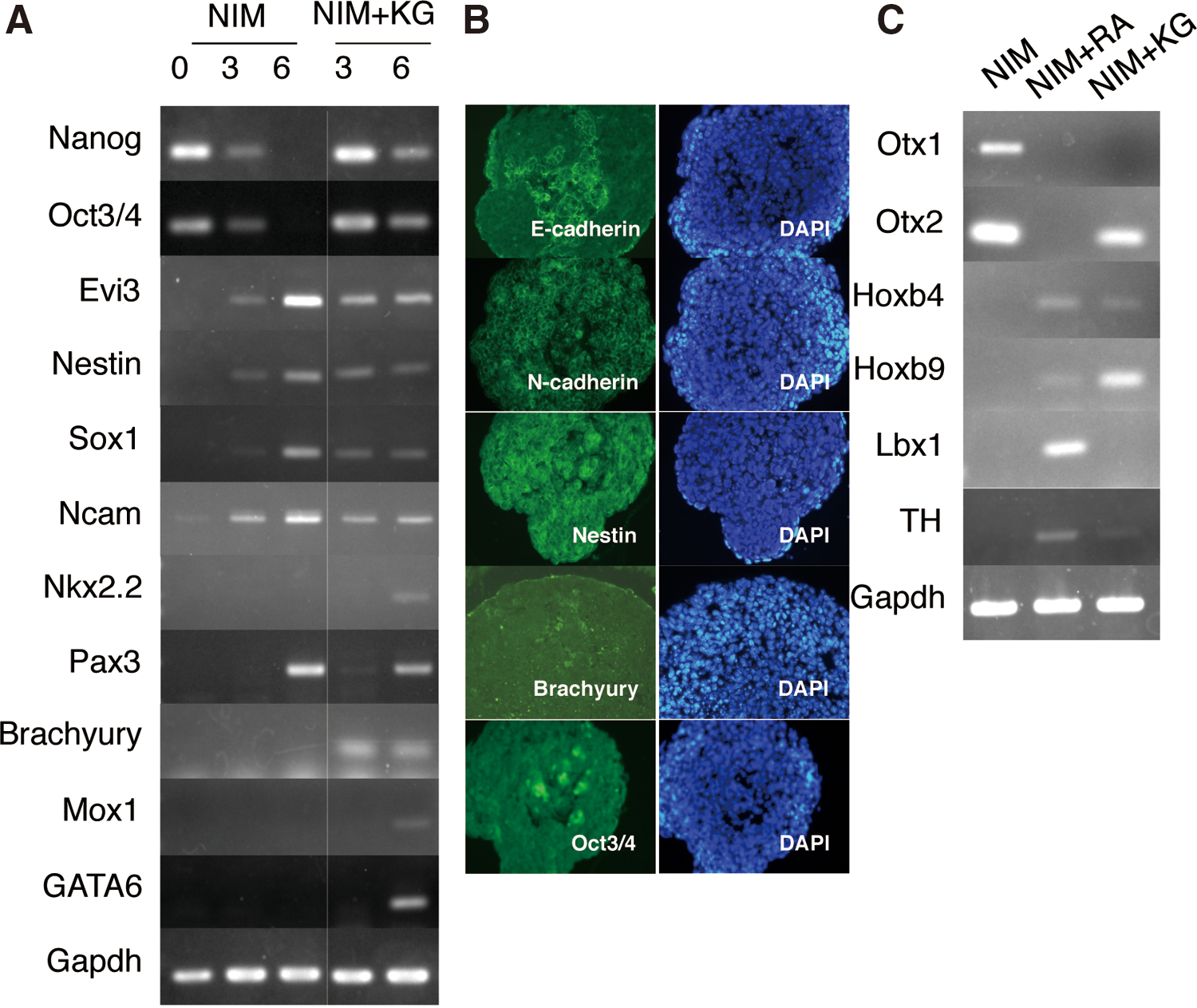
Neural differentiation of spheres cultured in NIM.
Several studies have demonstrated differentiation of ES cells to neuroepithelium using feeder- and serum-free suspension culture methods, in which the media were supplemented with
The expression levels of Evi3/zfp521, Nestin, Sox1, and Ncam were detected in day 3 and 6 culture with NIM+KG and in NIM-cultured neurospheres. However, the levels of neural markers were not upregulated in day 6 cultures with NIM+KG, and NIM+KG-cultured spheres expressed Nkx2.2 and Pax3, indicating that the spheres in both culture media differentiated into neuroepithelium, but NIM+KG-cultured spheres were not restricted to the dorsal neuroepithelium. In addition, the undifferentiated markers Nanog and Oct3/4, mesoderm markers Brachyury and Mox1, and endoderm marker GATA6 were detected in day 6 NIM+KG-cultured spheres. These results suggest that aggregates under the NIM supplemented with knockout serum replacement and
Therefore, immunostaining was performed to investigate the localization of the differentiation marker proteins (Fig. 2B). The undifferentiated markers Oct3/4 and E-cadherin were detected in small clusters of cells. Most cells expressed N-cadherin and Nestin. Brachyury was detected in a small number of cells in NIM+KG-cultured spheres.
We next examined the rostral/caudal identity of the neuroepithelium in NIM-cultured neurospheres using RT-PCR. The rostral markers Otx1 and Otx2 were detected, but the caudal markers Hoxb4, Hoxb9, and Lbx1 and the midbrain and hindbrain marker tyrosine hydroxylase (TH) were not detected (Fig. 2C), suggesting that the rostral neuroepithelium was selectively differentiated in NIM-cultured neurospheres.
The NIM+KG spheres were positive not only for Otx2 but also for Hoxb4 and Hoxb9 (Fig. 2C), which suggested that they differentiated into the neuroepithelium without the deflection of the rostral/caudal axis. However, the NIM+KG-cultured spheres did not express Otx1 and Lbx1, which were detected in caudalized spheres that were treated by the caudalizing factor retinoic acid (RA) [25] (Fig. 2C), suggesting that the rostralization and caudalization in the NIM+KG-cultured spheres was insufficient.
These data indicate that the cranial neural cell spheres can be differentiated in serum-free defined medium without serum replacement and
NC induction from NIM neurospheres
The mRNA expression of NC markers in the NIM neurospheres was analyzed by RT-PCR. AP2α, Sox9, p75, and Snail were expressed at low levels in day 6 NIM-cultured neurospheres. High-level expression of Slug and Twist was detected in day 6 NIM-cultured neurospheres (Fig. 3A). These results suggest that day 6 NIM-cultured neurospheres from ES cells largely contained the neuroepithelium with a small number of NC-like cells.

Neural crest (NC) cell differentiation of NIM-cultured neurospheres. NIM-cultured neurospheres were cultured for 6 days in NIM, transferred to fibronectin-coated dishes, and cultured for 4–5 days in medium containing bone morphogenic protein 4 (BMP4).
Several growth factors, including BMPs, have shown their potential roles in NC development [32]. To generate larger quantities of NC cells from NIM-cultured neurospheres, we transferred day 6 NIM-cultured neurospheres to fibronectin-coated plates and cultured them in the BMP4 containing serum-free medium for 4–5 days. Neurospheres were attached in a single day; the tegmental cells emigrated, and the spheres remained as clusters in central parts. To confirm NC differentiation of plated spheres, we examined the expression of NC markers using RT-PCR and immunostaining. RT-PCR analysis showed the upregulation of NC markers in the NIM-cultured neurospheres after the 4-day culture with BMP4 (day 10 neurospheres; Fig. 3A). The NC marker AP2α-positive cells increased in the cultured cells after BMP4 treatment based on immunostaining (Fig. 3B, C). These data indicate that the NC cells from the NIM-cultured neurospheres consisting of cranial neuroepithelium were induced in day 10 neurospheres.
The craniofacial skeleton originates mainly from the cranial NC. The possibility exists that NC cells from the cranial neurospheres are cranial NC cells which tend to be osteoblasts. To examine this possibility, we tested whether day 10 neurospheres could differentiate into osteoblasts.
After 4 weeks' culture of the day 10 neurospheres in osteogenic medium, only sphere clusters were positive for Alizarin Red S staining, and emigrating cells were not (Fig. 4A). Moreover, induced NC cells from the NIM-cultured neurospheres in medium without BMP4 also differentiated into Alizarin Red S-positive clusters (Fig. 4B). During osteogenesis, molecular markers, collagen I and Runx II were upregulated (Fig. 4C).

Osteogenic differentiation from NIM-cultured neurospheres. NIM neurospheres cultured in NIM supplemented with
Next, we mechanically separated the adherent spheres from the migratory cells after BMP4 treatment. The adherent spheres and migratory cells were dissociated by trypsinization, and each group of cells was analyzed. NC markers Sox9 and AP2α were expressed in both cell populations. Cells from adherent spheres expressed Pax7 and FoxD3. Migratory cells expressed Pax7, but not FoxD3 (Supplementary Fig. S1). Furthermore, both groups of cells were cultured in osteogenic medium under high-density monolayer culture conditions. The cells derived from spheres formed nodules, which were stained with Alizarin Red S (Fig. 4D), whereas migratory cells did not (Fig. 4E). In these experiments, almost all sphere clusters and nodule-like cell aggregates were differentiated into Alizarin Red S-positive clusters, but spreading monolayer cells were not. These results suggested that the cells within day 10 neurospheres differentiated into NC cells and cranial NC cells mainly existed in adherent spheres.
Isolation and expansion of NC stem cells from ES-derived neurospheres
We attempted to isolate NC stem cells in day 10 neurospheres, and subsequently propagate and differentiate them into various NC derivatives. Day 10 neurospheres were trypsinized, and the dissociated cells were expanded in expansion medium. We next determined whether these cells were able to give rise to a variety of NC derivatives. We confirmed that the expanded NC cells differentiated into adipocytes, smooth muscle cells, chondrocytes, and osteoblasts (Supplementary Fig. S2). These data indicate that day 10 neurospheres contain NC stem cells, which possess multipotentialities.
The isolation of stem cells from various tissues and tumors has been achieved using flow cytometry [33 –35] or sphere-formation assays based on the properties of the stem cells [36]. In these experiments, we isolated NC stem cells from expanded NC cells using their sphere-formation ability under the cultivation of defined serum-free medium with growth factors.
Spheres from single cells of expanded NC cells were transferred to low-adhesion U-shaped-bottomed 96-well plates and cultured for 2 weeks. Dissociated cells from 14 spheres were cultured on fibronectin-coated 96-well plate; nine clones were expanded successfully, and the ability to differentiate into osteoblasts was examined. Two of the nine expanded clones (clones #11 and #14) that differentiated into Alizarin Red S-positive osteoblasts were analyzed for NC stem cell characteristics. The expression of NC markers AP2α, Sox9, p75, Snail, Slug, and Twist and those of stem cell markers Nanog, Oct3/4, and Sox2 were detected in clone #11 (Fig. 5A), showing that clone #11 possessed characteristics of NC stem cells. Furthermore, clone #11 could be clonally amplified and maintained for >30 passages, and it was differentiated into peripherin-positive peripheral neurons, Pparγ-, and Oil Red O-positive adipocytes, smooth muscle actin-positive myocytes, Alcian Blue-positive chondrocytes, and Alizarin Red S-positive osteoblasts (Fig. 5B–G). These data indicate that we successfully established NC stem cell lines from ES cell differentiation.
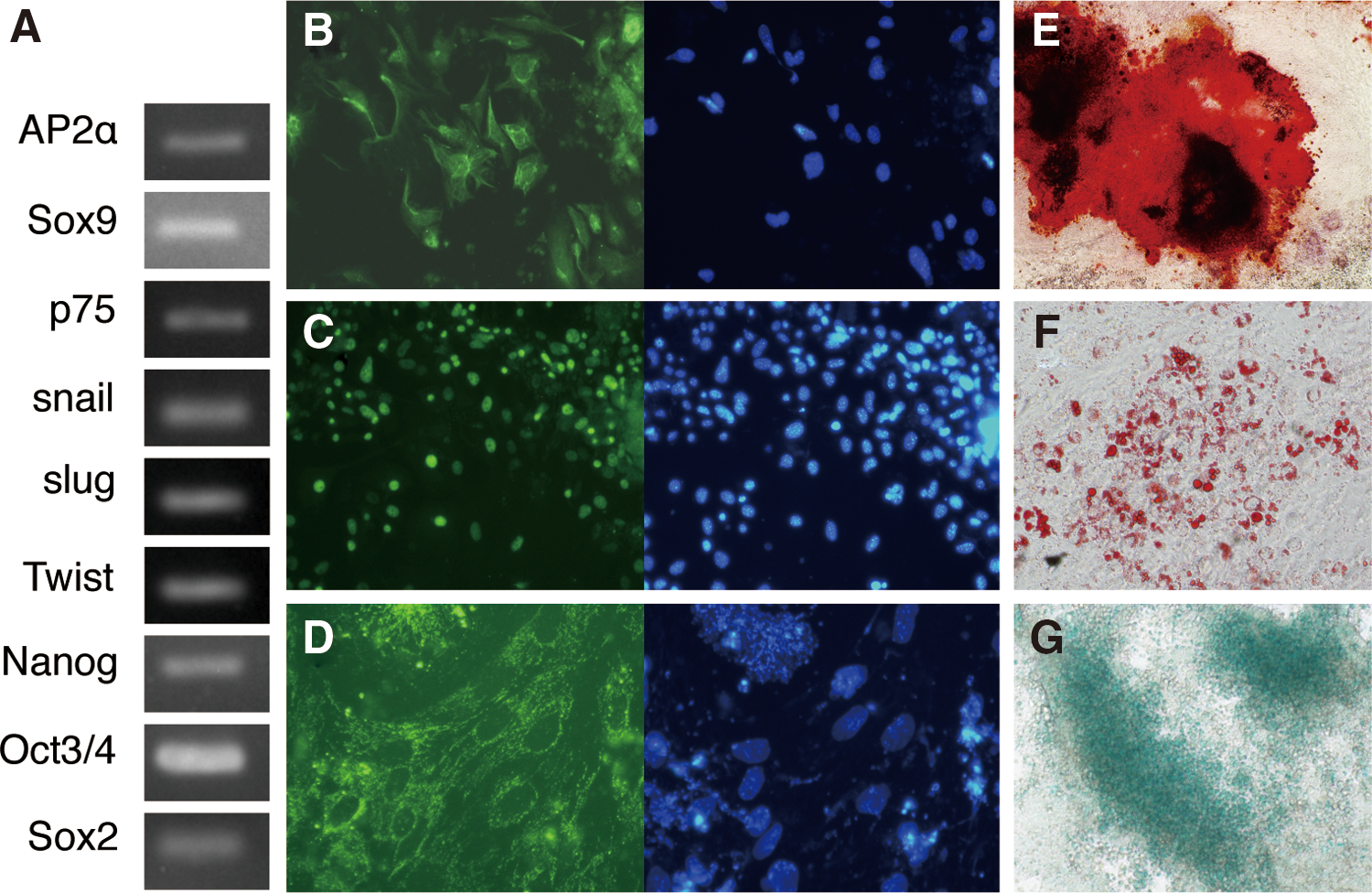
Characterization of neural crest (NC) stem cells in CKB-EK clone #11.
Discussion
In this study, we demonstrated that cranial NC cells were efficiently differentiated from mouse ES cells under culture conditions, in which ES cell aggregates were preferentially induced to cranial neuroepithelium in serum-free medium.
In this procedure, the aggregate size was reproducibly dependent on the number of cells seeded into each well of the low-adhesive U-shaped 96-well plates. Spheres formed from 1×103 ES cells selectively promoted neuralization, in which Nestin, Ncam, Evi3, and Sox1 were expressed. In larger-size aggregates, neuralization of ES cells was suppressed and mesoderm differentiation was promoted. These results are consistent with a previous study [30] reporting that the formation of small aggregates consisting of a small number of cells is associated with the efficient neuralization of ES cells. In this article, trypsin-dissociated undifferentiated ES cells were reaggregated in each well of the U-shaped low-cell-adhesion 96-well plates; a previous report selectively used aggregates composed of 50–100 cells in floating culture. Although these reports differ markedly in the initial seeding number of cells, ES cell aggregates consisting of less than 1×103 cells may be prone to differentiate into the neuroepithelium. Another study reported that the majority of neurospheres composed of Bf1+ telencephalic progenitors were formed using 3×103 cells under the same culture system using low-cell-adhesion 96-well plates [31]. Based on our results, these neurospheres might tend to differentiate into non-neural cells, because their neurospheres are composed of a comparatively large cell count and cultured in medium with serum replacement and
In this study, the ES cell-derived NIM neurospheres expressed the neuroepithelial markers Nestin, Sox1, and Ncam and the rostral markers Otx1 and Otx2, but not the caudal markers Hoxb4, Hoxb9, and Lbx; this profile suggests that the cranial neuroepithelium consisted of NIM-cultured neurospheres. In addition, the dorsal neuroepithelial marker Pax3 [37] was detected in NIM-cultured neurospheres, but the ventral neuroepithelial marker Nkx2.2 [38] was not. Taken together, these findings indicated that NIM-cultured neurospheres differentiated into the dorsal side of the cranial neuroepithelium. Developmentally, the cranial NC arises from the dorsal side of the cranial neuroepithelium and migrates extensively throughout the head; then, they differentiate into mesectoderm derivatives [39], including osteoblasts and chondrocytes, and contribute to the morphogenesis of the maxillofacial region, which is not derived from the trunk NC in vivo.
In this study, NC cells derived from NIM-cultured neurospheres consisted of the dorsal side of the cranial neuroepithelium and differentiated into osteoblasts. These results indicate that the cranial NC cells were selectively obtained. As shown in Fig. 2, RA treatment suppressed the rostral markers Otx1 and Otx2, whereas it induced caudal markers Hoxb4, Hoxb9, Lbx1, and TH in neurospheres. In addition, NC cells were generated from RA-treated caudalized neurospheres. NC markers expressed in NC cells from RA-treated caudalized spheres were distinct from those in NC cells from NIM-cultured neurospheres, indicating that region-specific NC cells may be obtained under appropriate culture conditions.
NIM-cultured neurospheres expressed low levels of NC markers Sox9 and p75. Histological and immunohistochemical analyses of the neurospheres showed that the neural tube-like structures with apical/basal polarity formed in the spheres, in which most cells were Nestin- and N-cadherin positive (Fig. 1C). In addition, the neurospheres showed high expression of Slug and Twist (Fig. 3A), which contribute to the epithelial–mesenchymal transition. Thus, a part of the NC cells may have arisen from already epithelialized neural cells. Moreover, the NC markers AP2α, Sox9, and p75 were markedly upregulated after 4 days' adhesion culture of NIM-cultured neurospheres on plates, showing that NC cells were differentiated. The neural tube-like structures in the NIM-cultured neurospheres are considered disorganized in adhesion cultures, resulting in the promotion of the neuroepithelium to the epithelial–mesenchymal transition; thus, NC cells are induced from neural cells.
Some emigrating cells from neurospheres that attached on the fibronectin-coated plates differentiated into Sox10-positive emigrating NC cells [28]. Replated sphere clusters without migrating cells gave rise to Sox10-positive emigrating NC cells again [28]. Here, we showed that osteogenesis which occurred within the sphere cluster of the NIM-cultured neurospheres formed nodules and that osteogenesis took place under osteogenic-dense culture conditions; however, emigrating cells from the NIM-cultured neurospheres expressed NC markers AP2α and Sox9, but did not differentiate into osteoblasts. These results indicate that the premigratory NC stem cells present in the clusters derived from the NIM-cultured neurospheres tended to differentiate into osteoblasts, whereas migrating NC cells from the NIM-cultured neurospheres tended to differentiate into various NC derivatives other than osteoblasts.
In this study, we attempted to propagate NC stem cells in clusters from the NIM-cultured neurospheres, which likely consist of premigratory NC stem cells. Alhough previous studies reported propagation of NC stem cells derived from ES cells via the neuroepithelium [28,40,41], most studies isolated NC stem cells from emigrating cells using FACS for the putative stem cell markers p75 and HNK1. In contrast, we succeeded in propagating cranial NC stem cells from premigratory NC cells in cranial neuroepithelium-rich clusters by taking advantage of their sphere-formation capability to assess the stem cell potential in combination with the osteogenic potential for assessment of NC differentiation.
In this study, we developed a system for reproducible differentiation and propagation of cranial NC stem cells. Since cranial NC stem cells tend to differentiate into mesenchymal derivatives, this system could be considered a useful tool for elucidating the pathogenesis of NC disorders and developing ways to treat diseases with poorly characterized differentiation processes in osteogenesis and adipogenesis.
Footnotes
Acknowlegment
Funding for this study was from the Osaka University and Osaka Dental University.
Author Disclosure Statement
The authors declare no potential conflicts of interest to the authorship and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.