
Abstract
Patient-specific induced pluripotent stem cells (iPSCs) are considered a versatile resource in the field of biomedicine. As iPSCs are generated on an individual basis, iPSCs may be the optimal cellular material to use for disease modeling, drug discovery, and the development of patient-specific cellular therapies. Recently, to gain an in-depth understanding of human pathologies, patient-specific iPSCs have been used to model human diseases with some iPSC-derived cells recapitulating pathological phenotypes in vitro. However, complex multigenic diseases generally have not resulted in concise conclusions regarding the underlying mechanisms of disease, in large part due to genetic variations between disease-state and control iPSCs. To circumvent this, the use of genomic editing tools to generate perfect isogenic controls is gaining momentum. To date, DNA binding domain-based zinc finger nucleases and transcription activator-like effector nucleases have been utilized to create genetically defined conditions in patient-specific iPSCs, with some examples leading to the successful identification of novel mechanisms of disease. As the feasibility and utility of genomic editing tools in iPSCs improve, along with the introduction of the clustered regularly interspaced short palindromic repeat system, understanding the features and limitations of genomic editing tools and their applications to iPSC technology is critical to expending the field of human disease modeling.
Introduction
H
While iPSCs provide promise for cell replacement therapies, they also represent a powerful tool for human disease modeling. As iPSCs are generated from patient cells, they can be used to generate specific cell types affected during disease. This would provide an unlimited source of cells for disease modeling and drug screening [14,15]. As iPSC generation has become more efficient, many patient-specific iPSCs have been derived to model disease [16]. Generally, monogenic diseases with clear causative mutations affecting well-characterized cell types have successfully recapitulated pathological phenotypes using iPSC technology [16,17]. For instance, long QT iPSCs with missense mutations in the KCNH1 and KCNH2 genes generate cardiomyocytes with increased depolarization and reduced potassium current, and spinal muscular atrophy iPSCs with SMN1 mutations generate fewer numbers of motor neurons with degenerated and diffuse synapses [18 –20].
On the other hand, other studies using disease-specific iPSCs have not been successful at modeling diseases [17]. There are many critical reasons as to why this may be the case. First, clonal variations caused by several factors during the reprogramming process, iPSC passage number, and culture conditions can affect the epigenetic status of individual iPSC clones [21 –25]. Second, in modeling diseases with sporadic or late onset, such as Alzheimer's and Parkinson's diseases (PD), in vitro assays often show insignificant differences between disease and control cells, suggesting that specific genetic variations between individuals may work as genetic modifiers that influence susceptibility to these diseases [26 –28]. It therefore becomes imperative that excess genetic variation between iPSC clones and controls should be removed to ensure more precise comparative and molecular analysis when modeling diseases [17]. To this end, generating isogenic sibling cell lines from patient iPSCs by altering only a few nucleotides is undoubtedly the most accurate way to establish a genetically defined condition.
Conventionally, homologous recombination (HR) has been a robust and frequently utilized method to modify genomic loci, most notably in the generation of diverse knockout mice. Despite requiring intensive efforts, HR has been used in hESC as well. In 2003, Zwaka and Thomson successfully disrupted the hypoxanthine phosphoribosyltransferase (HPRT) gene and also generated OCT4-GFP reporter hESC, which were the first knockout and knock-in hESC lines, respectively [29]. The conventional gene targeting methods used to generate the Oct4-GFP hESCs established the technical framework for the generation of many other transgenic hESC lines, and the cells themselves have allowed for monitoring cell fate specification in real time during differentiation [30 –33].
However, it is a prevalent idea that successful gene targeting with HR is particularly difficult in human pluripotent stem cells (hPSC). hPSCs tend to prefer to utilize nonhomologous end joining (NHEJ) to repair double-strand breaks (DSBs), as opposed to mouse ESC, which have a high propensity for HR. In addition, hPSCs do not grow well as single cells, and instead form colonies in culture making clonal selection challenging. Furthermore, they readily undergo anoikis, causing a dramatic drop in the efficacy of gene targeting when using electroporation or transfection reagents [34 –36]. To facilitate low frequency of HR in hPSC, bacterial artificial chromosomes (BACs) or adeno-associated viruses gene delivery systems have been applied [37 –39]. However, the most prevalent DNA delivery system in hPSCs remains intergrase-defective lentiviral vector (IDLV) [40 –42]. This system, developed from the human immunodeficiency virus type I (HIV-1), has been utilized in various cell types in vitro and in vivo, and delivers genetic information in the form of episomal DNA that confers transgene expression without affecting self-renewal or pluripotency in hPSCs.
Recently, besides gene delivery systems, genetic editing tools is considered a method to increase the efficiency of modifying genomic loci, and a recent wave of studies have applied the genetic editing tools to iPSC technology. This has led to the generation of isogenic iPSC disease and control lines that have been critical to advancing the utility of iPSCs to the human disease modeling field.
Rationale of Genomic Editing Tools: Utilizing Endogenous DNA Repair Systems
Genomic editing refers to techniques that are able to change one or more nucleotides in a given gene using engineered nucleases. These nucleases induce DNA DSBs in a sequence specific manner, but rely on the endogenous cellular DNA repair machinery to repair the breaks following DNA cleavage.
There are two endogenous DNA repair systems that work in conjunction with genomic editing tools: NHEJ and HR repair (Fig. 1) [43]. NHEJ joins two broken ends by synthesizing compatible overhangs that can then be ligated together [44]. When a DSB occurs, the two broken ends need to maintain proximity to induce subsequent repair processes. Ku proteins detect and bind to the broken region to recruit the DNA-dependent protein kinase (DNA-PK, ARTEMIS) that trims 5′ and 3′ overhangs to make two compatible ends to ligate together. Polymerase-μ then fills in any gaps remaining between the joined compatible ends, and finally, DNA ligase IV complex (XRCC) joins the break by ligating the two strands together. It is important to note that NHEJ is an imperfect process. Between ARTEMIS-DNA-PK recruitment and the final ligation, nucleotides may be added or deleted resulting in imperfect DNA repair (Fig. 1). When combined with genomic editing tools, NHEJ-mediated repair can induce small insertions or deletions in the target gene, which can consequently induce gene knockouts [45,46]. Additionally, using NHEJ-mediated repair large transgenes of up to 14 kb in size have been introduced into the genome [47].

Overview of nuclease-mediated double-strand breaks (DSB) repair system. When DSBs are made by a genomic editing tool, two endogenous DNA repair systems, either nonhomologous end joining (NHEJ) or homologous recombination (HR) repair are activated. NHEJ joins two ends by DNA ligase IV (XRCC) following the addition of random nucleotides by Polymerase-μ. On the other hand, HR requires a homologous template for repair. Recombinases are recruited to break points and guide the end of single strand into a homologous template, which can be a donor vector or strands of sister chromatid. The damaged region is then repaired by copying the sequence from the undamaged complementary template. Color images available online at
Contrary to NHEJ, HR repair is precise and accurate, utilizing a homologous DNA template to guide the repair process. HR is mediated by the highly conserved enzymes RAD51 and DMC1 that catalyze the pairing and switching of homologous DNA sequences [48]. When DSB occurs, double-stranded ends of breaks are resected to become single stranded with 3′ hydroxyl groups. RAD52 recombinases are then recruited and bind to the single-stranded DNA, enabling the broken DNA to invade into a homologous template. The invading sequence subsequently anneals to the complementary template forming a Holliday junction, a mobile junction between four strands of DNA. The damaged region is then extended, copying the sequence from the undamaged complementary template, thus repairing the damaged DNA strand, and the process completes with the resolution of the Holliday junction by specialized endonucleases (Fig. 1) [48]. To use HR repair with genomic editing tools, cotransfection of a donor vector containing homology arms is required. Usually, a linear donor vector also contains a selection marker such as GFP or an antibiotic resistance cassette. Additionally, single-stranded DNA oligonucleotides without selection markers are used to the target site of interest [49 –51].
Custom DNA Binding Domains of Zinc Finger Nucleases and Transcription Activator-Like Effector Nucleases
The specificity of genomic editing depends on the DNA binding domains of the tools of choice. Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are two common options for genomic editing studies. They share two fundamental characteristics in that they contain customized DNA binding domains and a FokI nuclease, which is able to cleave nucleotides in a nonspecific manner [52]. ZFNs are the synthetic fusion of multiple zinc finger domains, each recognizing specific target DNA sequences (Fig. 2C) [53]. The zinc finger domain is the most abundant DNA recognition domain in eukaryotes, and as such the diversity of known domains enables the targeting of a broad range of sequences. An individual zinc finger domain consists of 30 amino acids, which form two β-sheets and an α-helix that coordinate to stabilize a zinc ion (Fig. 2A) [54]. Each domain binds three sequential nucleotides in the major groove of double-stranded DNA through its unique α-helical motif (Fig. 2B). As multiple zinc finger domains are needed to confer ZFN specificity, the DNA binding domains of ZFNs are often composed of six to seven zinc finger domains that target sites of 18–21 nucleotides (Fig. 2C) [55]. Statistically, well-designed ZFNs should only target a single site in the genome, as a random 18 bp sequence should only be found once within 68 billion base pairs.
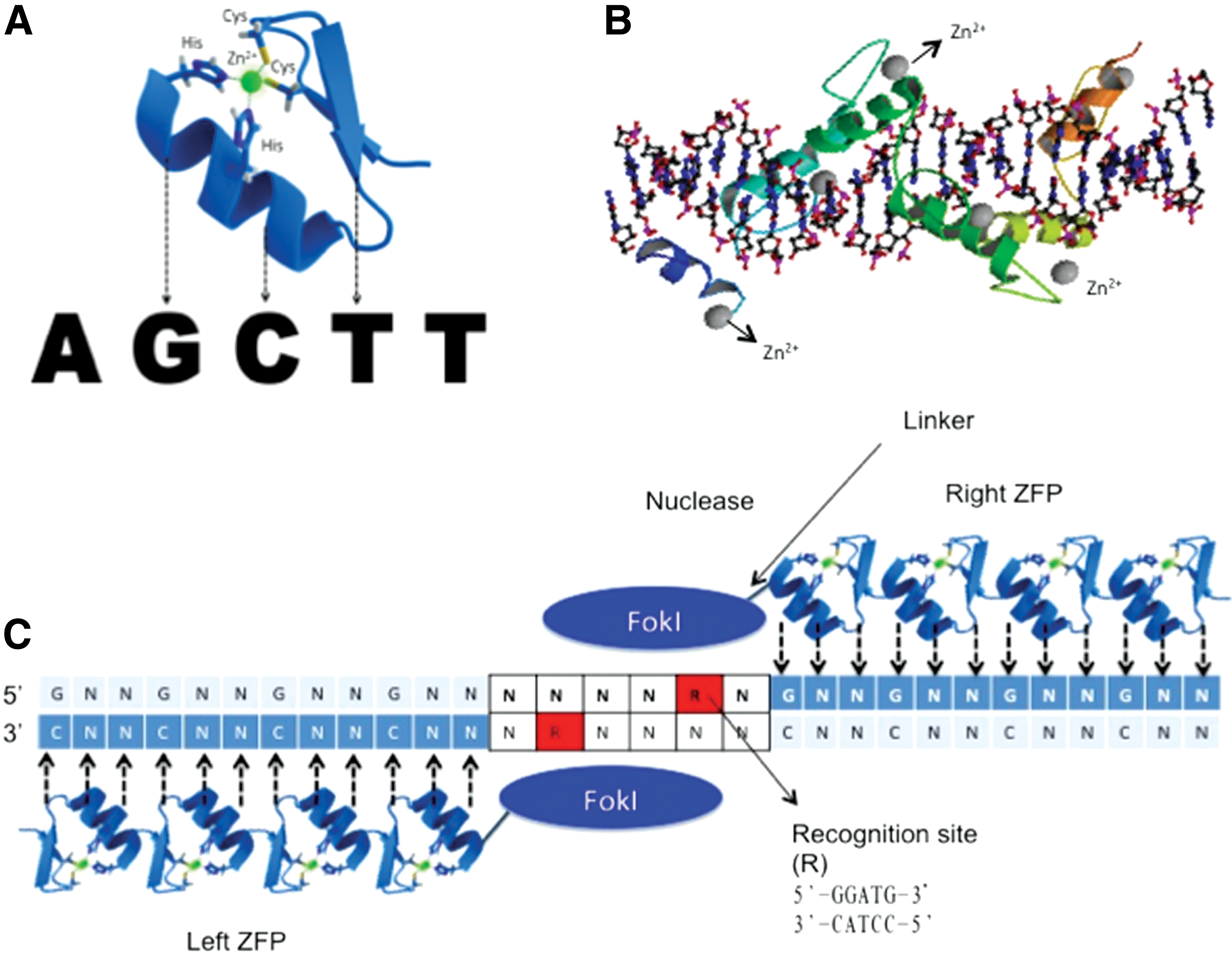
Structure of zinc finger nucleases (ZFNs) on target DNA.
To date, ZFNs have been widely used in many organisms and cell lines, and these have also been utilized for a variety of therapeutic purposes including a Phase I clinical trial to disrupt the C-C chemokine receptor type 5 (CCR5) gene as treatment for HIV infection [56 –60]. However, there are limitations to their applications. While the DNA binding regions of zinc finger domains are engineered to recognize most of the 64 possible nucleotide triplets, there are specific nucleotide triplets that do not have corresponding zinc finger domains because zinc finger domains preferentially bind guanine-rich sequences [61]. Additionally, linking together multiple zinc finger domains causes interactions between the domains that reduce their DNA binding specificity, and the construction process involved in synthesizing new ZFNs is complicated and tedious [62,63].
On the other hand, TALENs represent a new genomic editing system that is considered a convenient alternative to ZFNs. The DNA binding domains of TALENs consist of conserved repeated protein modules adapted from the transcription activator-like effectors (TALEs) in various strains of Xanthomonas, and these are more flexible and easier to assemble than those of ZFNs. Bacterial TALEs are secreted proteins that invade plant cell nuclei to activate gene transcription through binding to target gene promoters, aiding in the establishment of bacterial infection [64]. The TALEN DNA binding domain is a series of 33–35 amino acid repeats that form two short helices, with each repeat domain binding a single nucleotide through repeat variable di-residues (RVD) (Fig. 3A) [65]. RVDs are found at positions 12 and 13 of the amino acid chain repeat sequence and confer sequence specificity to the TALENs (Fig. 3B). There are four RVDs—Asn/Asn (NN), Asn/Ile (NI), His/Asp (HD), and Asn/Gly (NG)—that recognize the nucleotides guanine, adenine, cytosine, and thymine respectively [66]. TALENs work in pairs to target a genomic locus, like ZFNs, but maintain a 14–18 bp spacer between the two binding domains (Fig. 3B). Also like ZFNs, TALE repeats are linked together to recognize stretches of DNA sequences, but TALEs are easier to link together and linking multiple TALEs do not alter binding specificities, allowing TALENs to have longer domains with greater sequence specificity. More details regarding TALEN technology is available on the Addgene Website (
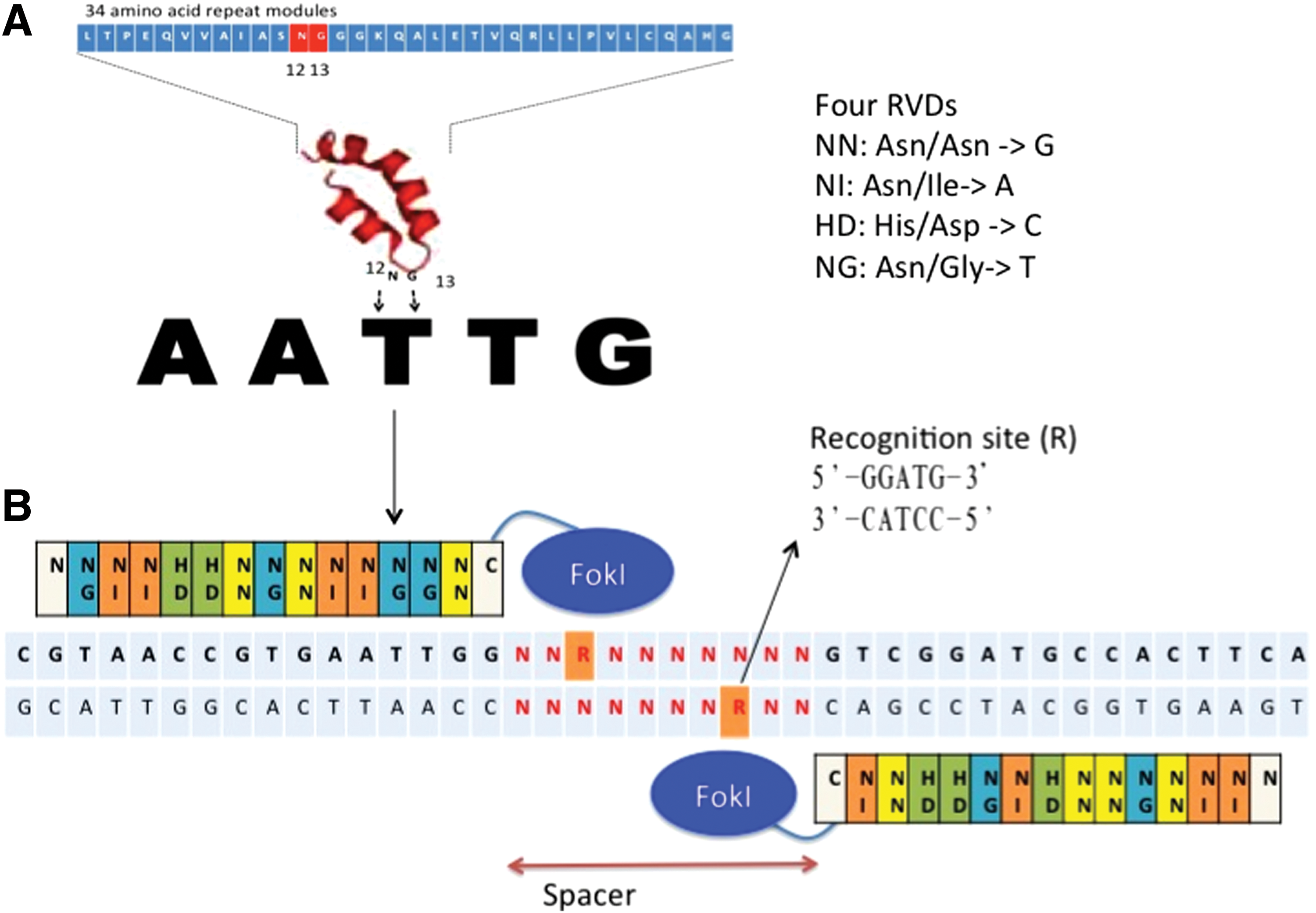
Structure of transcription activator-like effector nucleases (TALENs) on target DNA.
Clustered Regularly Interspaced Short Palindromic Repeat
Clustered regularly interspaced short palindromic repeats (CRISPRs) are a novel genomic editing tool that is rapidly gaining popularity. They are adapted from RNA-based adaptive immune systems that act by destroying viruses of Streptococcus pyogenes. CRISPRs are comprised of a Cas9 protein and short RNA sequences (Fig. 4A) [67]. Cas9 is an endonuclease that can make a break in DNA molecules guided by short RNAs. The short RNAs include two noncoding RNAs, a CRISPR RNA (crRNA), and a transactivating crRNA (tracrRNA). The endogenous CRISPR locus contains the Cas gene and a series of 21–47 bp repeats separated by unique spacer sequences called protospacers originating from viral parasites [67]. When viruses invade S. pyogenes, the CRISPR nuclease detects and cleaves protospacer adjacent motifs (PAMs) in target viral DNA. These cleaved protospacer sequences are collected and integrated into the endogenous CRISPR locus so that upon secondary infection by the same virus, this integrated sequence is transcribed and acts to guide the CRISPR to cleave the viral genome, which is known as a bacterial adaptive immune system [67,68].
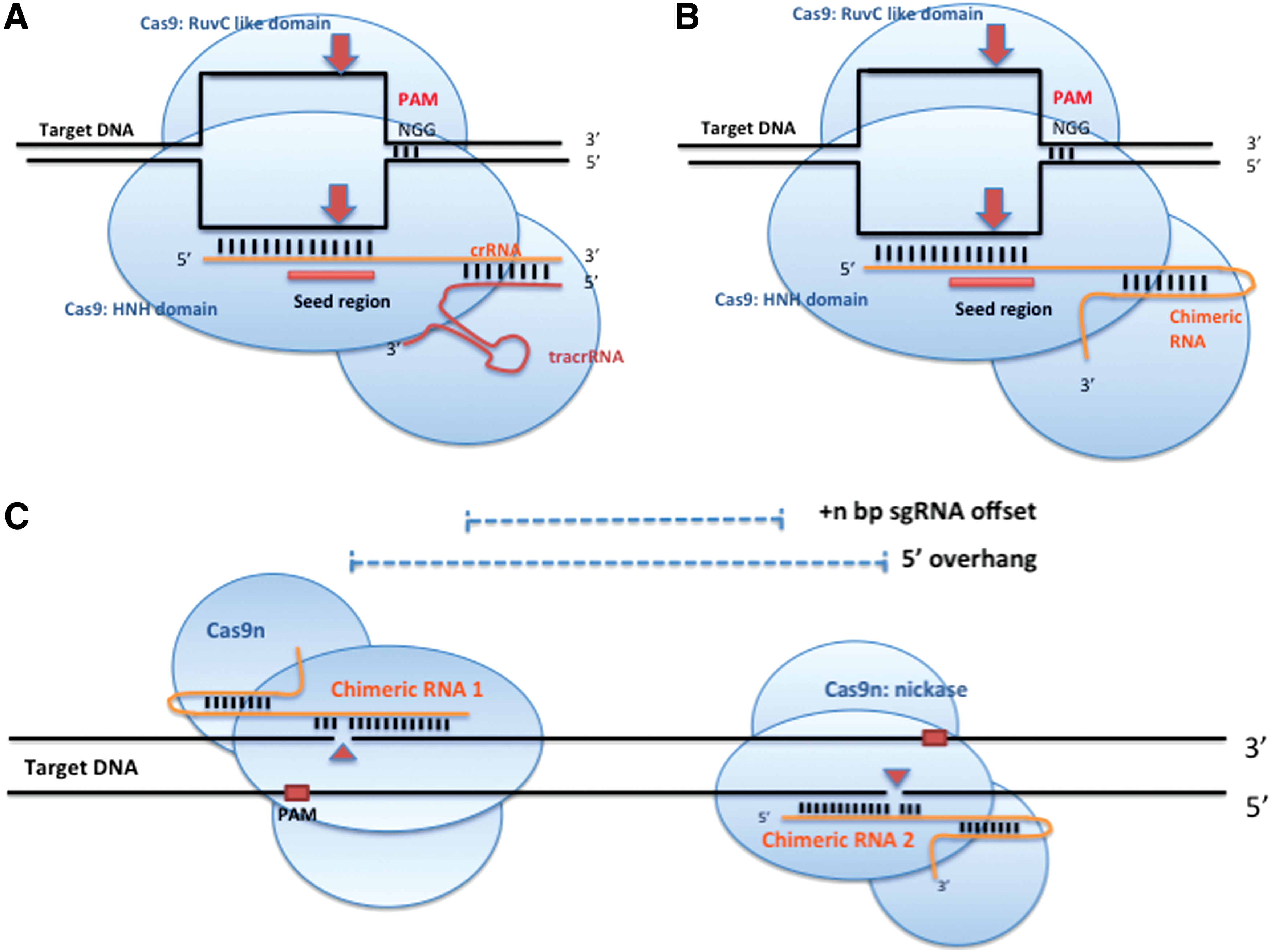
Schematic illustration of clustered regularly interspaced short palindromic repeats (CRISPRs) bound to target DNA.
In vitro, Cas9 of the CRISPR system requires complementary base pairing between crRNA, tracrRNA, and target DNA to efficiently cleave its target (Fig. 4A). A conserved PAM sequence, NGG, on the target DNA strand acts to recruit the Cas9-crRNA-tracrRNA complex to the targets seed region [69 –71]. PAMs are short, but when mutated the complex looses target affinity and the efficiency of cleavage is reduced [70]. The seed region consists of approximately seven bases near the PAM sequence, and is where pairing between the crRNA and target DNA occurs (Fig. 4A, B) [70,71]. The seed region helps to determine DNA binding specificity, and mutations in this region prevent Cas9-mediated cleavage of the target DNA. The PAMs specify the cleavage sites on each target DNA strand, which is cleaved by separate domains of Cas9—the HNH domain (a zinc finger domain having two pairs of histidines and one asparagine) cleaves the target strand complementary to the crRNA, and the RuvC-like domain (a conserved domain in Holliday junction resolvase) cleaves the noncomplementary strand, inducing a DSB in the target DNA (Fig. 4A, B) [69].
The CRISPR system has recently been reengineered to increase its specificity and feasibility of application. To this end, the crRNA and tracrRNA have been fused into a single chimeric guide RNA (gRNA) [70], which contains a protospacer and a hairpin loop to mimic the natural base pairing interaction between tracrRNA and crRNA (Fig. 4B). In cleavage assays that check the feasibility of the chimeric gRNA, it functions with efficiency similar to the smaller tracrRNA-crRNA duplex and the positions of the cleavage sites on target DNA are identical [70].
Limitations of Genomic Editing Systems
Despite the fact that each of the three genomic editing systems increases the efficiency of NHEJ or HR-mediated gene alterations, there is an inevitable disadvantage to engineered nucleases (Table 1). Ideally, DNA binding domain-based genomic editing systems have high specificity to target sites since the target sequence is flanked by two DNA binding domains that bind two unique sequences in the genome. However, the specificity of DNA binding domain relies on its binding affinity to DNA. It has been reported that high DNA binding affinities can cause imperfect specificity that can tolerate to up to three nucleotide mutations [72]. As a consequence, nucleases often make off-target DSBs leading to mutations in unwanted sites [72]. Pattanayak et al. tested the off-target effects of ZFNs in vitro to elucidate ZFN binding specificities and its mutational tolerance. Interestingly, they observed that ZFNs engineered with more zinc finger domains, allowing for greater sequence specificity, still generated mutations in multiple off-target sites, suggesting that different strategies are required for optimizing DNA binding affinities to reduce off-target effects [72].
CRISPR, clustered regularly interspaced short palindromic repeat; DSB, double-strand breaks; gRNA, guide RNA; PAM, protospacer adjacent motifs; ZFN, zinc finger nucleases.
Epigenetic modification also contributes to the specificity of genomic editing systems. It has been shown that sites where engineered nucleases can cleave may not be consistent from one cell line to another due to different epigenetic marks and chromatin structure of the disparate cells [73 –76]. Bultmann et al. investigated how epigenetic modifications affect the targeting efficiency of TALENs at the OCT4 locus, observing that the active OCT4 promoter in ESCs was successfully targeted by TALENs while the silenced OCT4 promoter in neural stem cells inhibited TALEN binding [77]. The use of small molecule epigenetic modifiers, such as valproic acid or 5-aza-2′-deoxycytidine, to alter epigenetic status has been suggested to overcome this limitation [78].
The recent introduction of CRISPRs as an effective and convenient tool for gene editing has shown that they are an advantageous alternative to ZFNs and TALENs. The specificity of CRISPRs relies on a single easily engineered gRNA, and Hsu et al. have demonstrated that CRISPR-mediated cleavage is resistant to DNA methylation [79]. Additionally, with the use of chimeric gRNAs, in conjunction with human-optimized Cas9 proteins, the CRISPR system now functions with a high degree of efficiency in human cells, and it can be manipulated to target multiple genetic loci [80,81]. However, this high Cas9 efficiency has demonstrated significant increases in off-target frequency when compared with ZFNs or TALENs due to the leniency of CRISPRs toward mismatched base-pairing between gRNA and target sequences [82,83].
Recently, a double nicking system was reported to reduce off-target activity. In a manner similar to dimerized ZFNs and TALENs, when two gRNAs designed with appropriate offsets between target sequences are used in combination with a Cas9 nickase, the two separate complexes can successfully induce DSBs and mediate NHEJ or HR repair at higher frequencies than wild-type Cas9 [70,84] (Fig. 4C). As mentioned above, the Cas9 enzyme has two enzymatic domains, HNH and RuvC, which cleave the complementary and noncomplementary strands to the gRNA, respectively (Fig. 4B). By inducing mutations in the catalytic residues of either the RuvC (Asp10Ala, D10A) or HNH (His840Al, H840A) domains, Cas9 becomes a nickase capable of cleaving only one strand of the target DNA (Fig. 4C). When two pairs of gRNAs that target opposite DNA strands are used, DSB can occur with considerably reduced off-target activity ranging from 50 to 1,500-fold depending on the cell line used [84]. Recently, Shen et al. demonstrated that both the D10A and H840A nickases, when used with two appropriately offset gRNAs, are also capable of reducing off-target effects in vivo [85].
While the new double nicking system does not completely eliminate all off-target effects of the CRISPR system, it is regarded as a promising genome editing tool with demonstrated widespread applications from gene manipulation in mouse embryos to systemic and genome wide screening in human cell lines [86 –88]. As CRISPRs show high targeting efficiency using both HR and NHEJ in ESC and iPSCs [89], it is anticipated that soon CRISPRs will also be utilized for modeling diseases.
Generation of Disease-Specific hPSCs with Genomic Editing Systems
Even though hPSCs are regarded as a powerful resource for cell replacement therapy, due to the limited feasibility of current cell replacement therapy options, disease modeling with iPSCs has recently become an especially attractive branch of research. To date, ZFNs have been the primary genomic editing tool used to model diseases with hPSCs, in part due to their availability before other genomic editing tools were established. Today there are currently only a few disease modeling studies that take advantage of TALENs, and no reports using CRISPRs (Table 2). The first report of ZFN application in hESCs was in 2007, and demonstrated that ZFN-mediated DSBs increase site-specific integration of vector sequences in hPSCs [90]. The CCR5 locus was chosen as the target site because its homozygous null mutations are tolerated in human cells. A donor vector with CCR5 homology arms containing the selection markers GFP or puromycin successfully targeted the CCR5 locus when delivered together with the ZFN expression vector into hESC through IDLV [90]. This demonstrated that ZFN-mediated HR efficiency is higher than standard HR-mediated integration alone [90]. GFP-positive clones maintained their pluripotent phenotype and were capable of generating GFP-positive neural progenitors in culture [90]. While this report was the first to show the utility of ZFNs for gene targeting in PSC, they did not demonstrate that their results were not due to the existence of random insertions. Additionally, they found that NHEJ-mediated mutations occurred in the CCR5 locus even if the donor vector was transferred [90]. These limitations raised by this article have been discussed in many subsequent articles using ZFNs or TALENs in hPSCs, which in turn has led to further advancement of ZFN and TALEN application methods.
Pathological phenotype is not clear due to lack of differentiation protocol.
The mechanism of pathological phenotypes was demonstrated using corrected patient iPSCs.
apoB, apolipoprotein B; HPRT, hypoxanthine phosphoribosyltransferase; HR, homologous recombination; iPSCs, induced pluripotent stem cells; HIV, human immunodeficiency virus; KO, knockout; NHEJ, nonhomologous end joining; TALENs, transcription activator-like effector nucleases.
Integration Methods of Genomic Editing Tools in hPSCs: Safe Harbor Locus
Most studies using genomic editing tools to alter gene function in hPSCs use one of three gene targeting methods: integration into a safe harbor locus, HR-mediated direct correction, and induction of gene mutations. Integration approaches that utilize a safe harbor locus refer to a method that targets a specific genomic site where integration of a transgene would allow for both active transgene transcription, and no downstream consequences caused by the disruption of the endogenous locus [91]. The AAVS1 locus (also known as PPP1R12C) on chromosome 19 in humans has long been used as a safe harbor region for gene therapy [92,93]. The AAVS1 locus has an open chromatin structure so that integrating transgenes can stably integrate into the AAVS1 site and show robust expression [94]. Taking advantage of the AAVS1 safe harbor locus to generate isogenic hPSCs for modeling diseases has been achieved and has been utilized most when mutations leading to disease are due to large deletions or other large genomic alterations. Zou et al. in 2011 used ZFN-mediated HR to insert corrected gp91phox (NOX2) into the AAVS1 locus to recover the enzyme deficiency for X-linked chronic granulomatous disease [95]. Another article using ZFNs to model α-thalassemia also utilized the AAVS1 integration method to compensate for large deletion sites found in endogenous α-globin [96].
However, there are significant concerns regarding the therapeutic use of ZFNs at safe harbor loci. iPSC could acquire genetic and epigenetic alterations from genomic editing through off-target integration, or generation of nicks in the DNA backbone [95]. Zou et al. showed that 25% of clones contained random insertions, and 50% contained NHEJ-mediated mutations. Second, safe harbor loci can be affected by integrated sequences. Sequences that integrate within close proximity to the AAVS1 gene locus are able to activate inappropriate AAVS1 expression in clones upon erythroid differentiation [96].
Correcting Mutations for Targeting Monogenic Diseases
The most prevalent disorders targeted by gene therapy and disease-modeling studies are thalassemia and sickle cell anemia (SCD) as these are well-characterized monogenic disorders [97]. Because these diseases are caused by missense mutations, HR-mediated gene correction is used to target the mutation. SCD is caused by a missense mutation in codon 6 of β-globin (HBB) causing a glutamine to valine change in the amino acid sequence, and two groups have reported the genetic correction of this mutation in human iPSCs (hiPSCs) through HR-based direct correction. Zou et al. showed homozygous correction of mutant β-globin alleles by a ZFN in conjunction with HR using vectors designed to integrate into intron 1 of the HBB gene, and demonstrated expression of wild-type β-globin in red blood cells derived from the corrected patient iPSCs [98]. A second group followed a similar strategy for modeling SCD, correcting the mutation with a cassette containing a drug resistance marker that allowed for easy selection of corrected clones [99]. While the second report found no evidence of off-target effects, Zou et al. raised the possibility that selection cassettes could affect corrected gene expression. Indeed, when the selection cassette was excised via Cre-mediated recombination, the expression of the corrected allele was diminished or lost during in vitro differentiation [98]. They reasoned that residual LoxP sequences left after Cre-mediated selection marker excision resulted in splicing defects, interfering with corrected HBB gene expression. Also, it has since been hypothesized that Cre-mediated excision of selection cassettes may also affect cis regulatory elements by mutating GATA binding sites found in the 3′ enhancer of the HBB gene, leaving questions and concerns for the use of HR-mediated ZFN repair [98 –101].
TALENs have also been applied to correct mutations for modeling SCD and thalassemia. β-Thalassemia iPSCs and their TALEN-corrected controls were used to recapitulate β-Thalassemia in vitro to demonstrate that restored β-globin expression reverses the phenotype [102]. Additionally, Sun and Zhao used TALENs to generate isogenic iPSC for modeling SCD instead of ZFNs to avoid off-target effects and potential genomic instability caused by the low specificity of ZFNs [103]. To avoid residual genomic scars left behind from removed selection markers, the authors used PiggyBack transposons as they do not leave and residual sequences after excision, unlike loxP sites [103]. The use of PiggyBack transposons resulted in 60% of clones with corrected mutations, with only two heterozygous clones out of 48 corrected clones [103]. Furthermore, no off-target effects, translocations, or alterations were found, confirming the specificity of TALENs and the advantage of PiggyBack vectors to target sites of interest [103 –105].
Generating Mutations in Wild-Type hPSC with Genomic Editing Systems
Genomic editing tools have been utilized not only for correcting mutations, but also for inducing mutations or deletions in hPSCs. In 2009, Zou et al. directly targeted the endogenous PIG-A locus, frequently found mutated in hematopoietic stem cells from patients with paroxysmal nocturnal hemoglobinuria. Using ZFNs and induced HR-mediated mutagenic insertion, they found a 200-fold increased targeting efficiency and a decreased random insertion frequency than would be found by HR alone [106]. Additionally, ZFNs have also been reported for the use of inducing genomic translocations. Brunet et al. used ZFN and TALENs in hESCs to generate genomic translocations, which are often associated with many cancers [107 –109]. While modeling of a specific cancer with chromosomal translocations was not performed, characteristics of translocations in human tumors such as deletions, insertions, and microhomology at t(19;X), t(11;22)(q24;q12), and t(2;5)(p23;q35) translocations were recapitulated, opening up the possibility of modeling cancer associated translocations with hESCs [108,109].
Most recently, Greber and coworkers used TALENs in hESCs to model Lesch Nyhan-Syndrom (LNS). To do so, TALENs were used to knock out the HPRT1 gene through NHEJ, as mutations in this region are known to cause the disease [110]. The targeting efficiency of HPRT1 mutant clones was found at 15% with the use of two selection markers. The use of the two antibiotic selection markers was critical, as targeted mutation rates without the additional selective pressure were low at 0.5%. As a result, out of five clones generated, three were homozygous and two were heterozygous for the induced mutation. Finally, when the HPRT1 mutant cell lines were differentiated into RN3A+central nervous system neurons, the knock-out cell lines successfully recapitulated phenotypes of LNS, including impaired neuronal differentiation and short neurite length. This demonstrated the usefulness of NHEJ-mediated KO system with TALENs in hPSCs for human disease modeling and also argued that conventional lipofection delivery methods are capable of generating sufficient numbers of clones [110].
As discussed above, there are some important concerns regarding the use of genomic editing tools. These include unwanted mutations in the genome due to off-target effect, the impact of selection markers on the expression of neighboring genes, or the effect of residual LoxP sites left after Cre-mediated excision. Thus, it is extremely important to ensure that there are no lesions or residues left behind by genomic editing tools, which can only be ensured through deep sequencing or global genomic analysis. However, it is nonetheless clear that ZFNs and TALENs significantly increase targeting efficiency either through the use of safe harbor loci, HR correction, or knock-out methods. Recent reports show that perfect isogenic cell lines can be successfully engineered, and they do not contain unexpected genomic mutations [99,102,103,107]. This implies that as ZFNs and TALENs continue to be optimized for use in hPSCs, perfect isogenic cell lines will be generated more efficiently and with greater ease, allowing for better models of genetic diseases.
Modeling Complex Diseases with Isogenic hPSCs
Unlike modeling early onset and monogenic diseases that manifest robust pathological phenotypes, when complex diseases such as PD are modeled with patient and wild-type iPSCs, the patient iPSCs tend to show subtle phenotypic changes or unpredictable background effects [111 –113]. This is a major challenge that stem cell technology needs to overcome for the comprehensive study of complex diseases. One conceivable solution is to generate perfect isogenic wild-type iPSCs directly from patient iPSCs. Genomic editing is inevitably required for modeling these diseases with iPSC technology, because, fundamentally, to create a disease in a dish an underlying mutation should be the only difference between disease and control cells. Indeed, this perspective has caused many to model complex diseases with isogenic iPSCs in combination with ZFNs or TALENs, with recent reports demonstrating mechanisms of pathogenesis or elucidating novel therapeutic targets, highlighting the unique therapeutic potential of iPSCs.
Previously, Soldner et al. applied ZFN technology to target a missense mutation in α-synuclein, a protein frequently mutated in familial PD [114]. They corrected A53T mutations in patient iPSCs and induced A53T or E46T mutations in wild-type iPSCs to model PD. Although these lines could successfully differentiate into dopaminergic tyrosine hydroxylase expressing neurons, they did not address a pathological phenotype at the time [114]. A recent report modeling PD with the same isogenic iPSCs was not only able to recapitulate a pathological phenotype, but also linked interactions between genetic mutations to environmental factors known to contribute to PD [115]. They found that the cells with the A53T mutation were more susceptible to toxin-mediated apoptosis than control cells. Furthermore, it was found that this sensitivity to cell death in A53T was mediated by the MEF2-PGC1α pathway [115]. They noted the accumulation of reactive oxygen species/reactive nitrogen species caused by A53T mutant-mediated protein aggregation, mitochondrial toxins, or both, resulting in the sulfonated nitrosylation of MEF2C that causes neurons to undergo apoptosis [115]. Importantly, they used hiPSCs and isogenic iPSCs for high throughput screening studies to resolve the pathway, highlighting the feasibility of using iPSCs for modeling multi-hit diseases [115].
Recently, modeling complex diseases using iPSCs in conjunction with genomic editing tools has successfully elucidated new mechanisms of disease irresolvable by mouse modeling or human autopsy samples alone [116 –118]. When modeling Tauopathy, a neurodegenerative disorder leading to Alzheimer's disease and frontotemporal dementia, isogenic TAU cell lines created by ZFNs elucidated a new pathological phenomena where the mutant microtubule-associated protein tau protein is predisposed to caspase cleavage, leading to axon degradation and shortened neurites [119].
Additionally, using TALEN-generated isogenic iPSCs, Ding et al. clarified data from conflicting reports about the effect of SORT1 on low-density lipoprotein and apolipoprotein B (ApoB) secretion from the liver, key components required for hepatitis C virus (HCV) infection [120]. While the SORT1 gene has been identified as a regulator of ApoB in human genetic studies, additional studies in mice and humans provide conflicting evidence as to whether SORT1 functions to increase or decrease ApoB expression [121,122]. Ding et al. demonstrated with isogenic lines that increased SORT1 expression resulted in decreased ApoB and lower cholesterol levels, which is consistent with human genomic studies, suggesting that modification of SORT1 may be a tractable therapy for HCV infection, and a promising tool to study the underlying mechanisms in a human context.
Conclusions
Since the initial generation of hiPSCs in 2007, the derivation of patient-specific iPSCs for modeling diseases has become more frequent and prevalent. Current studies have taken this a step further, and have focused on demonstrating the feasibility of genetic correction using patient-derived iPSCs. Isogenic cells generated by ZFNs, TALENs, and CRIPSRs will undoubtedly help to overcome variability found between iPSC clones and the intrinsic genetic background of patients. However, to generate perfect isogenic iPSCs, many factors need to be considered including selection markers, donor vectors, culture conditions, gene correction strategy, off-target effects, and genomic integrity. In addition, it is of note that ZFNs and TALENs each have their own strengths and limitations that should be considered (Table 1). Both are capable of producing off-target effects, and both show reduced efficiency when targeting methylated genomic loci. Furthermore, even though many isogenic iPSCs have been derived with ZFNs and TALENs, using isogenic patient-specific iPSCs generated by genomic editing tools to study molecular and pathological mechanism is still in the early stages, and continued evaluation of its feasibility is needed.
CRISPRs are the newest genetic editing tool for modeling diseases. Easy to construct and insensitive to DNA methylation, with increasing targeting efficiency in human cells, CRISPRs are rapidly gaining popularity for gene editing in human cells. Although off-target rates of CRISPRs cannot be disregarded, and their use has not been reported in hPSCs for disease modeling purposes to date, with the introduction of the new CRISPR double nicking system, CRISPRs may prove to be an effective genomic editing tool for modeling diseases with hiPSCs. Nevertheless, as time progresses and the technologies advance, ZFNs and TALENs, in addition to CRISPRs, will all aid in improving hPSC-based studies that will hopefully help to both elucidate biological mechanisms and provide novel clinical therapies.
Footnotes
Acknowledgments
This study was funded by grants from the National Institutes of Health (NIH) to I.R.L. (R01GM078465) and NIH Pathway to Independence Award (K99CA181496) to D.F.L.
Author Disclosure Statement
The authors declare they have no competing financial interests.