
Abstract
Induced pluripotent stem cells (iPSCs) have become the most promising candidates for in vitro modeling of motor neuron (MN) diseases, such as amyotrophic lateral sclerosis (ALS), and possibly for future therapeutic implementation in regenerative medicine. We here present for the first time the differentiation of human cord-blood-derived iPSCs (hCBiPSCs) into MNs, the cell type primarily affected in ALS. In contrast to iPSCs generated from adult tissue, the hCBiPSCs used in this study hold the promise of lower genetic mutation burden or epigenetic alterations, which makes them ideal candidates for transplantation studies. Small-molecule-derived neural precursor cells (smNPCs) were generated from hCBiPSCs and used for the following differentiation studies to substantially shorten MN differentiation time. Consequently, as early as 18 days of in vitro differentiation, the MNs stained positive for neuronal- and for MN-specific markers accompanied by respective gene expression patterns. To demonstrate that the hCBiPSC-derived neural precursor cells (smNPCs) can be differentiated into functional MNs, the cells were characterized by calcium imaging and patch-clamp analysis. Calcium imaging detected the expression of functional voltage-dependent calcium and ligand-gated channels of several important neurotransmitters. Using whole-cell patch-clamp recordings, we observed functional neuronal properties like sodium-inward currents and action potentials (APs). Some cells showed spontaneous APs and synaptic activity that are signs of essential functional maturation. Having established a rapid and efficient method to generate functional MNs from hCBiPSCs, we demonstrate the differentiation potential of genetically unbiased hCBiPSCs as promising source for transplantation studies and also create a framework for future in-vitro disease modeling.
Introduction
S
iPSCs can be used to improve our understanding of underlying processes and treatment of motor neuron diseases (MNDs), such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). Regarding the use of iPSCs for cellular therapies, a small number of studies established that transplantation of hESC-/iPSC-derived motor neurons (MNs) into central nervous system (CNS) tissue is feasible in animal models [3,4]. Substantial progress has been made with respect to iPSC in vitro modeling of MN disorders. Several recent studies have shown that MNs differentiated from patient-derived iPSCs indeed recapture relevant disease phenotypes comparable to changes in postmortem ALS [5 –7] or SMA [8,9] tissue. Given the fact that ex vivo studies of CNS tissue are not possible in MND patients, the differentiation of patient-derived iPSCs into MNs offers the possibility to better understand the underlying mechanisms and pathogenesis of MND. Further, identification of novel neuroprotective compounds using patient-derived iPSCs [5 –7] has shown the suitability of these in vitro models to serve as platform for high-throughput screening in drug discovery. However, interclonal variability and variability in differentiation outcome is a major drawback of the use of patient-derived iPSCs. Poorly defined differentiation conditions, including different cocultivation methods [3], may contribute to this observation and prolonged time frames [days in vitro (DIV) >60] [5,7,10,11] make the investigation of scientific questions cumbersome and cost expensive. Therefore, shorter, well-defined, and easily applicable protocols for MN differentiation of iPSCs are crucial to finally provide the promising field of iPSC-based in vitro modeling with an optimal starting point.
While the use of human cord-blood-derived iPSCs (hCBiPSCs) to directly model adult-onset diseases, such as ALS, is limited to overexpression studies and the introduction of other disease-relevant information, we present an efficient, time-saving, and easy-to-handle frame for the differentiation of patient-derived iPSCs into functional MNs, which can be used for more rapid differentiation of patient-specific IPSCs for drug screening and in vitro modeling in general.
Being reprogrammed from human cord blood (hCB) [12], these hCBiPSCs hold the promise of being free of epigenetic changes, chromosomal abnormalities, and point mutations that regularly occur during adult life. The resulting decreased risk of tumor formation upon transplantation consequently makes hCBiPSC-derived MNs superior candidates for implementation into future regenerative trials [13]. Further, the central aim of large-scale generation of allogenic neurons for drug-screening purposes or to develop novel strategies for allogenic transplantation approaches is facilitated by the proliferative power of hCB-derived endothelial precursor cells as source for reprogramming [12]. Starting with the expansion of several hCBiPSC lines, we used small molecules to generate an expandable neural precursor population [small-molecule-derived neural precursor cells (smNPCs)] [14] that was used throughout the whole study and differentiated into cells expressing neuronal- and MN-specific markers in <3 weeks. After DIV25 we observed functional ligand-gated ion channels by application of various neurotransmitters in calcium imaging experiments. Being held in maturation conditions until DIV30, the hCBiPSC-derived MNs further demonstrated basic neurophysiological characteristics like sodium inward currents, action potentials (APs), and spontaneous activity in whole-cell patch-clamp recordings, indicating essential neuronal functions.
Materials and Methods
iPSC culture
Characterized previously in detail [12], hCBiPSCs were derived by lentiviral transductions of hCB cells with the factors Nanog, Oct4, Sox2, and Lin28. hCBiPSCs were maintained in culture on a feeder layer of irradiated (60 gray) mouse embryonic fibroblasts. Aliquots were maintained for several months at −150°C, thawed upon demand, and seeded at a density of 1×105 cells per single well in a six-well plate previously coated with 1% gelatin (Sigma). iPSCs were cultured for up to 5 days before passaging in knockout medium consisting of DMEM supplemented with 20% knockout serum replacement, 1% penicillin-streptomycin, 0.1 mM MEM nonessential amino acids, 2 mM glutamax, and 0.1 mM β-mercaptoethanol. Media were replaced daily and supplemented with fresh 20 ng/mL basic fibroblast growth factor (bFGF). Unless specified, all media and supplements were purchased from Life Technologies. Cytokines were obtained from Peprotech.
smNPC generation and differentiation
For the generation of smNPCs, the hCBiPSC colonies of each clone (K1, K2, K106, and K120) were collected as whole, allowed to settle by gravity, and resuspended in iPSC media without bFGF but supplemented with 10 μM SB-431542 (Ascent Scientific), 1 μM dorsomorphin (DM; Sigma), 3 μM CHIR 99021 (Axon Medchem), and 0.5 μM purmorphamine (PMA; Alexis). Cell aggregates were kept on low-adherence, six-well plates (Falcon). On day 3, iPSC media were replaced with N2B27 media (DMEM F12 and neurobasal media in a 1:1 ratio, supplemented with 1:100 B2 supplement lacking vitamin A and 1:200 N2 supplement; both from Invitrogen). Two days later on day 5, DM and SB-431542 were replaced with 150 μM ascorbic acid (Sigma). On day 7 the cell aggregates were mechanically separated into pieces and replated onto Matrigel (MG; BD Biosciences)–coated dishes. MG was diluted 1:50 in Dulbecco's modified Eagle's medium (DMEM) F12 medium. MG-coated dishes could be stored up to 1 month at 4°C. As illustrated and described in detail elsewhere [14], homogeneous populations of smNPCs arose during the course of subsequent passages. At least once a week, the smNPCs were passaged in a 1:5 ratio using accutase for 5 min at 37°C. Final differentiation was induced by removing CHIR 99201 from the media for 3 days followed by the addition of 0.1 μM retinoic acid (RA) for another week. Maturation of the patterned cells was accomplished in N2B27 media enriched with 20 ng/mL brain derived neurotrophic factor (BDNF), 10 ng/mL glial cell-derived neurotrophic factor (GDNF), and 5 μM N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (dbcAMP). All in all 6 weeks of smNPC generation and expansion was necessary before final MN differentiation could be started.
Immunocytochemistry
For immunocytochemical staining, K2 cells were plated and differentiated in MG-coated, 35-mm petri dishes. On the day of evaluation they were fixed with 4% formaldehyde for 20 min at room temperature, washed three times with phosphate-buffered saline, and blocked for 60 min in blocking buffer (5% goat serum, 1% bovine serum albumin, and 0.3% Triton X-100). Primary antibodies (rabbit polyclonal anti-Oct4, 1:500, Cell Signaling; mouse monoclonal anti-Vimentin, 1:200, Abcam; mouse monoclonal anti-Nestin, 1:200, Abcam; rabbit polyclonal anti-Sox2, 1:200, Cell Signaling; mouse monoclonal anti-TuJ1, 1:500, Millipore; rabbit polyclonal anti-Map2, 1:500, Millipore; rabbit polyclonal anti-GABA, 1:500, Sigma; mouse monoclonal anti-SMI32, 1:500, Covance; mouse monoclonal anti-HB9, 1:50, DSHB; goat polyclonal anti-ChAT, 1:200, Millipore; and rabbit polyclonal anti-Islet1, 1:200, Abcam) were incubated overnight at 4°C. After additional washing steps, secondary antibodies [Alexa Fluor (488 or 555) goat anti-mouse, goat anti-rabbit, and donkey anti-goat] were applied for 2 h at room temperature. DAPI counterstaining in mounting solution was applied for 20 min at room temperature. Visualization was done by fluorescence microscopy (BX61; Olympus). Images were taken with an Olympus DP72 camera. The image-analysis software CellF (Olympus) was used for further evaluation. Cell counts were performed on three random visual fields from at least three independent experiments.
Quantitative real-time polymerase chain reaction
The RNeasy kit (Qiagen) was used for total RNA extraction according to the manufacturers' protocol. In total, 1 μg of RNA was reversely transcribed, using oligo-dT primer and Superscript II reverse transcriptase set (Life Technologies). The final quantitative real-time PCR (qRT-PCR) experiments were executed with cDNA from 50 ng of total RNA, 1.75 μM forward/reverse primer, and Power SYBR-Green PCR Master Mix (Life Technologies). The StepOnePlus Instrument (Applied Biosystems) was used for the following cycles: 90°C/10 min and 40 cycles of 95°C/15 s and 60°C/1 min. Final calculations were based on the cycle threshold (CT) normalized against the endogenous β2-microglobulin [Ct (target) – Ct (reference)=ΔCt]. Relative levels of gene expression were illustrated on logarithmic scales (log10) as ΔCt means±standard error of the mean (SEM) from three independent experiments for each of the four clones (K1, K2, K106, and K120).
Functional evaluations
Patch-clamp recordings of K2-derived MN cultures were performed as described previously (Stanslowsky et al. 2014, in press) at room temperature with an inverted microscope (Zeiss) and an EPC-10 amplifier (HEKA). Whole-cell currents were low-pass filtered and digitized at 2.9 and 10 kHz, respectively. Patch Master software (HEKA) was used for recording and final analysis. Borosilicate glass pipettes (Science Products) were pulled and polished to yield a resistance of 3–4 MΩ when filled with the internal solution (153 mM KCl, 1 mM MgCl2, 10 mM HEPES, 5 mM EGTA, and 2 mM Mg-ATP, calibrated to pH 7.3 with KOH; 305 mOsm). During pharmacological experiments, the external bath solution (142 mM NaCl, 8 mM KCl, 1 mM CaCl2, 6 mM MgCl2, 10 mM glucose, and 10 mM HEPES, calibrated to pH 7.4 with NaOH; 325 mOsm), 1 μM tetrodotoxin (TTX), 10 mM tetraethylammonium chloride (TEA), 20 μM mecamylamine, and 10 μM atropine (all Sigma) were applied via gravity through the modified SF-77B perfusion fast-step system (Warner Instruments) as described in Wegner et al. To record spontaneous activity patterns around DIV40–50, the cells had to be passaged one additional time at DIV18. For identifying MNs in our MN cultures, we applied strict selection criteria. We included only multipolar neurons with large cell bodies as indicated by cell capacitances of ∼20 pF [12,15 –17].
For calcium imaging, Fura 2-AM (Sigma)–loaded K1/K2 cells in standard bath solution (consisting of 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES, adjusted to pH 7.4 with NaOH) were recorded with an upright microscope (Axioskop 2 FS plus; Zeiss) connected to Till Vision Imaging System (TILL Photonics) and signals were taken up by a charge-coupled device camera as described previously [18]. Excitation of the cells at wavelengths of 340/380 nm and monitoring intervals of 300 ms at 510 nm allowed for visualization of intracellular [Ca2+] upon stepwise application of several neurotransmitters.
Statistical analysis
GraphPad Prism 5 Software (GraphPad Software) was used for statistical analysis. Statistical inferences were drawn from unpaired t-test and illustrated as mean±SEM. Results were regarded as statistically significant with P<0.05.
Results
Generation of expandable neural precursor populations using small molecules
To demonstrate that iPSCs derived from hCB can generate NPCs, we collected iPSC colonies from different hCBiPSC lines with passage numbers ranging from ∼50–96 (K1, K2, K106, and K120) [12]. Based on a previously developed protocol [14] iPSC colonies were maintained as spheres in suspension under neural induction conditions. During the first 2DIV, initiation of neural induction was evoked by the combined inhibition of bone morphogenic protein (BMP) and transforming growth factor beta (TGFβ) signaling pathways following the introduction of the small molecules SB43152 (SB) and DM [19 –21]. Further, the spheres were exposed to the ventralizing sonic hedgehog agonist PMA and the GSK3β inhibitor CHIR 99021, stimulating the canonical WNT signaling pathway in turn [14]. In the final smNPC media lacking factors SB and DM, spheres were passaged more than 10 times before further differentiation experiments (Fig. 1). The cells were stably passaged more than 40 times without losing their differentiation potential, which clearly illustrates their expandability. Variations in differentiation outcome due to different passage numbers were not observed. Immunocytochemical staining of the expanding cells revealed signs of basic NPC characteristics through expression of neural progenitor markers Nestin, Vimentin, and Sox1 (Fig. 2A). qRT-PCR further confirmed the observed expression of NPC markers Pax6 and Sox1 (Fig. 2B). Compared with their respective hCBiPSC line from which they were generated, the smNPCs very consistently demonstrated significantly higher expression levels of the NPC markers Pax6 and Sox1.
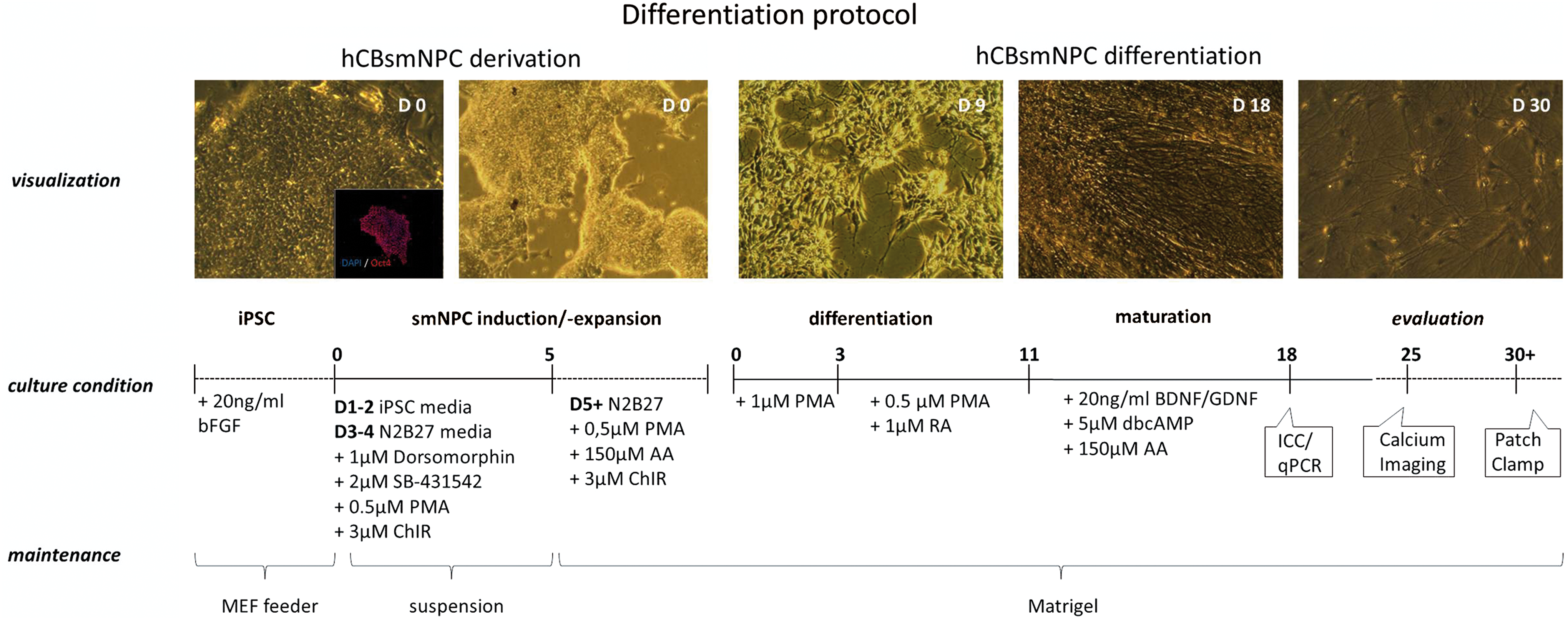
Schematic summary of small-molecule-derived neural precursor cell (smNPC) derivation and motor neuron (MN) differentiation. smNPCs are generated from human cord-blood-derived induced pluripotent stem cells (hCBiPSCs) that were positively stained for Oct4. These smNPCs grow in colonies (D0), are expandable up to passage number >40, and can be used for further differentiation experiments. In presence of purmorphamine (PMA) and retinoic acid (RA), the cells quickly change morphology (D9) and start to form extensive neuronal networks around ∼D18. Immunocytochemistry and quantitative polymerase chain reaction (PCR) were performed at days in vitro (DIV) 18. For calcium imaging at DIV25 and patch-clamp analysis at DIV30/DIV40–50, the cells were passaged once and replated onto matrigel-coated dishes. Color images available online at
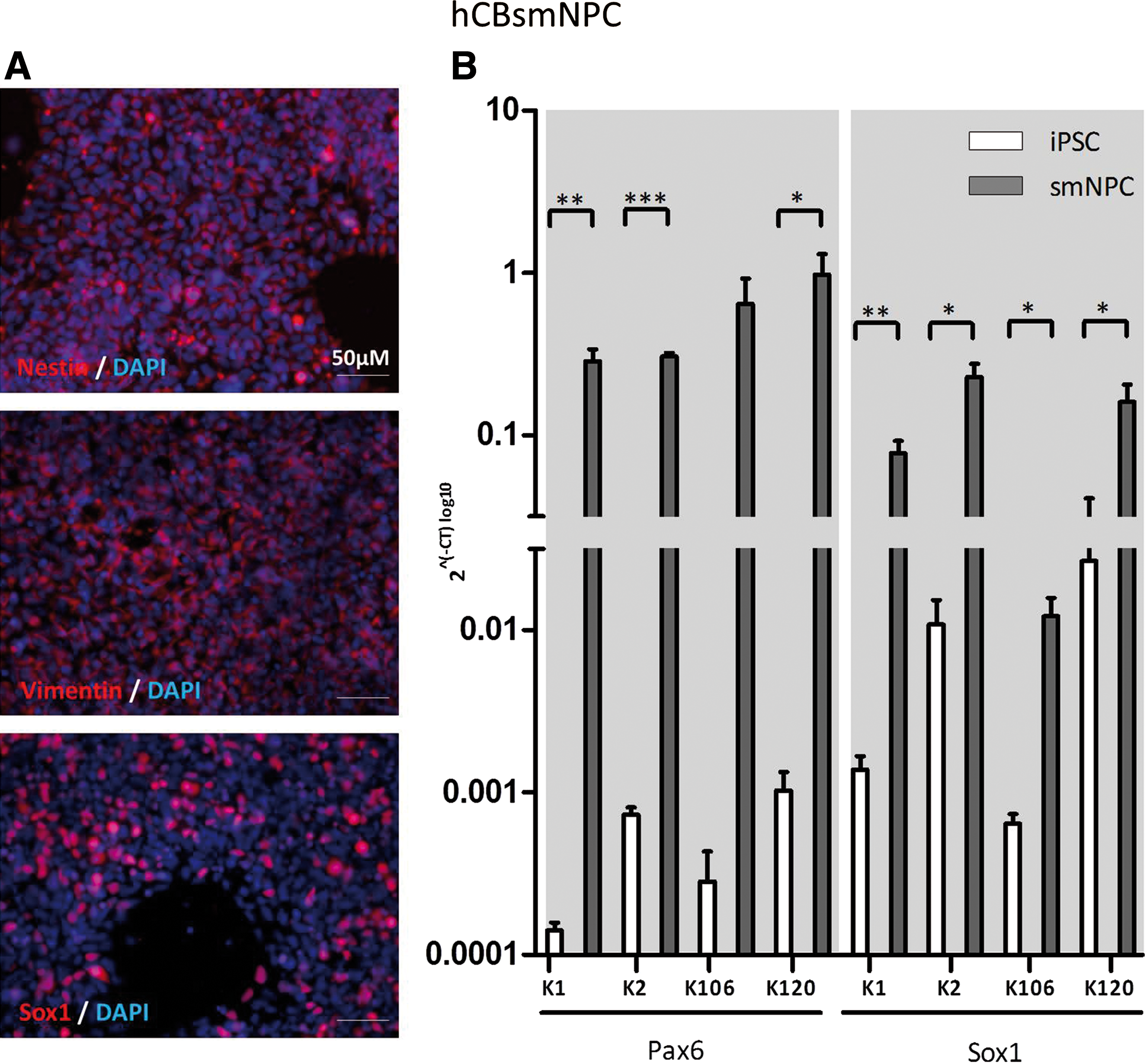
NPCs derived from hCBiPSCs homogeneously express various neural epithelial cell markers.
Rapid neuronal induction and MN specification
During differentiation of hCBiPSCs according to the protocol illustrated in Fig. 1, the combined action of the SHH agonist PMA and RA [22] resulted in rapid changes in morphology. In contrast to Reinhardt et al. [14] who used 1 μM RA and 1 μM PMA for the patterning process, we found that cells better tolerated doses of 0.1 μM RA and 0.5 μM PMA. Also the amount of dbcAMP was reduced by 10-fold (50 instead of 500 μM) in the final maturation phase. After 7–10 DIV, extensive branching and increased interconnectivity of cells was observed in our colonies (Fig. 1, D9 brightfield image), while earlier reports suggested this process to start from day 28 on [3,17]. After 1 more week under maturation condition (∼DIV18–20) a majority of the cells stained positive for the neuronal markers TuJ1 (71%) and Map2 (55%). Approximately 50%–60% of these TuJ1-positive cells coexpressed the neurofilament marker SMI32 (60%) or the LIM-homeodomain transcription factor Islet1 (49%), both indicative for MN fate (Fig. 3A). From the Map2-positive cells, 56% and 46% stained positive for ChAT and HB9, respectively. Earlier studies that differentiated hESCs/hiPSCs into MNs reported increases in neuronal and MN markers several weeks later starting from day 60 on [3,5 –7,10,11,17]. Just a small portion of our neuronal cells coexpressed the glial marker GFAP (6%). The unstained remaining cells presumably consist of undifferentiated smNPCs. To further confirm the immunocytochemical observations, MNs were analyzed for mRNA expression of neuronal-specific and rather MN-specific markers by qRT-PCR (Fig. 3B). In all four iPSC lines, a significant upregulation of neuronal-specific (TuJ1 and Map2) as well as more MN-specific markers (HB9, Islet1, and ChAT) was found (Fig. 3). As expected, downregulation of the pluripotency markers Oct4 and Lin28 was detected as well (Fig. 3B).
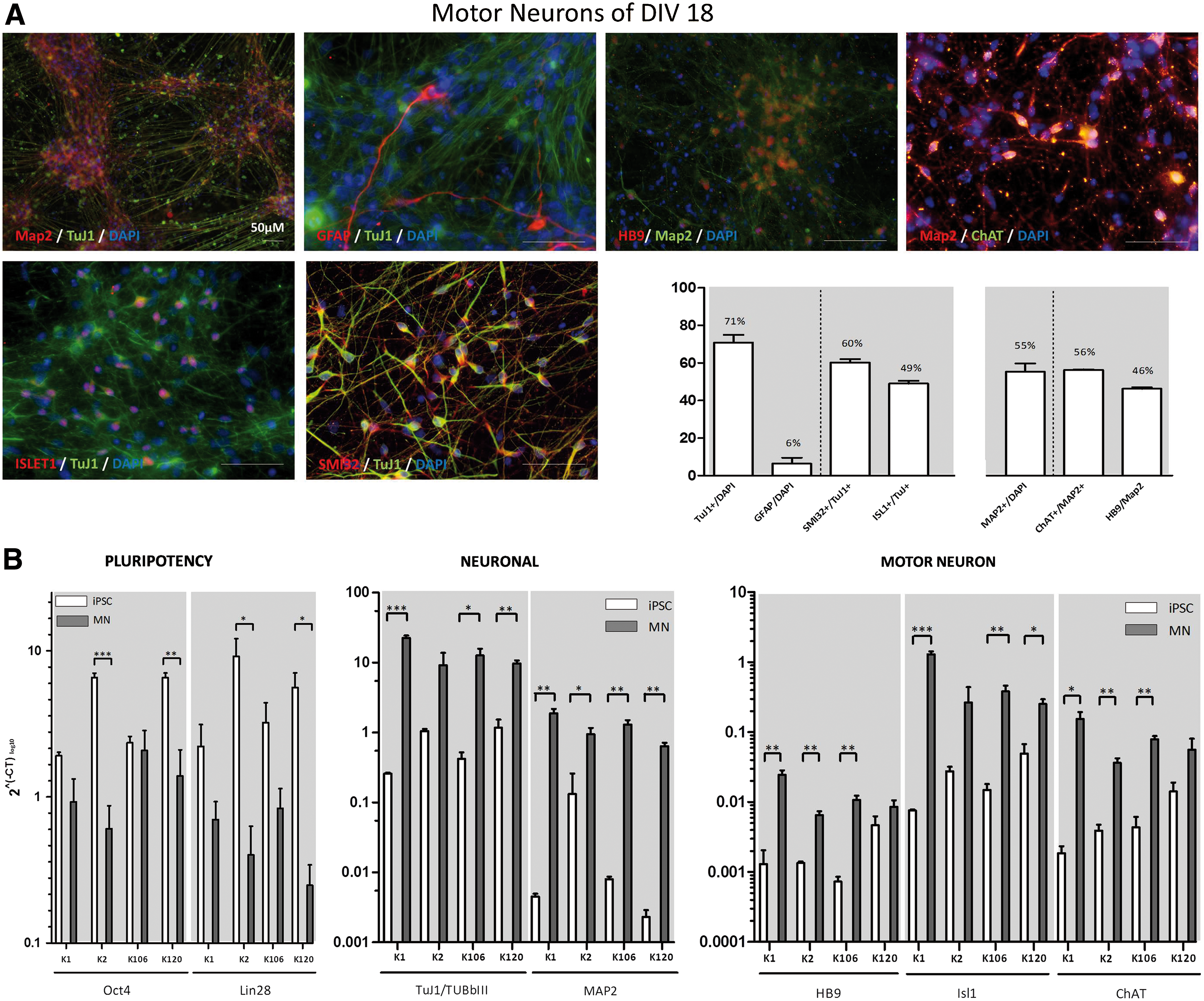
Differentiation of MNs.
Functional maturation in vitro
Having shown the expression of MN markers of the differentiated cells, we were interested in their functional properties. For this purpose we investigated the presence of ligand-gated ion channels by calcium imaging at DIV25 and of voltage-gated ion channels by patch-clamp recordings in the whole-cell mode at DIV30. We included only multipolar neurons with large cell bodies as indicated by cell capacitances of ∼20 pF in patch-clamp recordings.
In calcium imaging experiments we recorded the increase of intracellular Ca2+ concentration upon bath application of selected neurotransmitters and receptor agonists. Numerous Fura-2-loaded cells (39%–83%) (Fig. 4B) showed typical neuronal responses with rapid increases in intracellular Ca2+ levels (Fig. 4) evoked by stepwise application of GABA (100 μM), acetylcholine (100 μM), glutamate (50 μM), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic (AMPA; 100 μM), N-methyl-
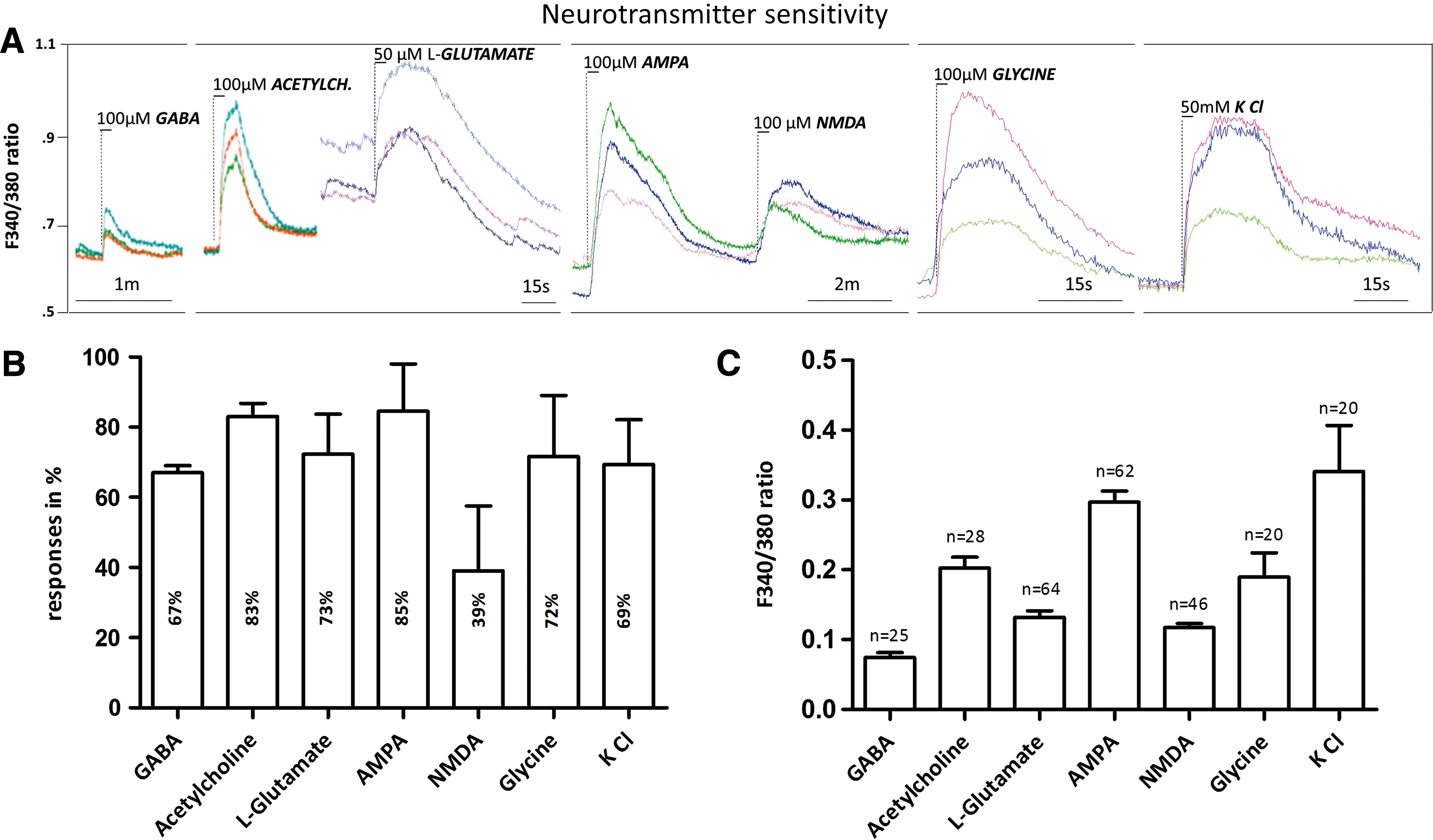
Effects of selected neurotransmitters on intracellular calcium concentrations at DIV25 MNs.
After 4 weeks of total differentiation, patch-clamp recordings of the hCBiPSC-derived MNs (n=40) had mean membrane potentials of −37.3±1.4 mV as well as membrane capacitances of 20.2±2.6 pF, indicating large cell bodies. Upon stepwise depolarization, all cells showed the emergence of voltage-dependent sodium inward and potassium outward currents (Fig. 5A). The application of the antagonists TEA (10 mM) and TTX (1 μM) effectively inhibited potassium and sodium channel currents, respectively (Fig. 5A). Further, APs were elicited in 80% of the measured cells by current injections in the current-clamp mode (Fig. 5B). Another critical feature of functional MNs is the occurrence of spontaneous APs and synaptic activity in the form of postsynaptic miniature events (Fig. 6). After 2 more weeks under maturation conditions at DIV40–50, roughly half of the cells (48%) presented postsynaptic miniature currents (>5 pA) that could be blocked by the coapplication of the acetylcholine receptor antagonists mecamylamine (20 μM) and atropine (10 μM) (Fig. 6A) [23,24]. Large spontaneous APs (>25 mV) and their respective inward currents (>100 pA) were abundant in 13%–20% of the DIV40–50 cells. APs were recorded during current-clamp and their evoked inward currents in voltage-clamp mode with frequencies of 0.41±0.18 Hz and 0.34±0.25 Hz, respectively (Fig. 6B). Taken together, our differentiated cells showed spontaneous activity that can be inhibited by acetylcholine receptor antagonists, which is typical for mature spinal MNs [3,25]. We summarized the electrophysiological membrane properties of the investigated MNs in Table 1.
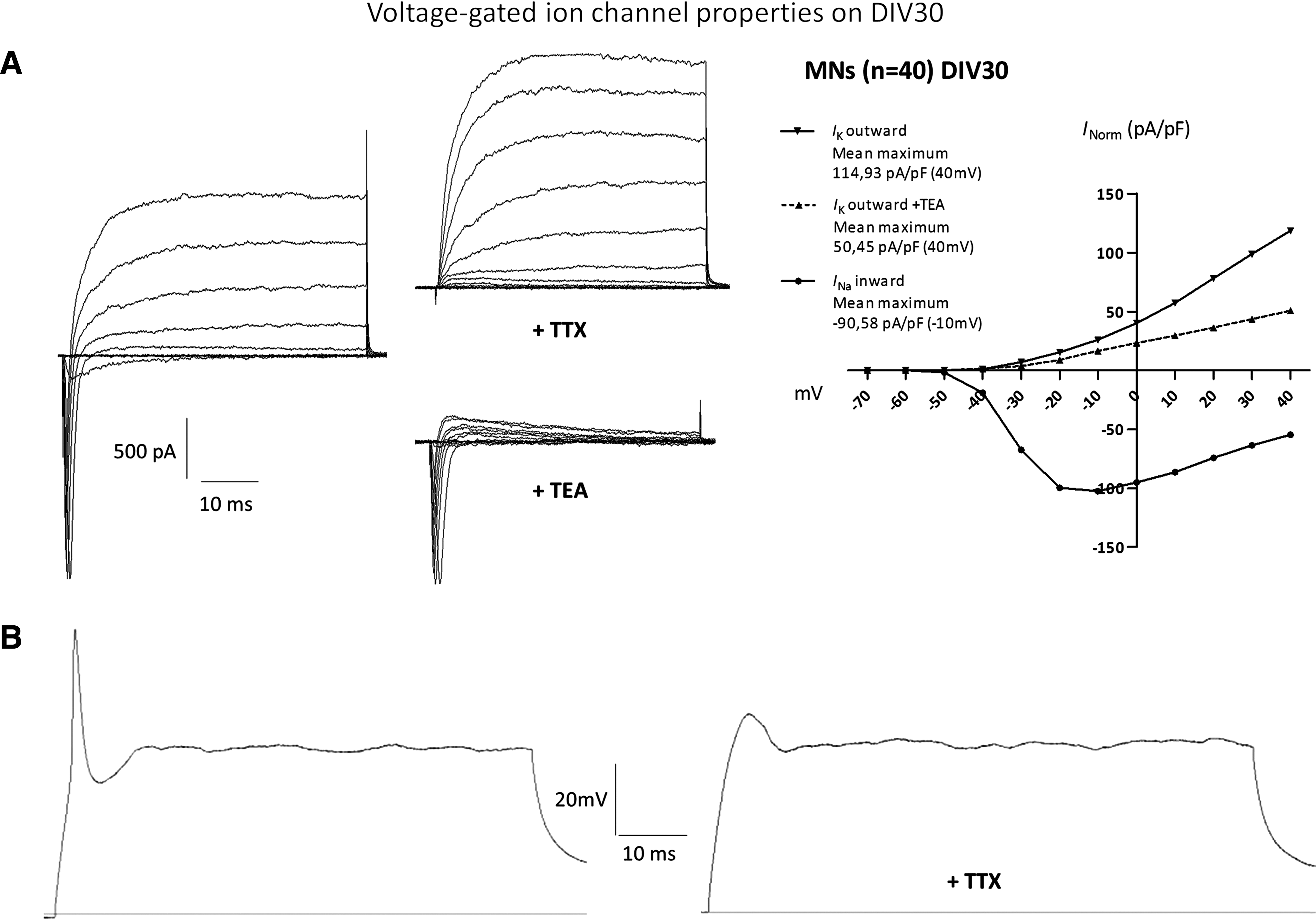
MNs differentiated from hCB-derived NPCs gain electrophysiological maturity at DIV30.

Spontaneous activity of MNs reflects maturation in vitro at DIV40–50.
Whole-cell patch clamp recordings of voltage-gated ion channel, passive membrane, and action potential (AP) capacities. AP amplitude was taken from the difference between spike onset and peak. The afterhyperpolarization (AHP) value reflects the time that lied between peak and plateau during current injection. The AP duration measurement was taken from half amplitude. Time to peak AHP was measured from spike onset.
Discussion
iPSC-derived MNs have become an important tool for the in vitro modeling of neurodegenerative diseases and a potential source for cell replacement therapies. In the present study, we demonstrate for the first time the potential of hCBiPSCs to be differentiated into functional MNs. We further present a rapid and efficient differentiation method. hCB-derived MNs are of particular interest to the field of regenerative medicine due to their juvenescent offspring.
As expected, we were able to differentiate hCBiPSCs via smNPCs into functional MNs within a substantially shortened period. After 18–20 DIV, the differentiated cells allow the observation of MN cell surface markers and increased expression of MN genes that usually are observed after 60+ DIV [3,5 –7,10,11,17]. The rapid differentiation and maturation time was made possible by starting from expandable smNPCs that homogeneously express the neural markers Pax6, Sox1, Vimentin, and Nestin. Starting MN differentiation from these NPCs circumvented classical differentiation protocols [5,10,18,26,27], which all rely on initial embryoid body formation during the first 2 weeks of the neural induction process. After <3 weeks of MN specification at DIV18, we can report the presence of 71% TuJ1+ and 55% Map2+ cells. With similar efficiency but longer duration, Lee et al. [3] observed 65% of TuJ1+ cells and Egawa et al. [5] ∼50%–70% of Map2+ cells after DIV >60. At the same time point these studies report more specific MN markers, such as HB9, in 43% [3], ∼50% [6], and 10%–23% [17] of counted neurons compared with 46% in our study. SMI32 expression was enhanced in 60% of our neurons in comparison to 15%–35% [5] and ∼20% [6] in the total cell number of previous studies. Despite the relatively short maturation period, we can report differentiation efficiency comparable to previous studies that differentiated MNs from iPSCs using conventional time-consuming protocols.
In line with the results from immunocytochemistry, qPCR revealed increases in typical neuronal (TuJ1 and Map2) and MN (Islet1, SMI32, and ChAT) markers in differentiated MNs from all four clones. We further observed the expected downregulation of the pluripotency markers Oct4 and Lin28 with still detectable construct-dependent [12] expression levels. As in any virally reprogrammed iPSC line, we cannot exclude persistent activity of the delivered factors even in functional MNs. To further reduce the risk of teratoma formation following transplantation [28 –31], nonintegral methods, such as the introduction of proteins [32], micro-RNA [33], or nonintegrating viruses [34], should be applied in future reprogramming of hCB cells. Nonetheless, it is important to note that in spite of the potential persisting activity of the integrated reprogramming factors, there was no interference with the differentiation potential of either hCBiPSCs or smNPCs.
To demonstrate that the differentiated cells also show basic electrophysiological properties typical for mature neurons, we performed calcium imaging analysis and patch-clamp recordings. As reported by Son et al. [35] who detected sensitivity of directly induced MNs to the application of GABA (73%, n=11), glycine (44%, n=9), and kainate (80%, n=15) during calcium imaging recordings, we observed intracellular Ca2+ concentration elevations upon the application of the same neurotransmitters, indicating excitatory GABAergic and glycinergic effects [36,37]. We also recorded responses from iPSC-derived MNs to the application of AMPA, NMDA, and acetylcholine, further manifesting their suitability for future drug-screening studies of therapeutic compounds working on either transmitter system. In whole-cell recordings the investigated neurons at DIV30 showed a depolarized mean resting membrane potential of −37.3±1.4 mV, which is in line with membrane potentials (−30 to −35 mV) recorded from human iPSC-derived neurons [14,18], human fetal midbrain-derived progenitor cells after 3 weeks of differentiation [37,38], and directly induced neurons derived from human fibroblasts with a membrane potential of −39.8±4.3 mV after 13 days of differentiation [39]. The presence of voltage-gated Na+ and K+ currents upon stepwise depolarization revealed current amplitudes of I Na=−98.14 pA/pF and I K=114.9 pA/pF after normalization for cell membrane capacitance. Observations from electrophysiological studies that recorded human iPSC-derived neurons ranged from I Na −70 to I Na −195 pA/pF and I K 42 to 208 pA/pF [14,18,40]. The observed variation between studies can be attributed to different maturation grades [15] but also to different neuronal lineages that were studied which traditionally differ in cell size. Whereas Hu et al. [17] observed single-peak APs in nearly all their cells, a vast majority (80%) of our neurons at DIV30 responded with APs upon current injection. This is similar to what has been reported in different studies that recorded APs in 75% [40], 50%–100% [15], and 40% [18] of measured neurons. In our study we did not observe large, multiple APs at DIV30. These repetitive APs typically do occur in mature neurons [41] and those differentiated from ESCs/iPSCs [3,7,11,27]. The reason for the reported discrepancy assumingly lied in an early maturation stage of our DIV30 cells. In line with this assumption Hu et al. [17] report having seen single APs after DIV40 and multiple APs after DIV60. Based upon these findings we additionally investigated the firing of APs in more mature cells and recorded multiple spontaneous APs at DIV40–50. As further indication of a more mature phenotype, we observed miniature postsynaptic events resulting from spontaneous vesicle releases in 48% of measured cells, which is in line with Johnson et al. [15] who reported postsynaptic currents in 50% of measured cells. Synapse formation therefore was not impaired by the reported low density of GFAP+ cells that are known to contribute to synapse formation in MN cultures [42]. As expected, the observed postsynaptic activity was reduced by the application of the nicotinic antagonist Mecamylamine, suggesting a cholinergic synaptic input. The presence of nicotinic acetylcholine receptors on functional spinal MNs has been reported earlier [25]. To our surprise, the muscarinic antagonist atropine caused a similar decrease in the postsynaptic events. This could be either explained by the rarely documented coexistence of muscarinic receptors on MNs [43] or rather by the competitive effects of atropine on nicotinic receptor subtypes [23]. Conclusively, following the rapid induction of neuronal morphology and MN identity at DIV18, our differentiated cells responded to applied neurotransmitters at DIV25, showed basic electrophysiological characteristics at DIV30, and acquired full neuronal maturation at DIV40–50.
In summary, we have established a fast protocol for the differentiation of hCBiPSCs into functional MNs. Starting with an expandable population of smNPCs, mature MNs expressing functional voltage- and ligand-gated ion channels arose within 30 DIV. Regarding the time line of the differentiation process and the convenient handling of the smNPCs, we can offer a novel platform for human iPSC-based in vitro modeling of MN disorders. Derived from hCB, these MNs are an attractive source for disease modeling and drug screening studies due to their juvenescence and easy accessibility.
Footnotes
Acknowledgment
The authors are indebted to Carola Kassebaum and Andreas Niesel for their excellent technical support.
Author Disclosure Statement
No competing financial interests exist.