
Abstract
The prospective isolation of defined contractile human pluripotent stem cell (hPSC)–derived cardiomyocytes is advantageous for regenerative medicine and drug screening applications. Currently, enrichment of cardiomyocyte populations from such cultures can be achieved by combinations of cell surface markers or the labor-intensive genetic modification of cardiac developmental genes, such as NKX2.5 or MYH6, with fluorescent reporters. To create a facile, portable method for the isolation of contractile cardiomyocytes from cardiomyogenic hPSC cultures, we employed a highly conserved cardiac enhancer sequence in the SLC8A1 (NCX1) gene to generate a lentivirally deliverable, antibiotic-selectable NCX1cp-EGFP reporter. We show that human embryonic stem cells (and induced pluripotent stem cells) transduced with the NCX1cp-EGFP reporter cassette exhibit enhanced green fluorescent protein (EGFP) expression in cardiac progenitors from 5 days into the directed cardiac hPSC differentiation protocol, with all reporter-positive cells transitioning to spontaneously contracting foci 3 days later. In subsequent stages of cardiomyocyte maturation, NCX1cp-EGFP expression was exclusively limited to contractile cells expressing high levels of cardiac troponin T (CTNT), MLC2a/v, and α-actinin proteins, and was not present in CD90/THY1+ cardiac stromal cells or CD31/PECAM+ endothelial cells. Flow-assisted cytometrically sorted EGFP
Introduction
H
Despite the development of increasingly efficient differentiation protocols, the final population inevitably contains a mixture of cardiomyocytes, smooth muscle cells, stromal cells, and endothelial cells [1,3,5,6,13,24 –27]. Isolation of pure functional cardiomyocytes and their committed precursors from heterogeneous populations is therefore of significant interest for further in vitro or in vivo applications and cardiac regeneration [28 –30]. To address this need, methods for flow-assisted cytometric isolation of cardiomyocytes and their precursors based on cell surface markers, such as SIRPα and HCN4, have been developed [31,32]. These protocols, however, require labeling and sorting steps, use cell surface markers that are also expressed in noncardiac cell types (eg, SIRPα) [31], and generate cardiomyocyte-enriched cultures that still contain low amounts of endothelial and stromal cells.
Alternatively, cardiomyocyte-specific fluorescent reporters can be introduced into pluripotent stem cell lines through homologous recombination. While the genetic tagging of cardiac developmental genes, such as NKX2.5, or usage of the promoter elements of the late cardiomyocyte structural protein genes, such as MYH6, MYL2, TNNI2, and TNNI3 [33,34], has created useful tools for identifying and characterizing cardiac differentiation stages in human and mouse pluripotent stem cells [35,36], their utility is limited by (1) a very limited number of hPSC lines that have been subjected to this technically challenging process, and (2) their limited accuracy, dictated by the temporal and tissue-specific expression of the gene. Due to the increasing number of human induced pluripotent stem cell (iPSC) lines that are generated from patients with cardiac and other genetic diseases, there is a need for a simple, portable technology that allows the isolation of pure functional cardiomyocytes from cardiomyogenic hPSC cultures.
The NCX1(SLC8A1) Ca2+/Na+ antiporter protein functionally contributes to Ca2+ handling of contractile cardiomyocytes [34,37,38] and is highly expressed in embryonic and fetal cardiomyocytes. Although NCX1 is widely expressed, cardiac-specific expression of NCX1 is thought to be directed by a highly conserved cardiac enhancer motif in its promoter, as suggested by the observation that the feline NCX cardiac enhancer motif is exclusively active in neonatal rat ventricular cardiomyocytes [35]. We therefore hypothesized that the human cardiac-specific enhancer of the NCX1/SLC8A1 gene would be an attractive candidate for selective marking of functional hPSC-derived cardiomyocytes.
Herein we report that lentiviral transduction of human embryonic stem cell (hESC) lines (H9 and HES3) and human iPSC lines, C32 and C11 [39], with a puromycin-selectable NCX1cp–enhanced green fluorescent protein (EGFP) reporter, based on the human upstream-most SLC8A1/NCX1 promoter and noncoding exon (NCX1cp), allows for the rapid establishment of a cardiomyocyte reporter line. We demonstrate that, independently of the cardiomyocyte differentiation protocol used, EGFP expression in every NCX1cp-hPSC reporter line identifies an early cardiomyocyte population that homogeneously differentiates into contractile cardiomyocytes, exhibiting high levels of cardiomyocyte-specific gene and protein expression, and cardiac electrophysiological signatures. The NCX1cp-EGFP+ fraction thus allows for the isolation of a pure, functional cardiomyocyte population that can be utilized for tissue engineering or pharmacological applications.
Materials and Methods
hPSC culture
Human embryonic (H9 and HES3) and induced-pluripotent (C11 and C32) stem cell lines were maintained as previously reported [39,40]. Prior to differentiation, hPSC lines were maintained in feeder-free conditions on Matrigel (BD/Life Sciences)–coated plates in mTeSR1 medium (Stem Cell Technologies) or MEF-conditioned medium supplemented with bFGF (16 ng/mL for hES and 80 ng/mL for iPS) and 100 μM β-mercaptoethanol (both from Life Sciences/Invitrogen).
Generation of the lentiviral reporter constructs
The upstream cardiac-specific promoter of the SLC8A1 gene (located at coordinates chr2:40739418–40741429 in GRCh37/hg19) was amplified from a BAC clone RP11-188E4 (supplied by the Australian Genome Research Facility, Melbourne). The promoter fragment was excised from the cloning vector using PmeI and AscI restriction endonuclease sites introduced with the primers used for its amplification, and cloned into EcoRV- and AscI-digested pRRLSIN.cPPT.PGK-EGFP.WPRE (Addgene plasmid No. 12252) lentiviral backbone for efficient delivery into primary rodent heart-derived cells (for functional testing), thus creating the pRRL-NCX1cp-EGFP-WPRE lentiviral vector. After successful verification of the activity and specificity of the promoter, the NCX1cp-EGFP-WPRE cassette was excised using ClaI and SacII restriction endonucleases and cloned into the digested, with same enzymes, pLenti6/V5-His backbone (LifeTechnologies/Invitrogen), resulting in a Blasticidin-selectable pL6-NCX1cp-EGFP-WPRE lentiviral vector.
Generation of stable hPSC reporter lines
Because transgene delivery via transduction or transfection of hPSCs can vary substantially depending on culture method and viral titers, the presence of a selectable cassette (driven by a ubiquitously expressed promoter) is essential for the rapid and reliable generation of stable, clonally derived pools of human pluripotent cells. The cassette consisting of the NCX1 cardiac promoters EGFP and WPRE was thus recloned into a backbone of a selectable and transcriptionally neutral (ie, devoid of extraneous strong transcriptional enhancer elements) vector of the pLenti6 family (Invitrogen/Life Sciences), resulting in generation of the final pL6-NCX1cp-EGFP-WPRE-Blastr lentiviral vector (see schematic in Fig. 1A).
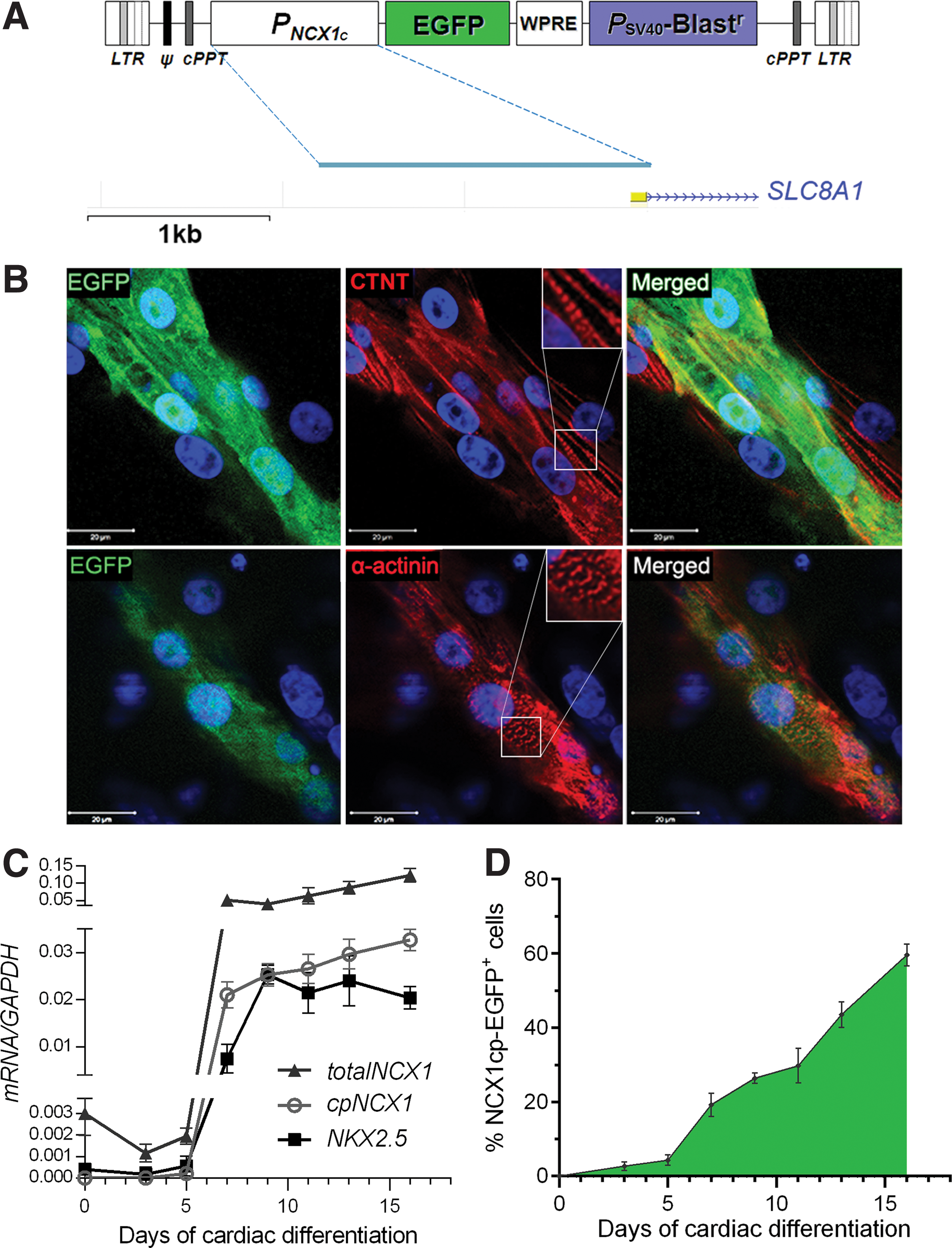
Design and characterization of the lentiviral NCX1cp–enhanced green fluorescent protein (EGFP) reporter.
Human ES (H9 and Hes3) and nonviral, integration-free iPSC lines (C32 and C11) [39] were transduced with the pL6-NCX1cp-EGFP-WPRE-Blastr lentiviral particles, and Blasticidin selection was applied 5 days after transduction in a ramping-up pattern (1 μg/mL on days 5–8 up to 2.5 μg/mL from day 10). Both pools and single-cell-derived clones were generated from two hES and two iPS lines, and at least six individual Blasticidin-resistant clones for each hPSC line were isolated, expanded, and cryopreserved. After subjecting each clone to cardiomyocyte differentiation, clones with lentiviral transgene integration site(s) favorable to cardiac-specific expression were selected. This approach allowed us to rapidly establish NCX1cp-EGFP reporter lines from both the hESC and iPSC lines outlined earlier.
HPSC cardiac differentiation
Human embryonic and iPSC lines were differentiated following two protocols. First, following as previously described by Hudson et al. [4]. Briefly, bulk cultures of pluripotent hES and iPS cells were differentiated toward mesodermal fates by culture for 3 days in RPMI medium with B27 supplement (Life Sciences/Invitrogen) and Activin A (20 ng/mL) and BMP4 (6 ng/mL). Next, cells were grown in RPMI/B27 medium supplemented with a WNT signaling antagonist IWP-4 for up to 13 days (a detailed protocol is described in the Supplementary Data; Supplementary Data are available online at
Immunofluorescence and confocal imaging
HPSC cultures were fixed with RostiFix 4% for 10 min at room temperature. Following that samples were blocked/permeabilized with 10% goat serum and 0.5% Triton-X100 in PBS for 30 min at room temperature. Samples were incubated with primary antibodies against myosin heavy chain/MYH (clone [3,42]; Abcam), CTNT (RV-C2, DSHB), or α-actinin (A7811; Sigma-Aldrich) for 30 min at 4°C. Secondary antibody labeling was performed at room temperature for 30 min.
Flow-cytometric analysis
Cells were detached and dissociated into single-cell suspension using tryptic or accutase digestion, and each sample was split 1:1 for isotype control and staining with antibodies (details in Supplementary Extended Experimental Procedures). Next, samples were washed twice and incubated with secondary antibody (goat anti-mouse IgG-Alexa Flour 488, 10 μg/mL [Invitrogen] and goat anti-mouse-Alexa Flour 633, 10 μg/mL [Invitrogen]) for 30 min at room temperature. FACS analysis was performed on a CFlow Accuri system and data were analyzed using the CFlow Sampler Software. FACS for isolation of the NCX1cp-EGFP cells was performed using the Influx Cell Sorter (BD Biosciences) at the FACS facility of the Queensland Brain Institute, University of Queensland.
Reverse transcription–quantitative polymerase chain reaction
RNA extraction and cDNA synthesis was performed as described previously [4], with use of additional random priming for cDNA synthesis when detection of upstream cardiac NCX1/SLC8A1 promoter was intended. For cDNA amplification, “SsofastEvaGreen supermix” (172–5200; BioRad) was used for the amplification, carried out on the BioRad CFX96 real-time PCR machine. Data analysis was performed as detailed in Supplementary Methods; primers used are listed in the Supplementary Table S1A.
Electrophysiological recordings on cardiomyocytes
Spontaneous action potentials were recorded from isolated individual cells using whole-cell patch-clamp configuration, as described previously [43]. After isolation via FACS, NCX1cp-EGFP-reporter-positive cells were plated on the Matrigel (Sigma-Aldrich)–coated glass or plastic coverslips. The patch-clamp pipette solution contained 120 mM KCl, 1 mM MgCl2, 3 mM ATP, 10 mM EGTA, and 10 mM HEPES (pH 7.4). The bath recording solution contained 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). Recordings were performed with whole-cell capacitance compensation and changes in membrane potential were measured under current-clamp condition (Axopatch 200A), digitized (Digidata1200 A-D converter), and recorded on a PC running pClamp8 software (Axon, Burlingame). All membrane potentials were corrected for liquid junction potential (−5.8 mV) determined via pClamp software, and the recordings were performed at room temperature (similar to methods described in Refs. [44 –46]).
Transcription factor binding site conservation analysis
Analysis of the promoter/enhancer overall sequence and transcription factor binding site conservation was performed using ECR browser (ecrbrowser.dcode.org), MULAN multiple-sequence alignment, and conservation analysis software (mulan.dcode.org), and transcription factor binding site conservation was analyzed using the CONREAL (Conserved Regulatory Elements anchored Alignment) tool (conreal.knaw.nl) and TRANSFAC and MATCH resources (available through
Statistical analysis
All data are presented as a mean±standard error of the mean (SEM). To determine statistical differences, two-tailed Student's t-tests were used with P<0.05 deemed as significant. Standard designation of confidence levels was used in figure labeling: * for P<0.05, ** for P<0.01, and *** for P<0.001.
Results
Identification, cloning, and validation of the human cardiac-specific promoter of the SLC8A1 gene
Based on our analysis of the features and evolutionary conservation of the neighborhood of the first cardiac-specific exon of SLC8A1/NCX1 gene, we identified a high degree of conservation in the proximal promoter, with 100% cross-species identity of >80% of bases in a
NCX1cp-EGFP expression marks functional early cardiac cells in heterogeneous populations derived from human stem pluripotent cells
We next subjected the H9, HES3, C32, and C11 NCX1cp-EGFP-transgenic hESC/iPSC lines to two different established cardiomyocyte differentiation protocols, using methods based on BMP/activin [4] and small-molecule modulators of Wnt signaling [3,41]. Initial NCX1cp-EGFP expression was observed from day 7 of differentiation. From days 11–12, EGFP intensity and the number of cells expressing EGFP increased (Fig. 1D), followed by initiation of beating at day 12 (Supplementary Movie S1). In all cultures, EGFP expression preceded contractions of individual cells or isolated foci by 24–48 h. By day 16 of differentiation, all beating clusters expressed EGFP (Supplementary Movies S1–S4), and all EGFP-expressing cells were beating synchronously (Supplementary Movies S1–S5). With both protocols, NCX1cp-EGFP expression was observed in cardiomyocytes derived from all four hPSC lines tested (two hES and two iPS), at levels correlating with the efficiency of cardiomyocyte differentiation.
While the SLC8A1/NCX1 protein itself is widely expressed and implicated in many important physiological processes, the upstream-most cardiac-specific promoter appears to be highly cardiomyocyte specific [47,48]. Analysis of the transcription from this promoter, as well as the profiles of induction of expression of genes associated with various stages of cardiac differentiation, revealed that the basal level of cardiac promoter expression is below detection limits until the day 5 of cardiac differentiation in hPSCs, while NKX2.5 and total SLC8A1/NCX1 transcripts are detectable from earliest time points (eg, day 0, Fig. 1C). Similarly, expression of the EGFP reporter is first seen from day 5, and reaches its maximum at day 16 (Fig. 1D) and precedes the expression of cardiac troponin T (TNNI2) that commences at day 7 and then steadily increases with differentiation (Supplementary Fig. S1C). Activity of the NKX2.5 promoter, on the other hand, peaked at day 9 without showing any further increase (Fig. 1C), while HCN4 was significantly upregulated between days 5 and 7 (Supplementary Fig. S1C).
To further define the nature of the subpopulation marked by EGFP expression, immunofluorescent staining for the late cardiac structural marker CTNT was performed at day 16 (Fig. 1 and Supplementary Fig. S1). As expected, beating foci expressed CTNT, which was colocalized with EGFP expression (Fig. 1B and Supplementary Fig. S1B and Supplementary Movies S1–S4). Remixing of the reporter-positive and -negative populations was performed to ensure presence of noncardiomyocytic cells and illustrate the cardiomyocyte specificity of the reporter (bottom panel in Fig. 2). The majority of EGFP

Colocalization of NCX1cp-EGFP expression with cardiac cell markers. Immunostaining for cardiomyocyte and fibroblast markers in replated, mixed, flow-cytometrically sorted, H9 NCX1cp-EGFP-negative and - positive populations at day 30 of cardiac differentiation. Analysis was performed at 2 days (top four rows) or 8 days (bottom row) after replating. Reporter expression correlates well with cardiomyocyte-specific proteins of the contractile complex (CTNT and α-actinin), marker of the early first heart field (HCN4), and pan-cardiac transcription factor NKX2.5 (note a number of NKX2.5+; NCX1cp-EGFP− cells). No coexpression of NCX1cp-EGFP and cardiac fibroblast marker THY1 could be detected in cultures of mixed, replated, EGFP-negative and -positive fractions (bottom row). A representative experiment of three is shown. Similar results were obtained with NCX1cp-EGFP-tagged HES3 and hiPS cell lines. Scale bar=20 μm. Color images available online at
To quantify cardiomyocyte induction efficiency and reporter specificity in our differentiation experiments, the distribution of CTNT and NCX1cp-EGFP expression was assessed by flow-cytometric analysis using costaining with the cardiac marker CTNT, the stromal cell marker THY1/CD90, and the endothelial marker CD31/PECAM (Fig. 3A, C, D). Flow-cytometric analyses performed at day 16 showed that all NCX1cp-EGFP+ cells express CTNT with a total percentage of double-positive cells of 43%±4.4% (n=3, mean±SD) (Fig. 3A) and that the NCX1cp-EGFP− fraction contained all of the stromal and endothelial fraction, with a total percentage of CD31/PECAM endothelial-like cells of 2.9%±1.3% (Fig. 3C), and CD90/THY1+cardiac fibroblast-like cells of 18.3%±4.6% (n=3, mean±SD (Fig. 3D). These observations, in combination with immunocytochemical evidence (Figs. 1 and 2), confirm that contraction-capable, CTNT-positive cardiomyocytes can be readily identified and isolated from hPSC cultures by NCX1/SLC8A1 cardiac-specific promoter-driven EGFP expression. Positional variegation of the lentiviral reporter in the pooled transgenic H9 population used in these experiments and its potential differentiation-associated silencing are likely contributors to emergence of the small fraction of CTNT-positive cells that no longer express the reporter (Fig. 3A, lower-right quadrant of the FACS plot).
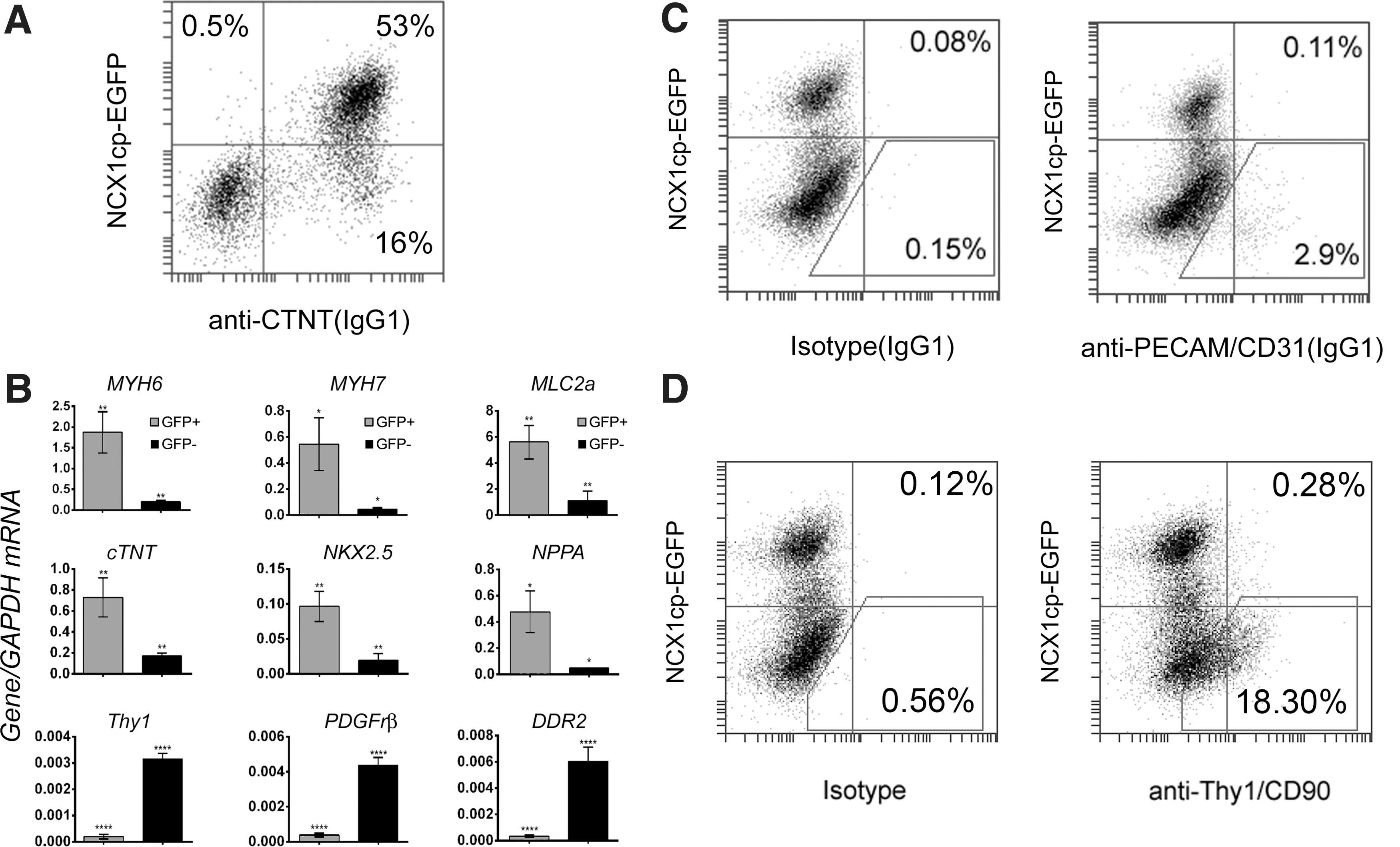
Characterization of the NCX1cp-EGFP-reporter-expressing populations in cardiac differentiation.
To further confirm cardiomyocyte commitment of the NCX1cp-EGFP-reporter-expressing population, EGFP-expression-based flow-cytometric cell sorting was performed at day 16, followed by analysis of gene expression at the transcript level. Separation of both populations was easily achieved due to the high intensity of the signal from the NCX1cp-EGFP
Electrophysiological signatures of NCX1cp-EGFP-reporter-expressing cardiomyocytes indicate presence of multiple cardiomyocyte phenotypes
To further exemplify the functionality and identity of the NCX1cp-EGFP+ cardiomyocytes, we FAC sorted EGFP+ cells from day-32 cardiomyocytes, and recorded action potentials using the whole-cell patch-clamp configuration. Three types of action potential signatures were observed in sorted NCX1cp-EGFP+ cells (Fig. 4A), namely, ventricular-like type, with a prolonged depolarization plateau phase, in agreement with the robust expression of MLC2v (Fig. 4B), as well as atrial-like and nodal-like types, displaying characteristic slower-onset (for atrial-like) and shorter-duration (for both types) depolarization phases (electrophysiological parameters listed in Table 1). These data suggest that the reporter allows for isolation of the three prevalent contractile cell types found in normal heart [49].
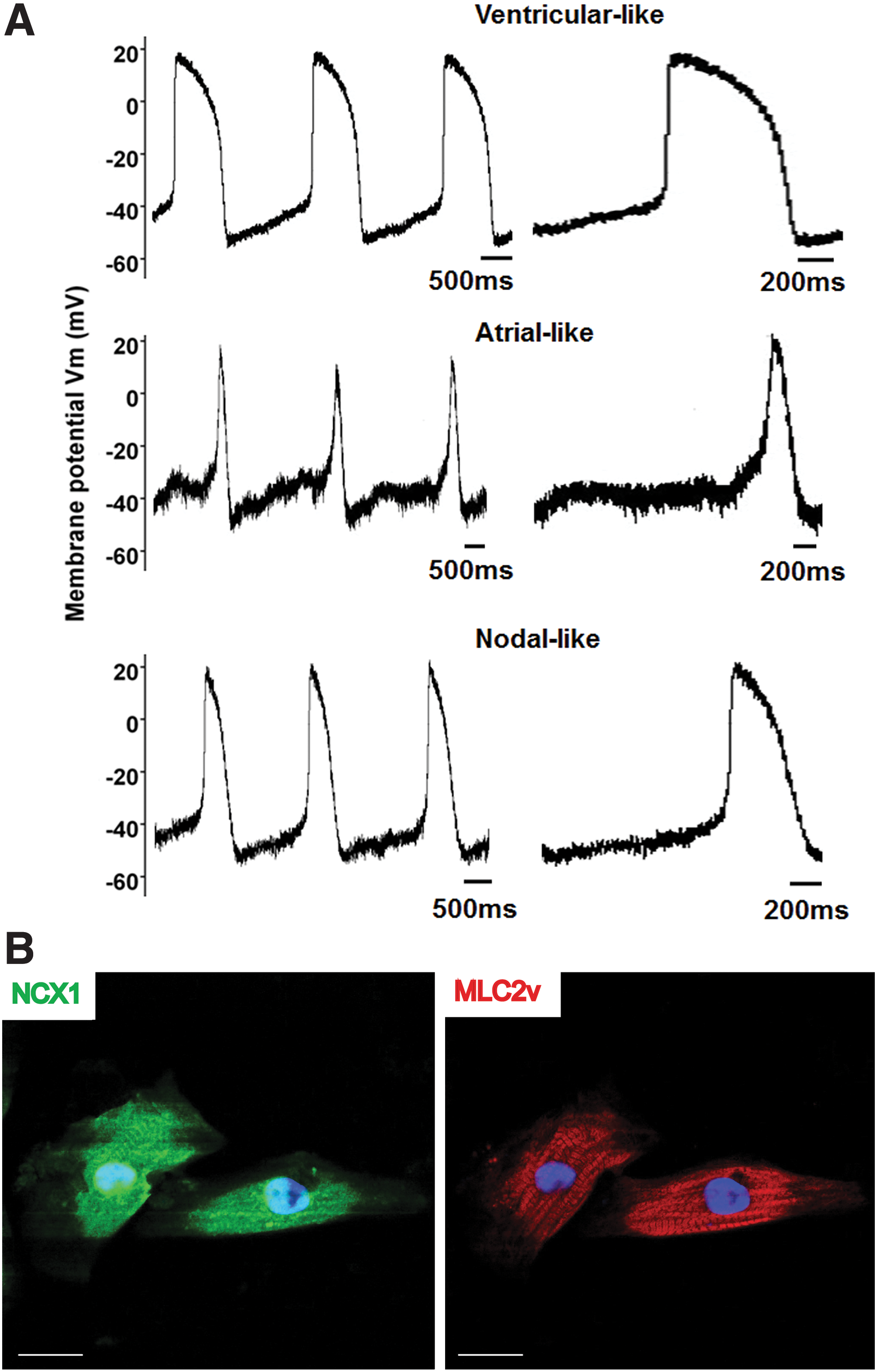
NCX1cp-EGFP+ cardiomyocytes display action potentials characteristic of the three major contractile cardiomyocyte types.
EGFP, enhanced green fluorescent protein; MDP, maximum diastolic potential; RP, resting potential; APD, action potential duration.
Discussion
This study exemplifies the utility of a portable cardiomyocyte-specific reporter that allows for the isolation of a committed functional cardiomyocyte population from human ES and iPS cell lines before and after commencement of beating activity. Using four cell lines and two cardiac induction protocols, the appearance of EGFP
We demonstrate that the NCX1cp-EGFP reporter facilitates the identification and isolation of functional cells from heterogeneous populations, even at early stages of the induction protocol, consistent with the observation that NCX1 is known to contribute to mediation of Ca2+ transients during the early stages of in vivo differentiation of hPSC-derived cardiomyocytes [34,49]. The reporter further permits monitoring of cardiomyocyte induction efficiency in real time, providing a useful tool to rank cardiac differentiation protocols. The breadth of the reporter expression (in all 3 major cardiomyocyte types: ventricular, atrial, and pacemaker/nodal like) is in keeping with the observation that in transgenic mice carrying the rat NCX1-EGFP promoter reporter, expression of EGFP is initially pan-cardiac and thereafter becomes progressively restricted to the pacemaker/nodal cardiomyocytes during late gestation and postparturition [50]. It remains to be determined whether the NCX reporter can be used for this purpose to specifically isolate human pacemaker cells from late cardiac differentiation cultures.
While many characteristics of the EGFP+ fraction at day 16 resembled developmentally immature functional cardiomyocytes described previously [4,51], this population already expresses mature cardiomyocyte markers, such as MYH6, MYH7, and NPPA (Fig. 3B) and MLC2v, at day 32 (Fig. 4B) (when compared with the starting cells and EGFP− population). Importantly, the EGFP− population expressed high levels of the cardiac stromal cell markers THY1/CD90, DDR2, and Vimentin, consistent with flow cytometry and immunofluorescence analyses for THY1/CD90 on mixed populations, and contained the entirety of the endothelial-like CD31/PECAM+ cells [52], another common contaminant in hESC-derived, cardiac-differentiated populations. In this respect, the NCX1cp-EGFP reporter appears to be at least as specific to cardiomyocytes as the NKX2.5-knock-in hESC lines. Indeed, while NKX2.5 is predominantly cardiomyocyte specific, it is known to also mark stromal cells in hESC-derived cardiomyogenic cultures [36,53]. We show that NCX1cp-EGFP does not mark the stromal compartment and that all NCX1cp-EGFP-positive cells express CTNT (Figs. 2 and 3) and NKX2.5 (Fig. 2 and Supplementary Fig. S3). While for some applications the presence of other cardiac cell types might be desirable, the NCX1cp-EGFP reporter allows for experimentation on a more defined population, and permits investigations into the interactions of the cardiomyocyte population with other (eg, stromal) cell types.
The current lentiviral delivery of the NCX1cp-EGFP reporter does have the drawback that it has the potential to lead to insertional mutagenesis. Therefore, individual clones should be assessed for appropriate upregulation of EGFP following cardiomyogenic differentiation (eg, through colabeling with CTNT and FACS).
Isolation of cardiomyocytes from cardiomyogenic cultures using cell surface antigens remains an attractive strategy [31,32], despite the fact that such FAC-sorted populations do not yield pure cardiomyocyte populations. As such, we envisage that NCX1cp-EGFP-reporter-sorted cardiomyocyte populations could indeed be used to identify and test additional cell surface markers that will further enhance the purity of such cultures.
We conclude that the ability to easily purify NCX1cp-GFP+ cells that display very high propensity to spontaneously contract and progress to long-term-beating cardiomyocytes is both a valuable tool for regenerative medicine approaches, as was recently exemplified by Huang and colleagues [22], and allows for testing of, for instance, cardiotoxic drugs on defined contractile cardiomyocyte populations.
Footnotes
Acknowledgments
The authors would like to thank the StemCore facility at the UQ AIBN for the provision of hES cell line (H9) and logistical support in karyotyping analyses. We would also like to acknowledge the Australian Stem Cell Centre and the ARC Excellence Centre “Stem Cells Australia” for the funding support.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.