
Abstract
The systematic localization of chronic lymphocytic leukemia (CLL) B-cells in the bone marrow (BM), together with the ex vivo protective effect of stromal cells on their spontaneous apoptosis, both indicate a specific role of the BM microenvironment. In vivo, the impact of CLL cells on mesenchymal stromal cells (MSCs) remains a source of debate. Here, we quantified and expanded colony forming unit-fibroblasts (CFU-Fs) from CLL-BM under standard conditions, analyzed the expression of selected genes, and studied secretion profiles. We observed failing of CLL-BM cultures in standard conditions (45.5% vs. <0.1%), and even after adding basic fibroblast growth factor (bFGF), there were fewer CFU-F than from normal BM (1.3 vs. 40/106 cells respectively; P<0.01). Furthermore, their polygonal aspect and low proliferative capacity, together with the expression of 384 selected genes and a secreted set of molecules related to senescence-associated secretory phenotype indicated a state of senescence, further confirmed by the higher proportion of senescence-associated β-galactosidase (SA-βGAL)-positive cells and p16INK4a overexpression. In our hands, hypoxic conditions (5% O2) did not rescue CFU-Fs. Given the role of MSC in BM tissue organization, we studied hematons that are generally considered to be elementary BM units. These structures were rare or had even disappeared completely. When hematons were present, we systematically observed nodular B-CLL cell invasion only. These data confirm that the B-CLL clone has a marked impact on MSC and disrupts BM organization in vivo, raising new questions about in vivo pathophysiology.
Introduction
B
Although the role of secondary lymphoid organ location versus BM location in the progression of CLL remains a source of debate, location of the CLL clone in BM appears to be a crucial step for B-cell survival owing to the fact that (i) the BM is systematically infiltrated; (ii) the infiltration pattern has a prognostic value [1,2]; (iii) residual BM B-CLL cells are considered to be at the origin of relapse [3 –5]; and (iv) a proliferative compartment was observed in the BM [6]. Together, these observations strongly suggest a major role of cell interactions within the BM microenvironment promoting CLL B-cell survival and progression.
B-cell expansion appears to be predominantly due to resistance to apoptosis maintained by external signals from the microenvironment [7]. Thus, CLL B-cells rapidly undergo apoptosis ex vivo, unless they are cocultured with accessory cells [8 –11]. Two of the most informative models of crosstalk between CLL cells and accessory cells are those using CLL blood monocyte-derived cells, known as monocyte-derived nurse-like cells [8,12,13] and those using mesenchymal stem cells from healthy donors [9,10]. These models helped identify several extrinsic signals involved in B-CLL cell survival and highlighted the possible involvement of stromal cells in chemotherapy resistance. However, the in vivo crosstalk between the counterparts of these accessory cells and the malignant clone may be significantly different.
Recently, several teams have attempted to understand the in vivo relationship between the B-CLL clone and mesenchymal stromal cells (MSCs). Thus, adherent cells derived from CLL bone biopsy explants were expanded as an in vitro model [14 –16].
More recently, two articles have documented the results concerning fresh mesenchymal cells from CLL-BM, but data were somewhat contradictory. The first study reported the rarity of mesenchymal stromal progenitors detected in vitro, related with their dependence on hypoxia [17], while the second study reported significant numbers of mesenchymal progenitors [18].
Aiming to be reproduce in vivo conditions as closely as possible, we started isolating primary colony forming unit-fibroblast (CFU-F) from CLL patients' BM 3 years ago and collected results that confirmed the fact that the B-CLL clone modifies the intrinsic properties of MSC, which become senescent in in vitro culture. However, in our experiments, low concentrations of O2 did not normalize cell behavior. We then evaluated the impact of the B-CLL clone on hematons. These elementary BM units are only partially conserved in the event of nodular infiltration, strongly suggesting a disrupting effect of the malignant cells on BM tissue.
Materials and Methods
Human BM cell source
Normal bone marrow (NBM) cells were obtained from two sources [19]: (i) the femoral head spongy bone (NSB) collected from the femoral head during hip arthroplasty (n=20), (ii) or from the left-over parts of iliac crest BM aspirates collected for cell therapy (n=37; age >50 years). To study hematons, a supplementary series of BM aspirates (n=28) was constituted from the left-over parts of sternal aspirates carried out for cytological analysis results of which were benign or indicated a peripheral etiology. CLL-BM was collected by sternal or iliac crest BM puncture from untreated CLL patients (n=40; Binet stage: 11 A (27.5%), 14 B (35%), and 15 C (37.5%); M/F: 31/9; median age: 63 years; Supplementary Table S1; Supplementary Data are available online at
Spongy bone was fragmented and adherent cells collected after collagenase (0.0125%; Sigma-Aldrich Chimie, Saint-Quentin Fallavier, France) incubation for 10 min at 37°C. Cell suspensions were then filtered on nylon mesh (Falcon; Becton Dickinson, Le Pont de Claix, France) and washed in phosphate buffered saline (PBS)/5% fetal calf serum (FCS).
BM harvesting collected macroscopic coherent tissue aggregates in a cell suspension diluted with blood. When these structures were observed, we counted the number of BM cell aggregates (hematons) per mL of BM; we then recovered the hematons as already reported [20]. Briefly, each BM sample was filtered through 100 μm nylon mesh (Falcon™; BD Biosciences, Le Pont de Claix, France). Retained hematons were washed thrice with PBS 1% bovine serum albumin and were then recovered for immunohistochemistry analysis.
CFU-F isolation and expansion
In a first series of experiments, CFU-F were isolated and amplified using routine in vitro culture techniques validated in a series of 205 NBM as previously described [19]. Briefly, cells were directly plated in primary culture at 2×104 cells/cm2 in flasks (Falcon; Becton Dickinson) in a standard expansion medium containing Iscove's Modified Dulbeco's Medium (Sigma-Aldrich) supplemented with 10% FCS (Biowest, Nuaillé, France), 1% L-Glutamine (Cambrex BioScience, Verviers, Belgium), and 1% Ciprofloxacin (Sigma-Aldrich) and cultured at 37°C with 5% CO2 in humidified air. In the aim of increasing CFU-F assay sensitivity, we seeded 10–16-fold more cells than usual by increasing the flask surface without modifying the seeding density per cm2, to avoid the reported negative effect of high cell seeding density on CFU-F proliferation [21] and to increase the sensitivity of the CFU-F assay (sensitivity: 1 CFU-F/4×107 nucleated cells).
After testing different culture conditions (10% human serum, 10% CLL serum, long-term culture medium, or bFGF; see Results), bFGF (1 ng/mL) was systematically added.
The adherent cells were fed at weekly intervals. Once cells were about 75%–80% confluent, they were treated with 0.25% trypsin-EDTA (Sigma-Aldrich), counted, and transferred to new flasks at 500 cells/cm2 (first passage or P1). In some CLL cases, when cells had difficulty in expanding, they were trypsinated while they were not confluent following 2 weeks culture.
The frequency of primary colony (CFU-F) was determined by microscopic examination at day 10. Hematopoietic BM content was evaluated by plating BM cells in short-term culture assay, as already reported [20].
Flow cytometry
The BM content in the CD45−CD14−/CD73+ mesenchymal progenitor-enriched cell subset was assessed as already described [19]. At least 1×105 viable events were acquired. CD200 and SPARC MSC expression was assessed by flow cytometry at the end of P1. For CD200 detection, MSC were first incubated with PBS/5% AB human serum for 15 min to prevent nonspecific binding, and they were then incubated with antiCD200-PE (Immunotech, Marseille, France), antiCD45-FITC, and antiCD14-FITC (Beckman Coulter, Roissy Charles de Gaulle, France) mAb for 15 min at room temperature. Cells were washed twice in PBS/4% FCS and again in PBS/4% FCS with 1 μg/mL propidium iodide. Acquisition and analysis of events were performed on an FACSCanto™ II cytometer (Becton Dickinson). Cells were gated to make a morphologic dot plot with forward and side light scattering. After gating viable cells, windows were set based on staining with combinations of relevant mAb and irrelevant mAb so that no cell was positive using irrelevant mAb. At least 1×104 viable events were acquired. For SPARC detection, fixation and permeabilization steps were performed before staining by anti-SPARC and anti-IgG1 mouse (FITC; R&D Systems, Lille, France). The CD34+ cell count was determined as recommended by the International Society for Hematotherapy and Graft Engineering (ISHAGE) guidelines.
Lymphocyte immunophenotyping was performed according to routine flow cytometry procedures using anti-CD45, anti-CD5, anti-CD20, and anti-CD38 antibodies (all from Becton Dickinson).
MSC in vitro differentiation
To induce osteogenesis, MSC were cultured in osteogenic medium consisting of Dulbecco's modified Eagle's medium (DMEM) with 10% FCS, 10 mM β-glycerophosphate, 0.1 μM dexamethasone, and 0.05 mM ascorbic acid (all from Sigma-Aldrich) for 3 weeks, renewing the media every 2 to 3 days. To assess osteoblast differentiation, Alizarin Red S [2% (pH 4.1) in ammonium hydroxide] was then used to stain calcium in the mineralized extracellular matrix. To induce adipogenic differentiation, MSC were cultured in DMEM-F12 containing 5% newborn calf serum, 1 μM dexamethasone, 50 μM isobutyl-methylxanthine, and 60 μM indomethacin (all from Sigma-Aldrich) for 2 weeks, renewing the media every 2 to 3 days. Evidence of adipocyte differentiation was shown by Oil Red O (Sigma-Aldrich) staining for lipids. Controls for osteoblast and adipocyte differentiation were conducted in parallel in standard expansion medium. Chondrogenic differentiation was induced by 21-day culture in micropellets. Cells were suspended in 15 mL polypropylene conical tubes [(2.5×105 cells), after centrifugation for 5 min at 600 g. The resulting pellets were cultured in 500 μL DMEM supplemented with 0.1 μM dexamethasone, 0.17 mM ascorbate-2 phosphate, 1% insulin-transferrin-sodium selenite supplement, and 10 ng/mL of recombinant TGFβ3 (R&D Systems). Media were changed every 2 to 3 days. Micromass pellets were then prepared for histology in paraffin sections. The sections were stained with toluidin blue.
TaqMan® gene expression assays
Expression of 384 genes was compared between five untreated CLL-MSC and four normal MSC using TLDA (Applied Biosystems, Foster City, CA). MSC were lysed directly in the culture flasks and total RNA was isolated using RNeasy Kit (Qiagen, Courtaboeuf, France). First strand cDNA was synthesized from 3 μg of total RNA using the High-Capacity cDNA RT Kit (Applied Biosystems) and real time-quantitative polymerase chain reaction (RT-qPCR) reactions were carried out in Micro Fluidic Cards using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). One hundred nanograms of cDNA combined with 1× TaqMan Universal Master Mix (Applied Biosystems) were loaded into each well. Microfluidic cards were thermocycled at 50°C for 2 min and 94.5°C for 10 min followed by 40 cycles at 97°C for 30 s and 59.7°C for 1 min. Data were collected with instrument spectral compensations by the Applied Biosystems SDS 2.2.1 software and analyzed using the threshold cycle (Ct) relative quantification method. GAPDH was determined as the appropriate internal housekeeping gene. For each sample, the Ct value for the gene of interest was determined, normalized to its respective value of GAPDH, and compared to the value obtained for normal MC, using the ΔCt method [22].
RT-qPCR analysis
p16INK4a expression was assessed by RT-qPCR. Total RNA was extracted from 0.2 to 1×106 trypsinized MC using the RNeasy Mini kit (Qiagen). Transcription of the corresponding cDNA was performed from 1 μg of RNA using the High Capacity cDNA RT Kit (Applied Biosystems), and p16INK4a expression was analyzed using predeveloped TaqMan Gene Expression Assays (p16: Hs00923894_m1; B2M: Hs00984230_m1; Applied Biosystems) on a Rotorgene 6000 (Qiagen) according to the manufacturer's instructions. Transcript expression levels were presented as log2-transformed expression normalized to β2 microglobulin.
Enzyme-linked immunosorbent assay
Quantification of IL-6, IL-8, VEGF, MCP1/CCL2, and SDF1/CXCL12 in the culture supernatant was assessed by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (Quantikine®; R&D systems).
Senescence-associated β-galactosidase activity
Senescence status was evaluated in comparison with age-matched normal MSC by senescence-associated β-galactosidase (SA-βGAL) activity according to the manufacturer's instructions (Biovision, Mountain View, CA) and, when possible, by p16INK4a gene expression.
BM cell culture under hypoxic conditions
In parallel to culture of BM cells under normoxic conditions, cells from the same suspension were exposed to 5% CO2 in a 3-gaz incubator (Binder, Tuttlingen, Germany), under the same culture conditions in terms of input cell density and medium. The number of CFU-F was counted.
Immunohistochemistry
Formalin-fixed or Dubosq-Brazil (alcohol-based Bouin)-fixed, paraffin-embedded tissue sections of 5 μm were placed on glass slides, deparaffinized at 56°C during 25 min, immersed in methyl for 10 min, and then rehydrated through graded concentrations of alcohol. Tissue sections were stained with hematoxylin-eosin-safran in an automate Microm HMS 740 (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's recommendations.
For immunohistochemistry, tissue sections were deparaffinized at 75°C in an automate Ventana BenchMark XT (Ventana Medical System, Tucson, AZ) according to the manufacturer's recommendations. Sections were incubated with mouse primary antibody against CD20 (clone L26; Dako, Carpintera, CA), CD5 (clone 4C7; Leica Biosystems, Newcastle, United Kingdom), and CD3 (clone LN10; Leica Biosystems, Newcastla, United Kingdom) for 24 min at 37°C. After blocking endogenous peroxidase, tissue sections were incubated with secondary peroxidase-conjugated antibodies and revealed by Ultraview Universal DAB Detection Kit (Ventana). All sections were counterstained with hematoxylin (Dako) for 3 min. Slides were then air dried and cover-slipped.
Statistical analysis
Numeric values were expressed as mean±SEM and compared by Student's t-test (paired or not) or the Wilcoxon test. The significance of differential expression (normal MC vs. CLL-MC) was tested using two statistical analyses: (i) Unpaired Mann–Whitney nonparametric test carried out with SPSS® software and gene selection with a |fold change|>1.2 and a P value≤0.05; and (ii) Significance Analysis of Microarrays software, using 1000 permutations, a fold change >1.2 or <0.8 and a false rate discovery of 5%. Given the low number of samples, we retained the 16 genes that were upregulated in CLL-MSC using the Mann–Whitney-based analysis and the 33 genes that were found downregulated using the two statistical methods.
Results
Routine CFU-F assay failed to detect mesenchymal progenitors
By using standard medium, that is, without b-FGF, we confirmed the higher frequency of mesenchymal progenitors in NSB than in NBM [median: 170 (n=20) vs. 22 (n=37)/106 BM cells; P<0.01], and consequently the necessity to compare CLL-BM data with those obtained from an equivalent BM source (Fig. 1A). In a first series of 11 experiments (4A, 3B, and 4C Binet stages), a significant proportion of cultures in standard medium failed [5/11; 45.5% (Fig. 1A)] in contrast with the NBM series (P<0.05) and our previously published series [19]. Furthermore, in productive cultures, the frequency of CLL-BM CFU-Fs was significantly lower (median: 0.2/106 BM cells; P<0.05).
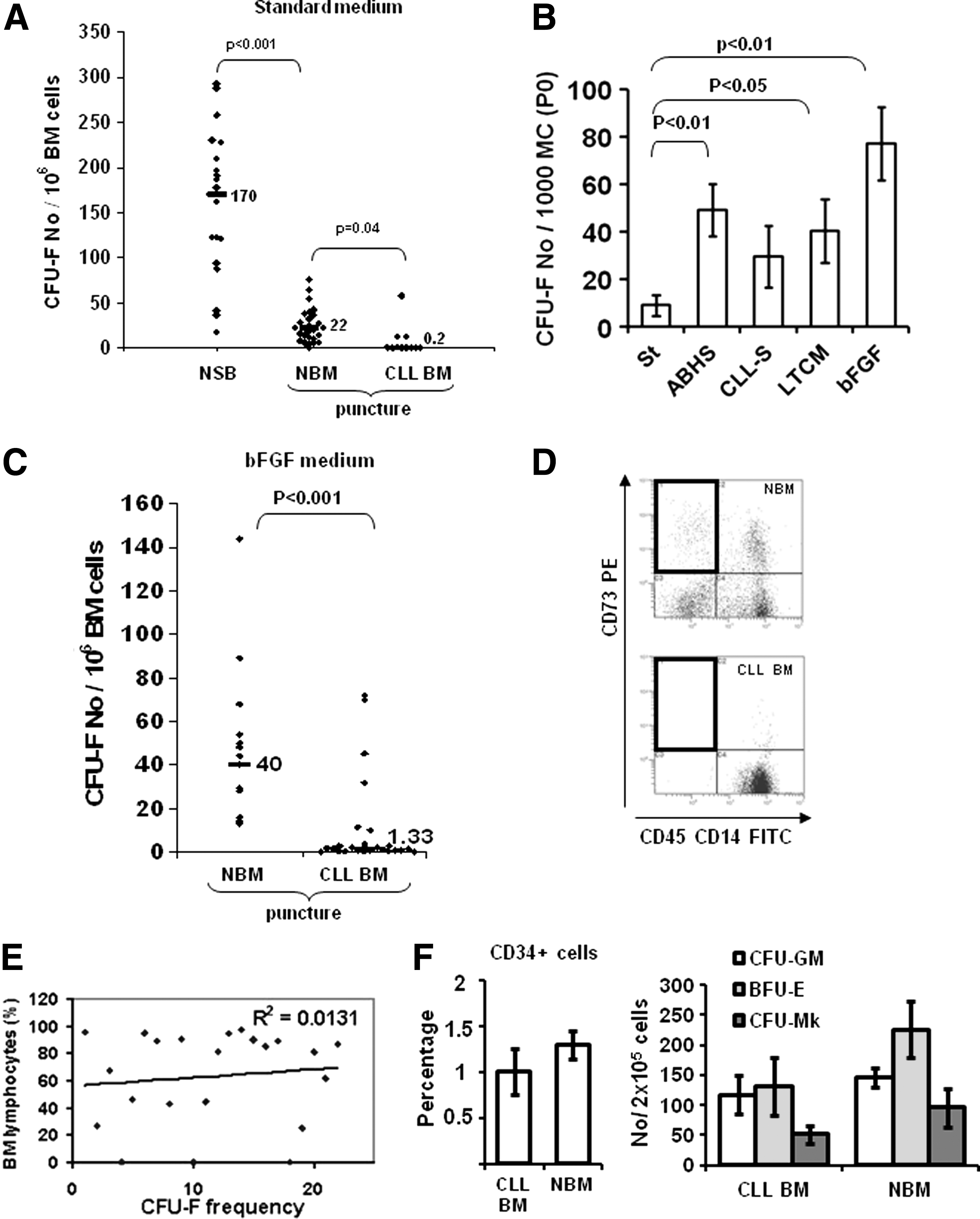
Mesenchymal progenitors are rare in BM from CLL patients.
CLL-BM mesenchymal progenitors remain rare after improving culture conditions, and show limited in vitro proliferating potential
In the aim of improving culture conditions, we also seeded CLL-MSC collected at the end of primoculture (n=7) in human serum-containing culture medium, in CLL serum, in long-term culture medium and in the presence of bFGF. We observed that bFGF was the most efficient inductor of MSC proliferation (P<0.01) (Fig. 1B). We then added bFGF (1 ng/mL) systematically in CFU-F assays.
CFU-F were thus detectable in 24 CLL-BM (82.7%) out of a new series of 29 CLL samples [Binet stage: 7 (24%) A, 11 (38%) B, 11 (38%) C]. No mesenchymal colony was detectable in five samples (17.3%). However, in productive culture, their frequency was about 30 times lower than in NBM (n=15) cultured in the presence of bFGF (median: 1.3 vs. 40/106 BM cells respectively; P<0.01; Fig. 1C), despite the fact that the number of fresh BM cells tested was much higher (>15×in most cases) than that required to systematically detect CFU-F in healthy BM [19]. Only rarely did CLL-BM show similar frequency as BM from healthy donors. The rarity of CFU-F was consistent with our recurrent (10/11) inability to detect the CD45−CD14−/CD73+ subset [19] in CLL-BM (Fig. 1D). We did not find any correlation between the frequency of CFU-F and the percentage of BM lymphocytes (72%±4%, extremes: 16% to 96%; Fig. 1E) and in CLL patients with partial BM invasion (less than 50%; n=11), the median number of CFU-F was under the median of the 40 CLL series and we did not detect any CFU-F (<1 CFU-F/4×107 nucleated cells) in three cases (27.2%). Furthermore, CD34+ cells and clonogenic hematopoietic progenitors were detectable in tested CLL-BM with only slightly lower frequencies (maximum two times) than in NBM (Fig. 1F). We found no correlation between the frequency of CFU-F and patients' age, sex, hemoglobin level, platelets, Binet stage, percentage of CD38+ lymphocytes, and cytogenetic alterations (data not shown).
In CLL productive cultures, we observed that most MSC mainly expanded in primary culture as shown by the number of population doublings (PD) at different passages (Fig. 2A), the negative PD values being the result of a lower number of CLL-MSC after culture than at the initial seeding. In parallel, we noted that most CLL-MSC had the large, polygonal appearance already described for a minor subset of slowly or nonreplicative cells in NBM (Fig. 2B) [23]. We observed that nondividing CLL-MSC remained alive a long time in culture (>3 months; data not shown). CLL-MSC from sufficiently growing samples were able to differentiate toward adipocytes, chondrocytes, and osteoblast lineages (data not shown).
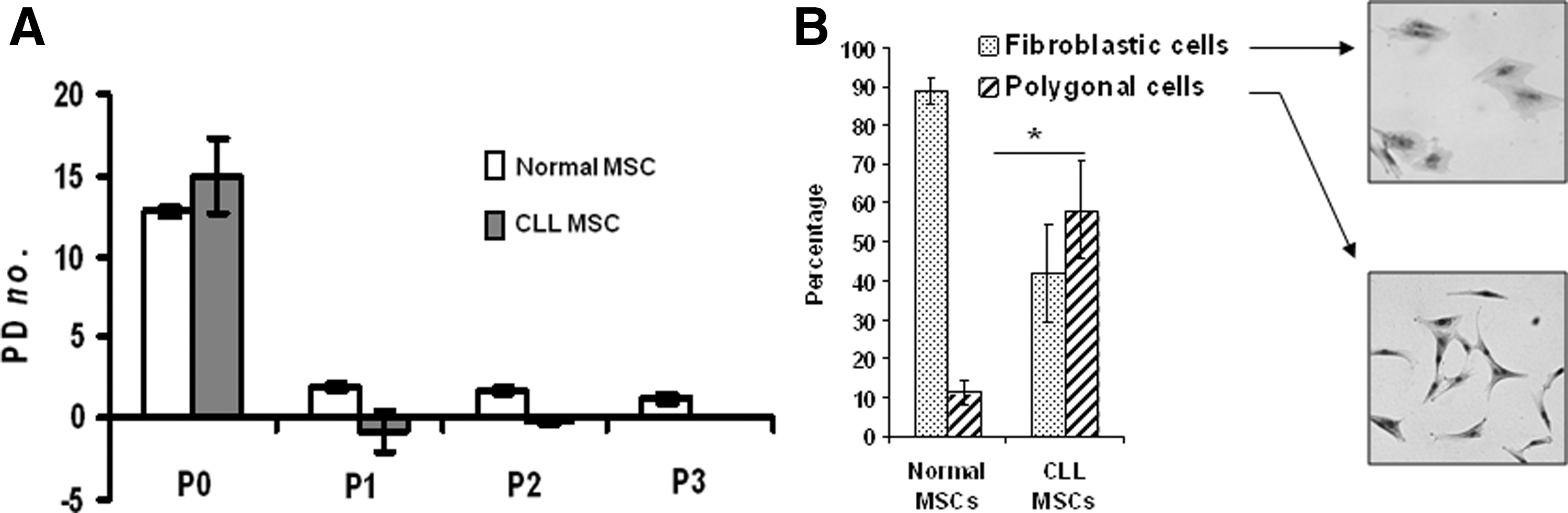
CLL mesenchymal progenitors poorly expand in standard culture conditions.
CLL-MSC differentially express selected transcripts and produce molecules related to a senescence-associated secretory phenotype
To better characterize MC from CLL-BM, the TLDA analysis of 384 genes designed by a French CSM network group statistically identified 16 upregulated and 33 downregulated genes (Fig. 3A, Supplementary Table S2). Among the upregulated genes, we first verified the increase of expression at protein level of molecules likely to play a role in the CLL lymphocytes—MSC relationship. We confirmed by flow cytometry the higher proportion of CD200-positive cells in expanded mesenchymal cells from CLL-BM than from NBM (10.6%±4% vs. 1.1%±0.5% respectively; P<0.05) and only a tendency for SPARC, of which the expression was more heterogeneous (15%±6.1% vs. 5.9%±1.5% respectively; P=0.06) (Fig. 3B). Intrigued by these nondividing CLL-MSC remaining alive a long time in culture, and given that several upregulated genes, that is, VEGF, IL-6, IGFBP2, and SPARC have been included in the recently described senescence-associated secretor phenotype (SASP) [24], we analyzed the capacity of CLL-MSC to secrete molecules corresponding to some of our upregulated genes and others included in SASP and/or in CLL physiopathology, such as IL-8, and SDF1/CXCL12 [8,25,26]. We observed a significantly higher level of IL-6, IL-8, and VEGF in the supernatants from CLL-MSC compared to age- and sex-matched healthy donor-derived MSC (Fig. 3C, D). Only a few samples produced high levels of MCP1 and SDF1 with no significant difference. We did not observe any correlation between the level of cytokines/chemokines and Binet stages. Thus, the association of (i) the polygonal aspect of MSC; (ii) their limited proliferative potential; and (iii) their secretion of several SAPS-related factors strongly suggested that CLL-MSC were mostly senescent. Indeed, a large proportion of CLL-MSC from early passages (P0 to P2) were SA-βGAL positive, equivalent to those of normal MSC matched in age at P5–P6 (Fig. 3E). These data were confirmed by an early upregulation of p16INK4a considered a sign of irreversible senescence status (Fig. 3F).

CLL-BM mesenchymal cells are mostly senescent ex vivo in standard culture conditions.
Hypoxia partially improved the frequency of mesenchymal progenitors but did not induce normalization
All the previous data are consistent with those from Kipps' team who observed the dependence of mesenchymal cells on hypoxia [17]. In consequence, we tested in parallel 10 BM cells in culture (21% and 5% O2) (Fig. 4). However, in our hands, the increase in CFU-F was variable, predominantly concerning the two most productive cultures. Out of five unproductive cultures in 21% O2, only two were able to proliferate in 5% O2 atmosphere and without reaching normal values. Thus, any corrective effect of hypoxia remains limited.
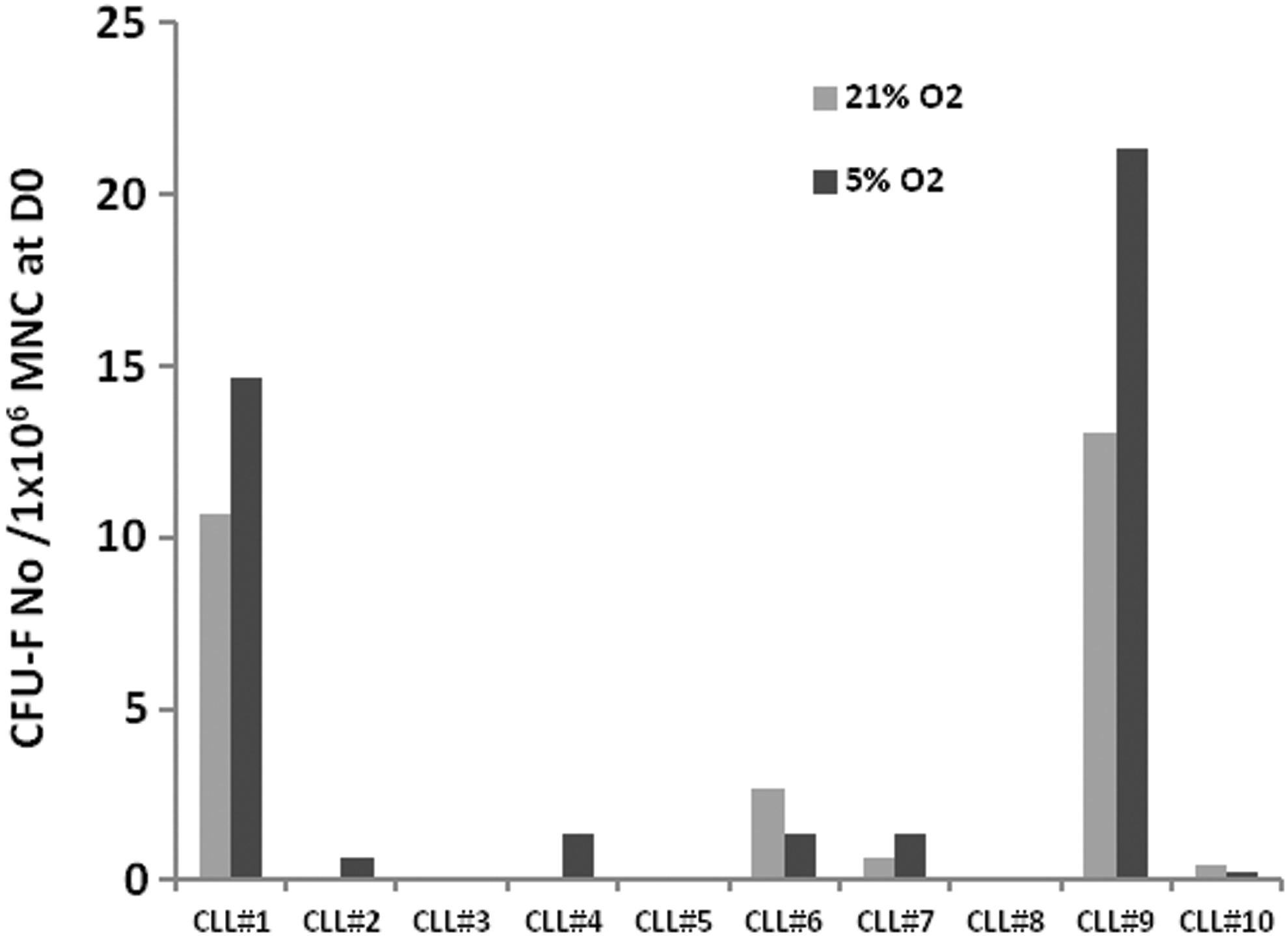
Hypoxia slightly improves CFU-F detection. We investigated the impact of 5% O2 concentrations by performing parallel tests on BM cell cultures under normoxic and hypoxic conditions (n=10). We observed a moderate increase in CFU-F in 3 cases out of 10 under hypoxic conditions. In two cases, cultures were negative under normoxic conditions but a few CFU-F were detected under hypoxic conditions. However, frequencies did not return to normal. Finally, in two samples, CFU-Fs could not be detected in either normoxic or hypoxic conditions. Results were normalized in relation to the same initial number of BM cells (1×106).
Invasion of BM by the CLL clone induces a disruption of BM elementary units
BM puncture withdraws cell aggregates considered to be elementary units, and containing both hematopoietic and stromal cells [20,27,28] (Fig. 5A, B). We noticed that the BM harvest from 22 CLL patients appeared to contain fewer hematons, as seen by their number per mL of BM (Fig. 5C). Nine out of 22 CLL-BM (41%) did not contain any hematons while we systematically observed hematons in the BM from 28 BM aspirates from patients with benign or peripheral blood etiologies of blood disease. Furthermore, when we analyzed the hematons by immunohistochemistry, we observed that all CLL-BM containing hematons exclusively corresponded to nodular infiltration (Fig. 5D, E), confirmed after CD20 and CD5 staining. In all cases, the nodules appeared to push other cells away, especially cells belonging to the BM microenvironment such as adipocytes, normally distributed through the depth of the structure.
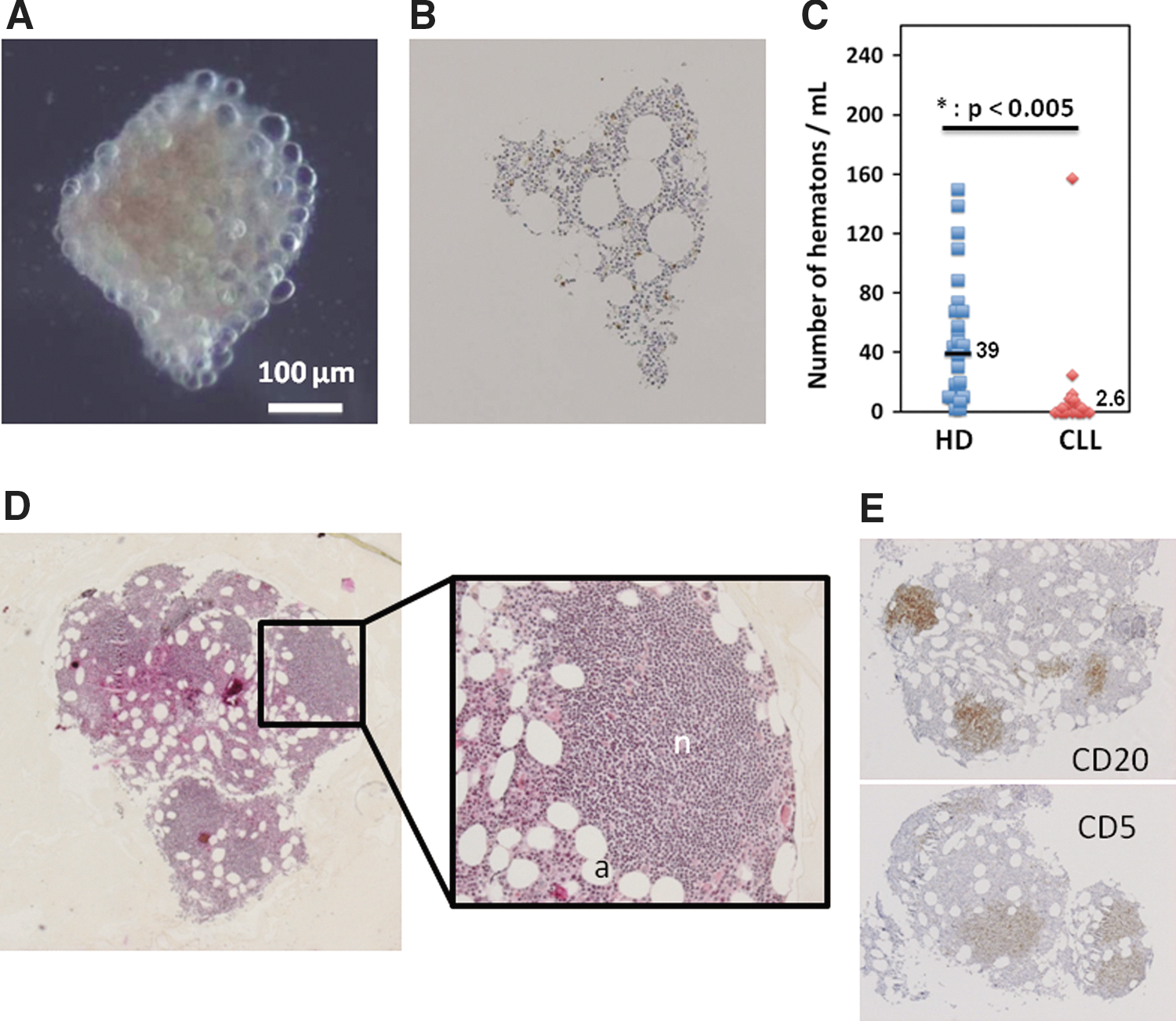
The CLL clone disrupts BM tissue organization. We evaluated the number of hematons, cell aggregates considered to be elementary units of BM. In healthy donor BM, these tiny coherent structures (less than 1 mm in diameter) contain all the elements of medullary stroma as shown by the demonstrative photograph in contrast phase
Discussion
Improved understanding of the relationship between the B-CLL clone and the tissue microenvironment is crucial if we are to develop new therapeutic strategies. The dependence of CLL cells on extrinsic signals has been largely documented with in vitro models [8 –11,13,29] but in vivo information is lacking.
In this study, assaying native mesenchymal progenitors usually referred to as CFU-F in standard culture conditions; we observed that CLL-BM CFU-Fs were rare and modified. Their low frequency cannot be explained only by the dilution effect induced by the B-cell clone because (i) no significant inverse correlation was observed between BM lymphocytosis and CFU-F frequency; (ii) the number of fresh BM cells tested was much higher than that required to systematically detect CFU-F in healthy BM [19] and largely compensated for the dilution of healthy cells; and (iii) the richness in CD34+ cells and clonogenic progenitors showing a diluting effect of healthy BM progenitor cells by a maximum two-fold factor only. Furthermore, in the hypothesis that CFU-F could simply result from NBM dilution, we should observe normal in vitro expansion of any detected mesenchymal progenitors. All these observations appear to argue against the hypothesis of simple dilution of CFU-F and demonstrate mesenchymal progenitor depletion. Furthermore, CLL-BM mesenchymal progenitors had a polygonal aspect and limited proliferative potential as already reported for NBM [23], and was associated here with prolonged survival (several months, data not shown) with no argument for apoptosis (data not shown). They differentially expressed some transcripts; most of the downregulated genes were involved in differentiation pathways. Furthermore, they secreted higher levels of molecules included in a SASP. The expression of SA-βGAL and p16INK4a confirmed that from the first passages, the proportion of senescent mesenchymal cells was similar to healthy donor-derived ageing cultures (>4th passage).
All these observations confirm previously reported studies [17,18] and are similar to those made by Fecteau et al. who did not detect any CFU-F in usual in vitro conditions in a smaller series. Because Fecteau et al. obtained satisfactory production of mesenchymal cells in a 5% O2 atmosphere and demonstrated the potential role of low O2 concentration in CLL cell survival [30], we tested hypoxic conditions with a series of 10 CLL-BM, but we only obtained a slight and variable increase in CFU-F and never obtained normal values. In our hands, the rarity of mesenchymal progenitors was not explained by hypoxia.
However, we cannot extrapolate this result to in vivo CLL-BM mesenchymal cells because senescence of CLL-BM-derived mesenchymal cells could result from inappropriate culture conditions. Thus, this observation asserts only that BM-derived mesenchymal stem cells are abnormal when compared with healthy BM cultured in the same conditions [19]. In the hypothesis that in vitro senescence corresponds to unsuitable culture conditions, we can speculate that this MSC behavior may correspond to either a subset of normal MSC [23] selected by the B-CLL clone or to an abnormal subset of stromal cells impaired by malignant proliferation. We observed partial bFGF dependency of amplified MSC that could be related to selection of a mesenchymal cell subset sensitive to bFGF secreted from CLL cells [31]. But since the detection of CFU-F was not improved in the media containing human sera or the media conditioned with CLL cells (Fig. 1B, C), and given the fact that in the absence of any malignant clone cell cultures failed, an intrinsic growth defect would appear to be involved.
In the hypothesis that MSC may be senescent in vivo, these cells could play an important role. It has been demonstrated that relatively few senescent cells have far-ranging effects within tissues, since they can stimulate premalignant cells to proliferate and/or can promote a particular environment by secreting large quantities of molecules such as cytokines for example, as already reported for other malignancies [32 –35]. Here, some of these molecules are known to favor lymphocyte survival (IL-6, IL-8, and VEGF) and others are involved in the immunotolerance of cancer cells (CD200, SPARC) [36,37].
Considering the role of MSC in BM tissue organization, the rarity of CFU-F should have an impact on BM tissue organization. Thus, we evaluated BM content in hematons, which are considered to be the elementary units of BM [20,27,28]. MSCs play a cohesive role since, in vitro, hematons break down once the MSC they contain adhere to the surface of the container and proliferate. Thus, the fact that hematons decreased or even disappeared in 41% of BM from CLL samples might be explained by the rarefaction of MSC or even a decrease in their proliferative capacity, which reinforces our results. Furthermore, when some BM tissue subunits were preserved, they systematically showed partial nodular infiltration, suggesting that diffuse infiltration (an indicator of progressive disease [1,2,38]) may dissociate hematons. In summary, in our knowledge, we have collected, for the first time, convincing evidence of the disrupting effect of the B-CLL clone on BM tissue organization.
But all these observations appear to contradict the studies conducted by Kay's team [14 –16]. However, significant differences should be noted in these studies in comparison with ours: (i) cells were recovered from osteomedullar biopsy explants and it is likely that this origin favors a higher harvest of mesenchymal cells [19,39]; (ii) mesenchymal cells isolated from explants might correspond to the progeny of cells more distant to CLL cells or at least from a different location than those collected by puncture; it has been reported that B-CLL cells are not located in paratrabecular BM [38]; (iii) explant culture does not allow for quantifying adherent cells and comparing with normal spongious bone; (iv) stromal cells were isolated and amplified using a long-term culture protocol that favors a heterogeneous cell composition [40 –42] and substantially modifies cells; (v) the failure rate of the cultures in these studies is unknown; and (vi) the patient series was different from ours; first, the patients used in Kay's studies presented all stages of CLL, whereas we enrolled a majority of patients with aggressive forms of the disease, since BM puncture is only indicated in these aggressive forms in France; second, they recruited treated and nontreated patients, even if treatment had been stopped between 4 weeks to 3 months before sample processing, while we mostly retained untreated patients since treatment during the year preceding analysis can modify the frequency of CFU-F. Moreover, we observed the recurrence of hematons in patients after treatment (data not shown).
The results of our study and of others [17] are in favor of an impact of the CLL clone on the mesenchymal cell compartment, which results in a decrease in the frequency of mesenchymal progenitors and significant damage to the remaining progenitors, in relation to a profound modification of BM tissue. These observations open up new pathophysiological hypotheses. The direct interaction between B-cells and marrow stromal cells is believed to play a decisive role in B-CLL survival but it should be noted that most of the results reported in the literature were obtained using expanded MSC from healthy donors or CLL bone biopsy explants [8 –16], which is not exactly the in vivo situation. First, merely putting MSC in contact with a rigid surface like a culture flask induces their proliferation and favors their differentiation, cell behavior that is very different to in vivo behavior [43]. Second, no in vivo situation corresponds to contact between a malignant clone and a purified mesenchymal population. Third, the protective effect of expanded mesenchymal cells appears to be a robust “ex vivo” behavior in all tested systems [44], consistent with data collected for all adherent cells toward nonadherent cells [29,45], and in favor of a nonspecific mechanism. Consequently, we should be prudent when extrapolating data from in vitro models using expanded mesenchymal cells.
On the other hand, several studies have reported the possibility of the stimulating effect of CLL cells on MSC in models using healthy donor derived-MSCs [46,47]. We also observed an increase in healthy donor-derived MSC proliferation and emperipolesis (data not shown), apparently contradictory to our study. But this is an allogenic model, which may modify the interactions between CLL cells and MSC, and this interaction occurs very early. We must be aware that this stimulating effect of B-CLL cells could correspond to the initial and localized steps of disease development, when expanding CLL cells encounter normal mesenchymal cells. But the natural history of CLL stretches over several years, and the time a BM puncture is performed corresponds to a different disease phase, after several years of B-CLL and MSC crosstalk.
Consequently, it is difficult to apply the in vitro protective model of MC for the CLL clone, and the issue of the in vivo crosstalk between malignant B cells and stromal cells, particularly in the BM, remains relevant. Indeed, while several recent studies have showed the critical role of lymph nodes in CLL cell proliferation and trafficking [48,49], the role of the lymph node microenvironment versus BM microenvironment in keeping alive the cells responsible for relapse remains unknown. Furthermore, the relationship between BM residual disease and the risk of relapse was observed [50]. Further knowledge about in vivo crosstalk between the CLL clone and the microenvironment is now crucial.
Our observations demonstrate an in vivo disintegration of CLL-BM tissue, notably due to the impact of the malignant clone on the mesenchymal compartment, suggesting that normal MSC are not appropriate cellular partners for B-CLL survival or proliferation in vivo. These data are akin to an equivalent concept regarding other hematological malignancies such as acute myeloid or lymphoid leukemia [51,52]. Complementary studies are necessary to better understand in vivo crosstalk between the CLL clone and BM microenvironment.
Footnotes
Acknowledgments/Funding
The authors would like to thank Dominique Chadeyron for secretarial assistance, Jean-Jacques Guérin, Laurent Guillouard, Pascale Pigeon, Frédérique Lioret, Caroline Jamot, Nathalie Chaudagne, and Lucie Lafontaine, Clermont-Ferrand University Hospital, for technical assistance, the staff from the Centre de Ressources Biologiques (CRB) Auvergne for cryopreserving human samples and Lemlih Ouchchane for participating in statistical analysis. This work was supported by grants from the Comité Départemental (Puy-de-Dôme) de la Ligue Contre le Cancer [Anti-Cancer League Regional Committee (Puy-de-Dôme, France)] and the Association Laurette Fugain, France.
Author Disclosure Statement
The authors reported no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.