
Abstract
Human bone marrow stromal/stem cells (hBMSCs) have an inherent tendency to undergo hypertrophy when induced into the chondrogenic lineage using transforming growth factor-beta 1 (TGFβ) in vitro, reminiscent of what occurs during endochondral ossification. Surprisingly, Indian Hedgehog (IHH) has received little attention for its role during hBMSC chondrogenesis despite being considered a master regulator of endochondral ossification. In this study, we investigated the role that endogenously produced IHH plays during hBMSC chondrogenesis. We began by analyzing the expression of IHH throughout differentiation using quantitative polymerase chain reaction and found that IHH expression was upregulated dramatically upon chondrogenic induction and peaked from days 9 to 12 of differentiation, which coincided with a concomitant increase in the expression of chondrogenesis- and hypertrophy-related markers, suggesting a potential role for endogenously produced IHH in driving hBMSC chondrogenesis. More importantly, pharmacological inhibition of Hedgehog signaling with cyclopamine or knockdown of IHH almost completely blocked TGFβ1-induced chondrogenesis in hBMSCs, demonstrating that endogenously produced IHH is necessary for hBMSC chondrogenesis. Furthermore, overexpression of IHH was sufficient to drive chondrogenic differentiation, even when TGFβ signaling was inhibited. Finally, stimulation with TGFβ1 induced a significant and sustained upregulation of IHH expression within 3 h that preceded an upregulation in all cartilage-related genes analyzed, and knockdown of IHH blocked the effects of TGFβ1 entirely, suggesting that the effects of TGFβ1 are being mediated through endogenously produced IHH. Together, our findings demonstrate that endogenously produced IHH is playing a critical role in regulating hBMSC chondrogenesis.
Introduction
I
Previous studies have shown that TGFβ ligands tend to differentiate hBMSCs into hypertrophic chondrocytes [12], a more mature cell type than articular chondrocytes that orchestrates many key stages of bone formation during endochondral ossification. Hypertrophic chondrocytes are characterized by the expression of RUNX2 and production of collagen type 10 (COL10), matrix metalloproteinase 13 (MMP13), and alkaline phosphatase (ALP), as opposed to that of articular chondrocytes, which express high levels of SOX9 and produce collagen type 2 (COL2) and aggrecan (ACAN) in great abundance. Notably, recent evidence has found that hBMSC-derived hypertrophic cartilage possesses all the cues necessary to ultimately form bone when implanted subcutaneously in vivo through a process reminiscent of endochondral ossification [13]. In addition, chondrocyte hypertrophy occurs during the progression of osteoarthritis [14,15]. Thus, the cartilage tissue produced in vitro may actually be more diseased than healthy and, as such, holds little clinical utility. Thus, a better understanding of the molecular mechanisms driving hypertrophic differentiation is needed if hBMSCs are to have clinical utility in articular cartilage tissue engineering applications.
Endochondral ossification is one of the two essential processes driving bone formation during vertebrate skeletal development. For limb formation, endochondral ossification begins when mesenchymal progenitor cells aggregate into limb buds at the sites of future skeletal element formation [16]. In developing long bones, the mesenchymal progenitor cells undergo chondrogenesis to form the cartilage anlagen and subsequently undergo sequential phases of proliferation and differentiation. Specifically, slowly dividing periarticular chondrocytes differentiate into highly proliferative columnar chondrocytes before exiting the cell cycle and undergoing hypertrophy. Hypertrophic chondrocytes secrete MMPs and vascular endothelial growth factor (VEGF) to degrade the cartilaginous extracellular matrix (ECM) and promote blood vessel and osteoblast invasion before eventually undergoing apoptosis. Thus, chondrocyte hypertrophy is an essential step coordinating several steps of endochondral bone formation during development and also postnatally during growth plate maturation [17].
A number of secreted proteins have been shown to cooperatively regulate the transition toward chondrocyte hypertrophy. Of these, Indian Hedgehog (IHH) has been studied most extensively and has even been dubbed “the master regulator of endochondral ossification [16].” During endochondral ossification in the growth plate, IHH is expressed most abundantly by prehypertrophic chondrocytes and regulates the onset of hypertrophic differentiation by forming a negative feedback loop with parathyroid hormone-related peptide (PTHrP) [18,19]. Specifically, IHH activates the expression of PTHrP in periarticular chondrocytes, which in turn signals through its receptor, PTH1R, to suppress IHH expression and inhibit chondrocyte hypertrophy [19,20]. The induction of PTHrP expression by IHH may be indirect, however, as TGFβ2 (and perhaps BMP2 and 4) expressed in the perichondrium have been shown to serve as a signal relay between IHH and PTHrP [21,22]. Taken together, IHH indirectly represses hypertrophy by inducing PTHrP expression, perhaps through a signal relay involving various TGFβs and BMPs. However, in the absence of PTHrP, IHH has been shown to have the exact opposite effect by promoting chondrocyte hypertrophy [23]. Furthermore, IHH has been shown to be required for chondrocyte proliferation and osteoblast differentiation independent of PTHrP signaling [19,24,25]. Thus, IHH has been shown to be a critical signaling component within the vast network of signals responsible for controlling chondrocyte hypertrophy during endochondral ossification.
Given the inherent tendency of hBMSCs to undergo hypertrophy in response to TGFβ stimulation and the critical role of IHH in orchestrating hypertrophic differentiation during endochondral ossification, it is interesting that IHH has not received more attention for its role during hBMSC chondrogenesis. In this study, we investigated the role that endogenously produced IHH is playing during TGFβ1-mediated chondrogenesis of hBMSCs in vitro. We hypothesized that IHH acts to promote hypertrophy during hBMSC chondrogenesis but, to our surprise, found that endogenously produced IHH is actually essential for the successful execution of the overall chondrogenic differentiation program.
Materials and Methods
Ethics statement
Written consent was obtained from patients by the University of Wisconsin-Madison Hospital to obtain bone marrow-derived hBMSCs before proceeding with total hip replacement surgeries.
Isolation and expansion of bone marrow-derived hBMSCs
With approval from the Institutional Review Board at the University of Wisconsin-Madison, hBMSCs were isolated from femoral heads of patients undergoing total hip replacement surgery, as described previously [26], and frozen for future studies in DMSO-containing medium. Cells were thawed and expanded for two passages in DMEM with 1 g/L glucose (DMEM-LG) supplemented with 10% FBS (Invitrogen), 1% antibiotics (10,000 IU/mL penicillin, 10,000 μg/mL streptomycin, 25 μg/mL amphotericin B; Mediatech), and 5 ng/mL FGF2 (Peprotech) [27] at 37°C in a humidified environment with 5% CO2 before every study. Medium changes were made every 3 days. Upon 80% confluence, hBMSCs were harvested for pellet culture. Three different patients' cells were used interchangeably to validate our findings. Specifically, each experiment was carried out using one donor's cells and then repeated a second time using another donor's cells to confirm the reproducibility of experimental data. The results of each assay are representative and presented in triplicate.
Chondrogenic differentiation of hBMSCs in pellet culture
Upon 80% confluence, hBMSCs were trypsinized, counted, and resuspended in serum-free chondrogenic medium composed of DMEM with 4.5 g/L glucose (DMEM-HG; Gibco) supplemented with 1% antibiotics, 1% ITS (BD Biosciences), 0.9% sodium pyruvate (Sigma), 50 μg/mL ascorbic acid, 40 μg/mL
Plasmid construction
HuSH retroviral shRNA plasmids against IHH (shIHH) were purchased from OriGene, along with the control shRNA plasmid containing a nonspecific scrambled DNA sequence in the HuSH pGFP-V-RS vector. To construct a plasmid for IHH overexpression, the coding sequence of the IHH gene was isolated by digesting the pCMV6-Entry vector (Origene) with BamHI and FseI. The BamHI and FseI fragment was PCR amplified to add an EcoRI restriction site after the stop codon of the coding sequence using the Phusion High-Fidelity DNA Polymerase (Fisher) and subsequently inserted into the BamHI and EcoRI sites of the pMCs-Puro retroviral vector (Cell Biolabs) through a single ligation reaction. To prepare a control construct, pMCs-Puro was digested with EcoRI and XhoI and the linearized vector was re-ligated after blunting the digestion sites using Klenow DNA polymerase. The plasmid for IHH overexpression was verified by restriction digestion and sequencing of the insert.
Transfection and transduction for IHH misexpression
The IHH overexpression or shIHH plasmids were cotransfected with the pCMV-VSV-G envelope vector (Cell Biolabs) into the Plat-GP (Cell Biolabs) packaging cell by calcium phosphate precipitation. Briefly, 5 μg of the IHH overexpression or shIHH plasmid and 5 μg pCMV-VSV-G were diluted in 250 mM CaCl2 solution with a final volume of 250 μL, and then slowly mixed with 250 μL of 2×HEPES-buffered saline [2×HBS, 50 mM HEPES, 280 mM NaCl, 1.5 mM Na2HPO4 (pH 7.0)]. The plasmid/vector mixture was incubated for 20 min at room temperature and slowly added to Plat-GP cell culture treated with chloroquine at 50 μM for 15 min. Five hours after transfection, the medium was replaced with fresh medium, and 48 h after transfection, virus-containing medium was collected from the transfected packaging cell culture and filtered through a 0.45-μm PES filter to eliminate packaging cell contamination from the collected supernatant. The viral supernatant was then concentrated using Retro-X Concentrator (Clontech) by following the manufacturer's instructions and added into hBMSC culture at 70%–75% confluence. To enhance infection efficiency, hBMSCs with virus were spun at 1,200 g for 5 min at room temperature before incubating the cells in a culture incubator. Successfully transduced cells were selected through treatment with 1 μM puromycin.
RNA isolation and quantitative polymerase chain reaction analysis
Total RNA was isolated, quantified, reverse transcribed, and amplified, as described previously [28]. The following oligonucleotide primers were used to amplify the cDNA: SOX9—F: 5′-TAAAGGCAACTCGTACCCAA-3′, R: 5′-ATTCTCCATCATCCTCCACG-3′; COL2—F: 5′-GGAAACTTTGCTGCCCAGATG-3′, R: 5′-TCACCAGGTTCACCAGGATTGC-3′; ACAN—F: 5′-CACGATGCCTTTCACCACGAC-3′, R: 5′-TGCGGGTCAACAGTGCCTATC-3′; COL10—F: 5′-CCCAGGAAAACCAGGTCTCG-3′, R: 5′-CAGCCTCTCCATTGTGTCCG-3′; RUNX2—F: 5′-GGTTCCAGCAGGTAGCTGAG-3′, R: 5′-AGACACCAAACTCCACAGCC-3′; IHH—F: 5′-AAGGACGAGGAGAACACAGG-3′, R: 5′-AGATAGCCAGCGAGTTCAGG-3′; MMP13—F: 5′-TTGACCACTCCAAGGACCC-3′, R: 5′-ACATCGTCATCAGGAAGCAT-3′; and ALP—F: 5′-CAAAGGCTTCTTCTTGCTGG-3′, R: 5′-GGTCAGAGTGTCTTCCGAGG-3′. The following Ubiquitin C (UBC) primers were used to normalize the amount of cDNA synthesized per sample: UBC—F: 5′-TGAAGACACTCACTGGCAAGACCA-3′, R: 5′-CAGCTGCTTTCCGGCAAAGATCAA-3′. Values were compared to relevant control pellets using the 2−ΔΔCt method.
Histological staining
Cartilage pellets were fixed with 4% PFA (methanol-free; PolySciences), serially dehydrated in alcohol, and paraffin-embedded. Paraffin-embedded cartilage pellets were sectioned with a microtome, and 8-μm sections were deparaffinized in xylene, serially hydrated, and prepared for either immunofluorescence or immunohistochemical staining of COL2 (Millipore), COL10 (Abcam), IHH (Abcam), cleaved Caspase-3 (Cell Signaling), or Ki67 (Abcam). Antigen retrieval was performed either through digestion with 0.1% pepsin (Calbiochem) in 10 mM HCl for 15 min or with the Retrievagen A solution (BD Pharmingen) and heated according to the manufacturer's protocol. For immunofluorescence staining, sections were then blocked with 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 20 min at room temperature. The primary antibody or control IgG was then applied in 1% BSA in PBS for 45 min. The secondary antibody was FITC-conjugated goat anti-mouse IgG (eBioscience) or TRITC-conjugated goat anti-rabbit IgG (Abcam) and was applied in 1% BSA in PBS for 45 min. DAPI was applied as a nuclear counterstain during mounting with ProLong Gold antifade mounting reagent (Molecular Probes). For immunohistochemical staining, sections were exposed to 3% hydrogen peroxide after antigen retrieval to eliminate endogenous peroxidase activity before incubation in primary antibody. Sections were incubated with biotin and a horseradish peroxidase (HRP)-conjugated streptavidin using the Stat Q staining kit (Innovex Biosciences). The bound antibody complex was visualized using diaminobenzidine. Stained sections were dehydrated, cleared, and coverslipped. Slides were documented with a 10× or 20× objective using a Benton Dickinson Pathway fluorescence microscope.
Protein isolation and western blot analysis
Protein was isolated from chondrogenic pellets, quantified, subjected to SDS-PAGE, and transferred onto PVDF membranes for western blotting, as described previously [29]. Membranes were incubated with the following primary antibodies overnight at 4°C: rabbit monoclonal to SMAD3 (Cell Signaling), rabbit monoclonal to pSMAD3 (S423/425; Cell Signaling), rabbit polyclonal to SOX9 (Abcam), rabbit polyclonal to pSOX9 (S181; Abcam), rabbit monoclonal to ERK1/2 (Cell Signaling), and rabbit monoclonal to pERK1/2 (T202/Y204). GAPDH was used as an internal loading control and detected with a rabbit monoclonal antibody (Cell Signaling). The secondary HRP-conjugated antibody was goat anti-rabbit (Cell Signaling). The membrane was developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce) on Kodak Image Station 4000R Pro.
Statistical analysis
All quantifiable studies were performed in triplicate and reported as means±standard deviations. Both analysis of variance (ANOVA) and Student's two-tailed t-test were performed to determine the level of statistical significance. When ANOVA showed significance across groups, the Tukey post hoc test was performed to determine statistical significance between any pairs of groups. P values of <0.05 were considered significant.
Results
IHH expression positively correlates with the expression of chondrogenesis- and hypertrophy-related genes
Previously published data have demonstrated a critical role for IHH in orchestrating endochondral ossification [18,19]. More recent evidence has shown that hBMSCs recapitulate many key features of endochondral ossification upon chondrogenic induction in vitro [12,30,31]. In looking at the gene expression profile of differentiating hBMSCs every 3 days throughout chondrogenic differentiation in pellet culture, we found a particularly interesting pattern of expression for IHH. IHH expression was virtually undetectable in undifferentiated hBMSCs before showing greater than a 7,500-fold upregulation upon chondrogenic induction (highlighted with a bar graph in Fig. 1A). Subsequently, IHH displayed a sharp 6.3-fold peak in expression from days 9 to 12, which coincided with an increase in the expression of both chondrogenesis-related (Fig. 1B) and hypertrophy-related genes (Fig. 1C). The expression of the ECM molecules, COL2, ACAN, and COL10, all showed a marked and sustained increase in expression from days 9 to 12 (Fig. 1B, C). SOX9 expression peaked by day 12 of differentiation (Fig. 1B), and the expression of ALP showed a transient 2.3-fold peak from days 9 to 12 (Fig. 1C). RUNX2 displayed a peak in expression from days 6 to 9, whereas MMP13 expression showed a sustained increase beginning at day 12 (Fig. 1C). In addition, there was a strong correlation between the expression of IHH and the expression of COL2 and COL10 (R 2=0.8089 and 0.8666, respectively), regardless of the treatment employed or time point of differentiation (Fig. 1D). Together, these data suggest that perhaps the endogenous production of IHH is playing a role in promoting chondrogenesis, or more specifically hypertrophy, during differentiation in pellet culture.
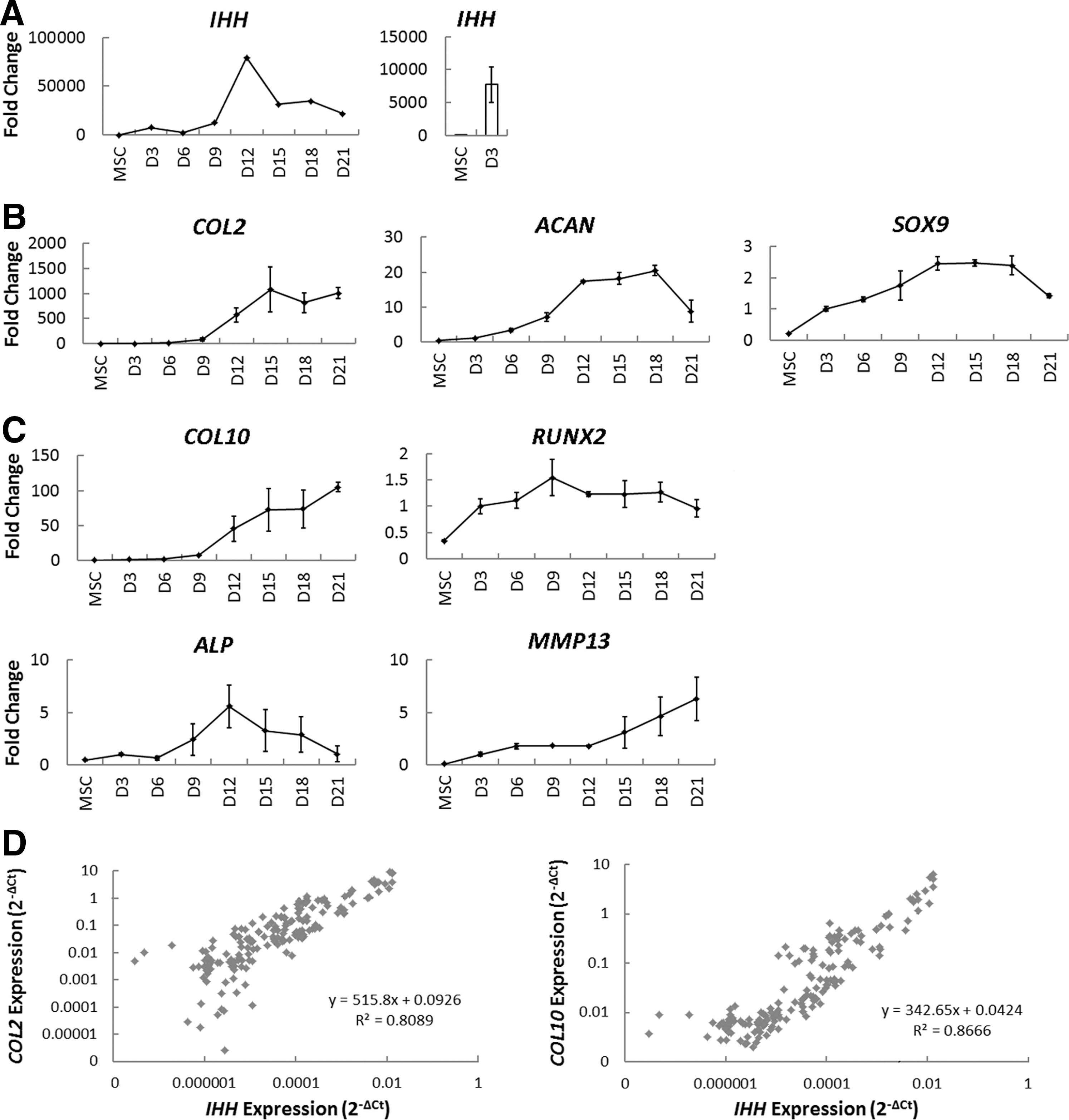
Dynamics of cartilage-related gene expression and IHH correlations throughout chondrogenic differentiation of human bone marrow stromal/stem cells (hBMSCs).
Endogenously produced IHH is necessary for TGFβ1-mediated chondrogenesis of hBMSCs
To determine if endogenously produced IHH is in fact playing a role during hBMSC chondrogenesis, we first blocked Hedgehog signaling with the pharmacological inhibitor cyclopamine. To do so, we treated differentiating hBMSCs with chondrogenic medium containing TGFβ1 in the presence or absence of 10 μM cyclopamine for 21 days of differentiation in pellet culture and analyzed the expression of markers of chondrogenesis and hypertrophy using qPCR. Indeed, cyclopamine induced a significant decrease in the expression of the Hedgehog-responsive gene GLI1, demonstrating that the treatment was effective (Fig. 2A). Interestingly, repression of Hedgehog signaling significantly reduced the expression of both cartilage- and hypertrophy-related markers. Most notably, cyclopamine reduced the expression of COL2, ACAN, SOX9, and COL10 by 99.3%, 92.7%, 62.5%, and 96.8%, respectively, by day 21 of chondrogenic differentiation (Fig. 2A). In addition, while recombinant PTHrP exposure had no effect on hBMSC chondrogenesis (Supplementary Fig. S1A; Supplementary Data are available online at
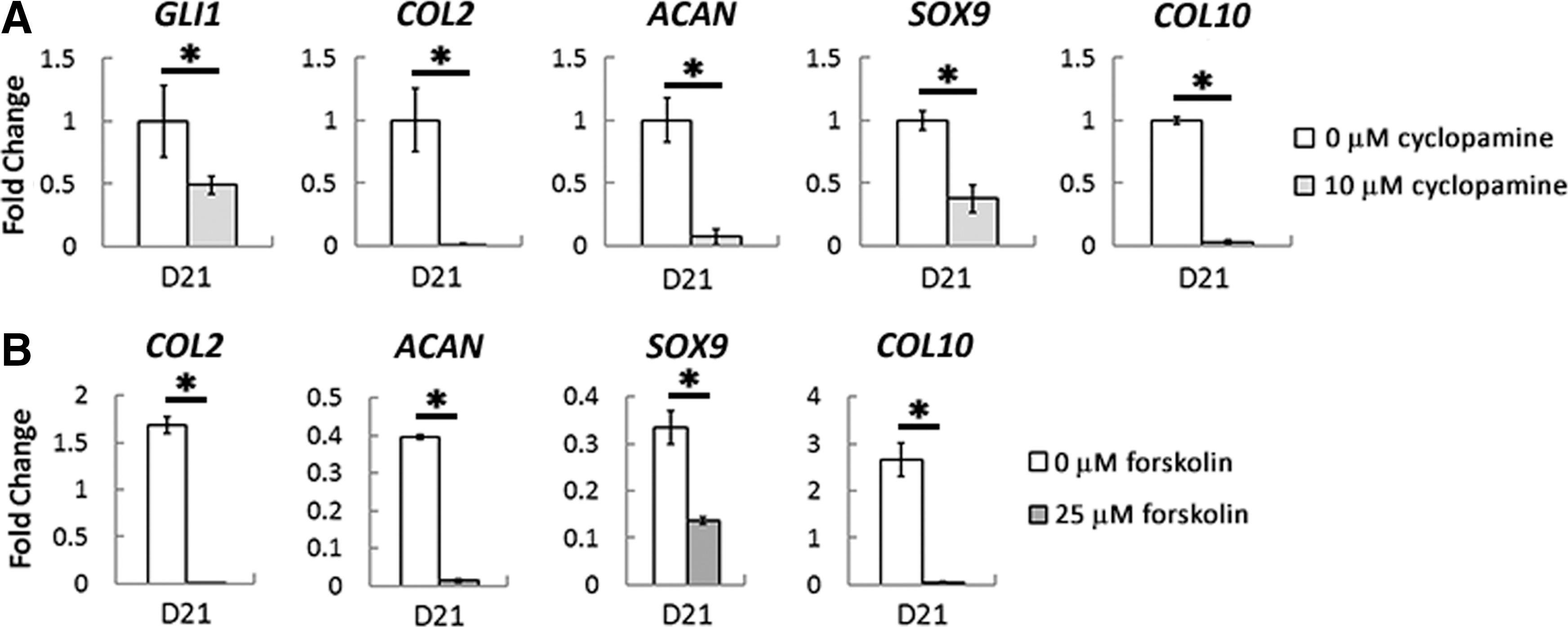
Chondrogenesis of hBMSCs during pharmacological inhibition of Hedgehog signaling or activation of parathyroid hormone-related peptide (PTHrP) signaling. hBMSCs were treated with cyclopamine
To further confirm these findings and narrow our findings to IHH specifically, we next used shRNA to stably knockdown the expression of endogenous IHH. We first examined the success of IHH knockdown and found that IHH expression was knocked down 92.7%, 52.3%, 99.4%, and 99.9% at days 0, 3, 12, and 21, respectively, as measured by qPCR (Fig. 3A). Furthermore, it reduced the intensity of immunofluorescence staining for IHH within our cartilage pellets at day 21 (Fig. 3B). Remarkably, knockdown of IHH almost entirely blocked the TGFβ1-mediated induction of hBMSC chondrogenesis instead of hypertrophy specifically, as evidenced by at least a 99.9% reduction in COL2 gene expression by days 12 and 21 (Fig. 3C) and near absence of COL2 immunofluorescence staining within cartilage pellets (Fig. 3D). Furthermore, knockdown of IHH led to an 89.7% and 89.2% reduction in ACAN expression and a 64.7% and 52.1% reduction in SOX9 expression at days 12 and 21, respectively (Fig. 3C). Similarly, IHH knockdown led to an 88.3% and 97.1% reduction in COL10 expression and a 45.3% and 62.6% reduction in RUNX2 expression at days 12 and 21, respectively (Fig. 3C). There was likewise a reduction in COL10 immunostaining at day 21 (Fig. 3D). Taken together, these data demonstrate that endogenously produced IHH is necessary for TGFβ1-driven chondrogenesis.
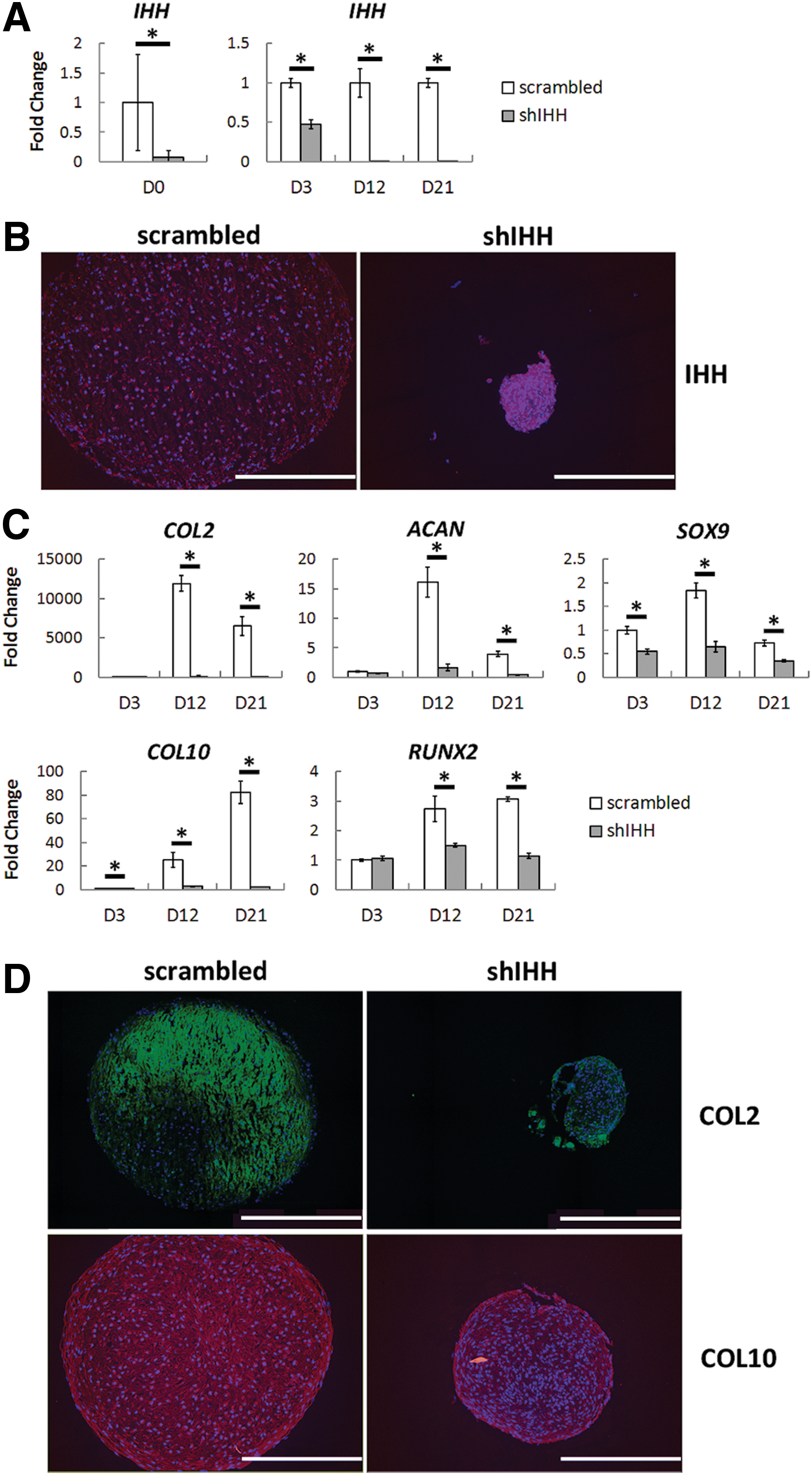
Chondrogenesis of hBMSCs upon knockdown of endogenous IHH. After knockdown of IHH expression using shRNA, hBMSCs were differentiated into the chondrogenic lineage in the presence of TGFβ1 and analyzed for their cartilaginous properties using qPCR and immunofluorescence staining. IHH expression was successfully knocked down at the mRNA
To confirm that knockdown of IHH had a direct impact on hBMSC chondrogenesis without indirectly affecting chondrogenesis through attenuation of apoptosis or proliferation, we stained for cleaved Caspase-3 and Ki67, respectively. In doing so, we found no profound difference in cleaved Caspase-3 immunostaining upon knockdown of IHH, and there was a little to no Ki67 immunostaining in either condition (Fig. 4). Thus, knockdown of IHH appears to be reducing hBMSC chondrogenesis directly and not indirectly through apoptosis or proliferation.
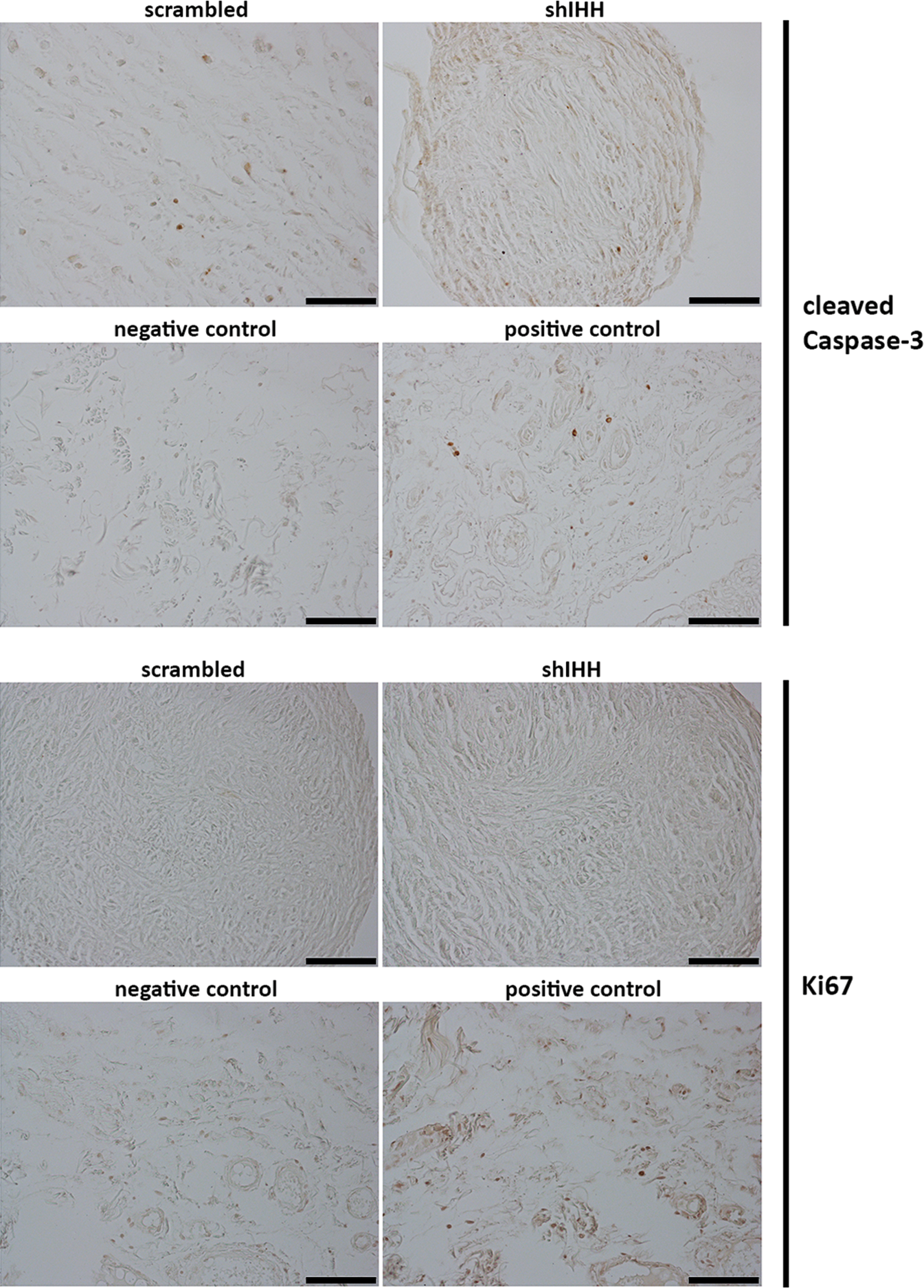
Apoptosis and proliferation of hBMSCs upon knockdown of endogenous IHH. After knockdown of IHH expression using shRNA, hBMSCs were differentiated into the chondrogenic lineage in the presence of TGFβ1 and stained for cleaved Caspase-3 and Ki67 to observe its effect on apoptosis and proliferation, respectively. Knockdown of IHH did not induce a substantial amount of apoptosis or proliferation. Scale bar=100 μm. Color images available online at
Endogenously produced IHH is sufficient for hBMSC chondrogenesis
To determine whether IHH is sufficient to induce hBMSC chondrogenesis without the necessity for exogenous TGFβ stimulation, we examined the effect of Hedgehog signaling activation. To do so, we first administered recombinant IHH but found that IHH treatment had no effect on the expression of COL2, COL10, or IHH (Supplementary Fig. S1B, C) despite showing bioactivity (Supplementary Fig. S1D). Next, we administered a pharmacological activator of Hedgehog signaling, SAG, to differentiate hBMSCs in the absence of exogenous TGFβ ligands. After determining an effective SAG concentration (Supplementary Fig. S2A), we found that SAG had no effect when administered alone throughout 21 days of differentiation, but when coadministered with the TGFβ inhibitor SB431542, SAG induced a significant 4.5-fold upregulation in COL2 expression without affecting the expression of COL10 (Fig. 5A).
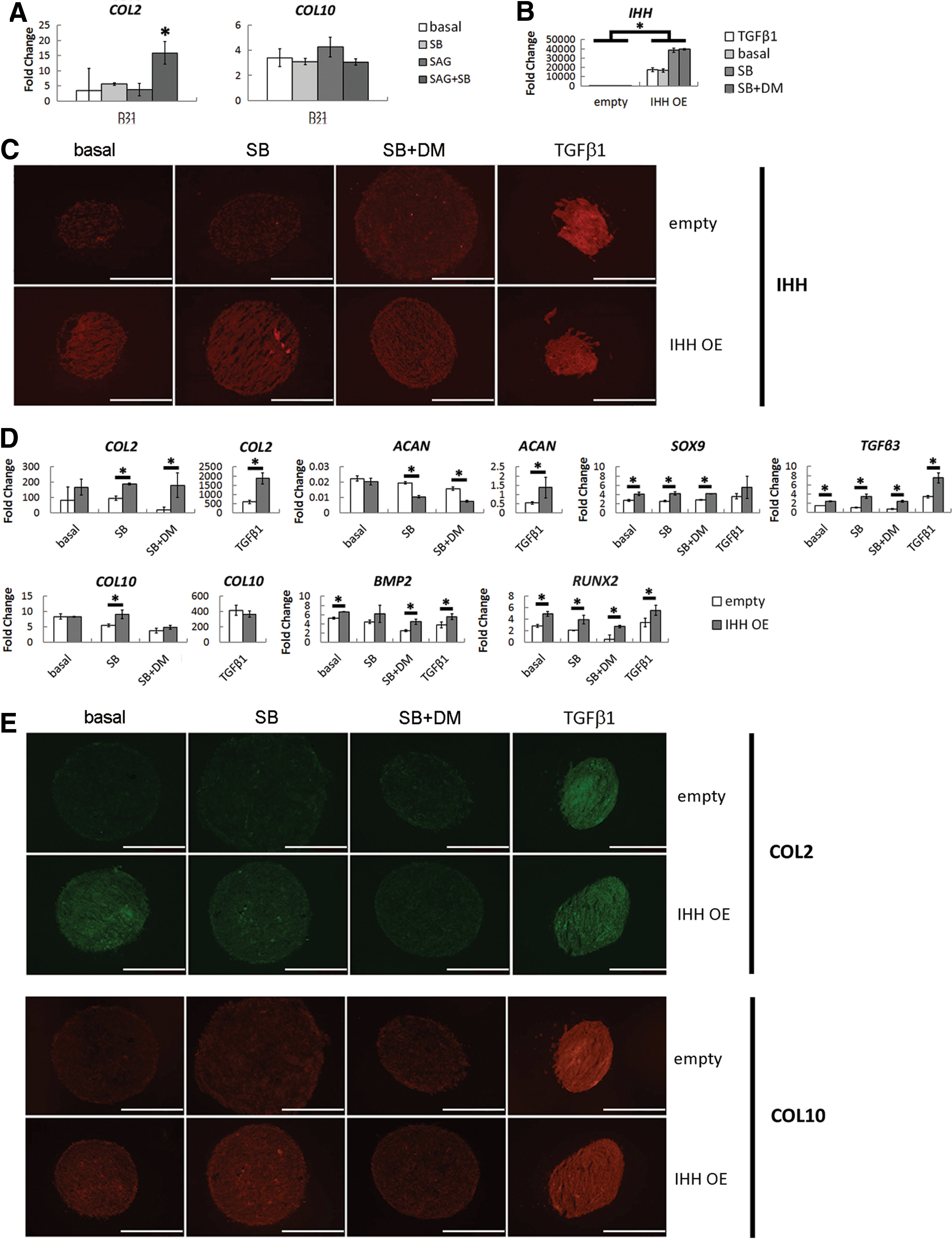
Chondrogenesis of IHH-overexpressing hBMSCs.
We then overexpressed IHH in the presence of pharmacological inhibitors of TGFβ and BMP signaling to determine if IHH can drive hBMSC chondrogenesis without exogenous or endogenous TGFβ and BMP signaling. The rationale for doing so was even if overexpression of IHH was capable of driving chondrogenesis in the absence of recombinant TGFβ1, it may be doing so by promoting the production of endogenous TGFβ and BMP ligands, which are classically believed to be the primary inducers of hBMSC chondrogenesis. Thus, our aim was to tease out whether IHH was a more direct inducer of chondrogenesis than TGFβ or BMP signaling. We first confirmed that SB431542 and dorsomorphin were able to effectively block hBMSC chondrogenesis in response to TGFβ1 and BMP7, respectively (Supplementary Fig. S2B, C). Subsequently, we confirmed the success of IHH overexpression. Quantitative PCR analysis demonstrated a robust increase in IHH expression at days 3, 12, and 21 of differentiation (Fig. 5B and Supplementary Fig. S3A), and immunofluorescence staining showed more intense staining in pellets containing IHH-overexpressing cells than control pellets (Fig. 5C), thus demonstrating successful overexpression of IHH.
At days 3 and 12, IHH-overexpressing cells generally showed an increase in cartilage-related gene expression (Supplementary Fig. S3B), and by day 21, IHH-overexpressing cells demonstrated a 2.1-, 2.0-, 9.7-, and 3.1-fold increase in COL2 expression compared to control cells when cultured in basal chondrogenic medium, basal medium with SB431542, basal medium with both SB431542 and the BMP inhibitor dorsomorphin, or TGFβ1-containing medium, respectively (Fig. 5D). Similarly, immunofluorescence staining generally showed more intense COL2 staining in pellets containing IHH-overexpressing cells compared to control pellets (Fig. 5E). However, inhibition of TGFβ and BMP signaling progressively reduced COL2 immunostaining in IHH-overexpressing pellets, suggesting that all three signaling pathways are ultimately important for hBMSC chondrogenesis. In addition, SOX9, RUNX2, TGFβ3, and BMP2 expression were all upregulated in IHH-overexpresssing cells, whereas ACAN expression was downregulated (Fig. 5D). COL10 expression was largely unaffected, but immunostaining for COL10 was generally more intense in pellets containing IHH-overexpressing cells (Fig. 5E). Finally, positive control pellets receiving recombinant TGFβ1 showed dramatically higher levels of COL2, ACAN, COL10, and TGFβ3 expression (Fig. 5D), as well as markedly greater IHH, COL2, and COL10 immunostaining than IHH-overexpressing pellets (Fig. 5E), suggesting that while IHH may be sufficient to drive hBMSC chondrogenesis, recombinant TGFβ ligands perhaps initiate a cascade of events that cooperatively drives more robust chondrogenesis.
Given that COL2 immunostaining was negligible upon inhibition of BMP signaling, we also wanted to investigate the relative importance of endogenous BMP signaling in mediating the effects of TGFβ1. To determine if endogenous BMP signaling is critical for hBMSC chondrogenesis, we simply differentiated hBMSCs into the chondrogenic lineage using TGFβ1 in the presence or absence of dorsomorphin. In doing so, we found no significant effect on the expression of any cartilage-related gene analyzed (Fig. 6), suggesting that endogenous BMP signaling is not necessary for hBMSC chondrogenesis.
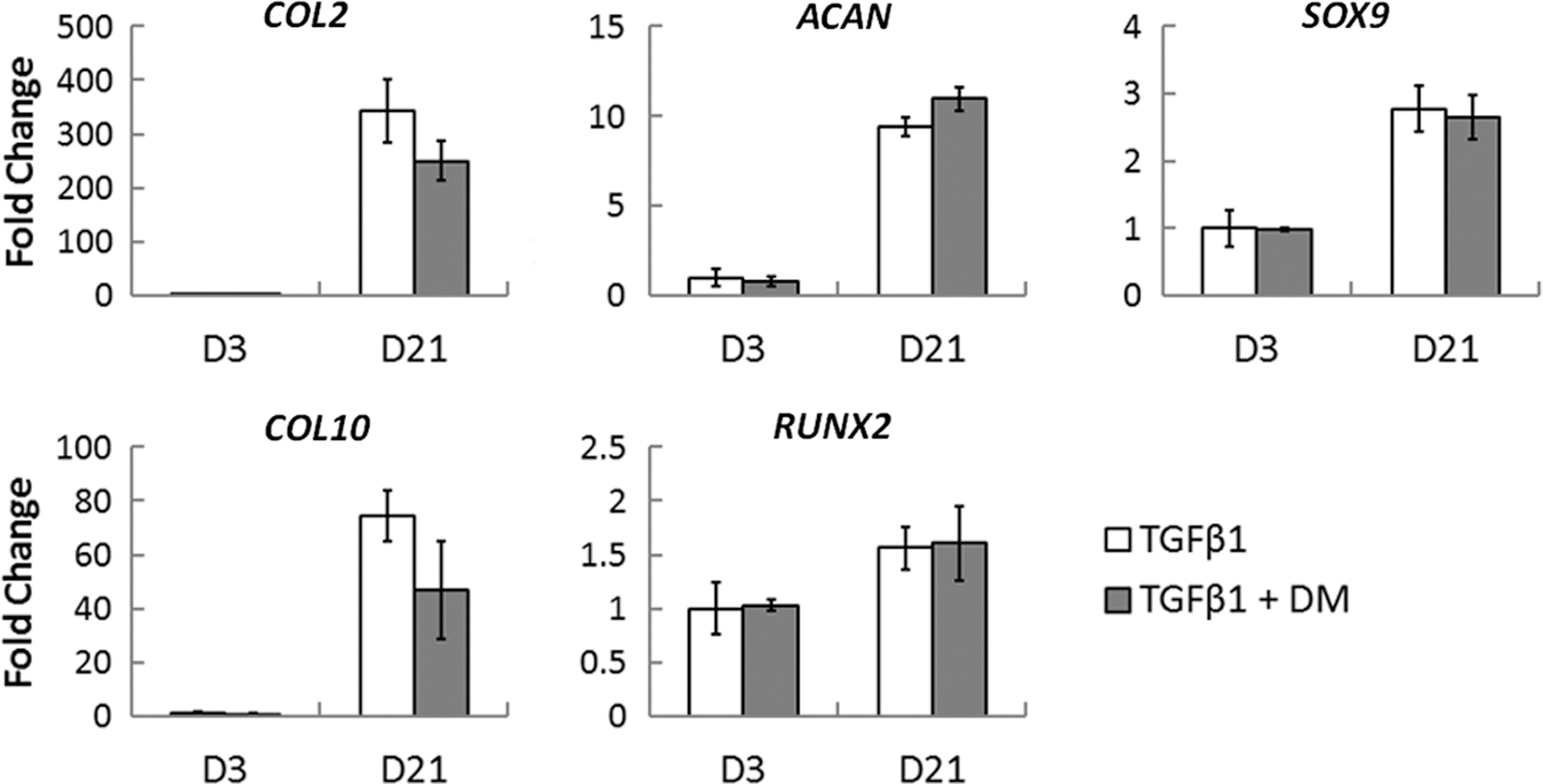
Effect of inhibition of endogenous BMP signaling on TGFβ1-mediated chondrogenesis. hBMSCs were differentiated into the chondrogenic lineage using TGFβ1 in the presence or absence of dorsomorphin (DM) to inhibit endogenous BMP signaling. Inhibition of BMP signaling had no significant effect on TGFβ1-mediated chondrogenesis. Values are represented as mean±SD; n=3.
Endogenously produced IHH mediates the effects of TGFβ1
Given that exogenous TGFβ1 induced more profound chondrogenesis than overexpression of IHH, and knockdown of IHH was able to completely block hBMSC chondrogenesis in response to TGFβ1, we last aimed to gain deeper insights into the relationship between exogenous TGFβ stimulation and endogenous IHH expression. To do so, we treated differentiating hBMSCs with chondrogenic medium supplemented with or without TGFβ1 at day 3 of differentiation and analyzed the expression of a variety of genes at various time points throughout the next 3 days using qPCR. In doing so, we found that differentiating hBMSCs showed a peak in IHH expression beginning at 3 h postfeeding that declined by 12 h (Fig. 7A). TGFβ1 significantly amplified this spike in IHH expression (1.72-fold increase) and led to a significant and sustained upregulation in its expression throughout 72 h. Subsequently, cells showed a significant increase in SOX9 expression by 6 h postfeeding in response to TGFβ1 and the prochondrogenic growth factors, TGFβ3 and BMP2, by 12 h postfeeding (Fig. 7A). Finally, the expression of COL2 and COL10 showed a significant upregulation by 24 h postfeeding (Fig. 7A). At the protein level, western blot analysis showed elevated pSMAD3 levels within 30 min of exposure to TGFβ1 that gradually declined by 12 h before increasing again around 24 h (Fig. 7B), presumably as a consequence of endogenous TGFβ3 secretion (Fig. 7A). Total SOX9 protein levels did not increase substantially until 12 h post-TGFβ1 treatment, whereas pSOX9 levels increased transiently at 2 h before showing a more sustained increase in abundance from 12 to 24 h post-TGFβ1 treatment (Fig. 7B), which coincided with the point at which COL2 and COL10 finally showed significantly increased expression, suggesting that SOX9 is playing a crucial role in driving cartilage-related gene expression.

Dynamics of cartilage-related gene expression and intracellular protein activation in response to TGFβ1.
Interestingly, in performing similar gene expression analysis as in Fig. 7A with hBMSCs after IHH knockdown, we found that the dynamics of gene expression activation were remarkably similar between IHH-knockdown hBMSCs receiving TGFβ1 (Fig. 7C) and uninfected hBMSCs treated with chondrogenic medium without TGFβ1 supplementation (Fig. 7A), suggesting that the effects of recombinant TGFβ1 are being mediated by endogenously produced IHH. Of note, the endogenous expression of TGFβ3 and BMP2 were significantly reduced in IHH-knockdown hBMSCs, as well (Fig. 7C), suggesting that IHH is likewise important for maintaining endogenous TGFβ and BMP signaling, and collectively, these three signaling pathways may be working together to drive optimal chondrogenesis.
Discussion
IHH has been studied extensively for the several roles it plays during endochondral ossification, but very few studies have sought to analyze the role of IHH during hBMSC chondrogenesis [3]—a process that recapitulates many key features of endochondral ossification. Here, we showed that IHH expression is upregulated dramatically upon chondrogenic induction, peaked from days 9 to 12 of differentiation, and was strongly correlated to the expression of COL2 and COL10. More importantly, knockdown of IHH almost completely blocked TGFβ1-induced chondrogenesis, and overexpression of IHH was sufficient to drive chondrogenic differentiation, even when TGFβ signaling was blocked, thus demonstrating a critical role for endogenously produced IHH during hBMSC chondrogenesis. Furthermore, stimulation with recombinant TGFβ1 induced a significant and sustained upregulation of IHH expression within 3 h that preceded an upregulation in all other genes tested, suggesting that TGFβ1 is driving chondrogenesis through endogenously produced IHH. Finally, knockdown of IHH significantly reduced the expression of endogenous TGFβ3 and BMP2, and overexpression of IHH significantly increased the expression of those same growth factors. Together, these data allow us to build a model whereby recombinant TGFβ1 induces the expression of endogenous IHH, which mediates the effects of TGFβ1 to drive hBMSC chondrogenesis (Fig. 8). Concomitantly, endogenous IHH maintains the expression of endogenous TGFβ3 and BMP2, which further potentiate chondrogenesis in a cooperative manner.
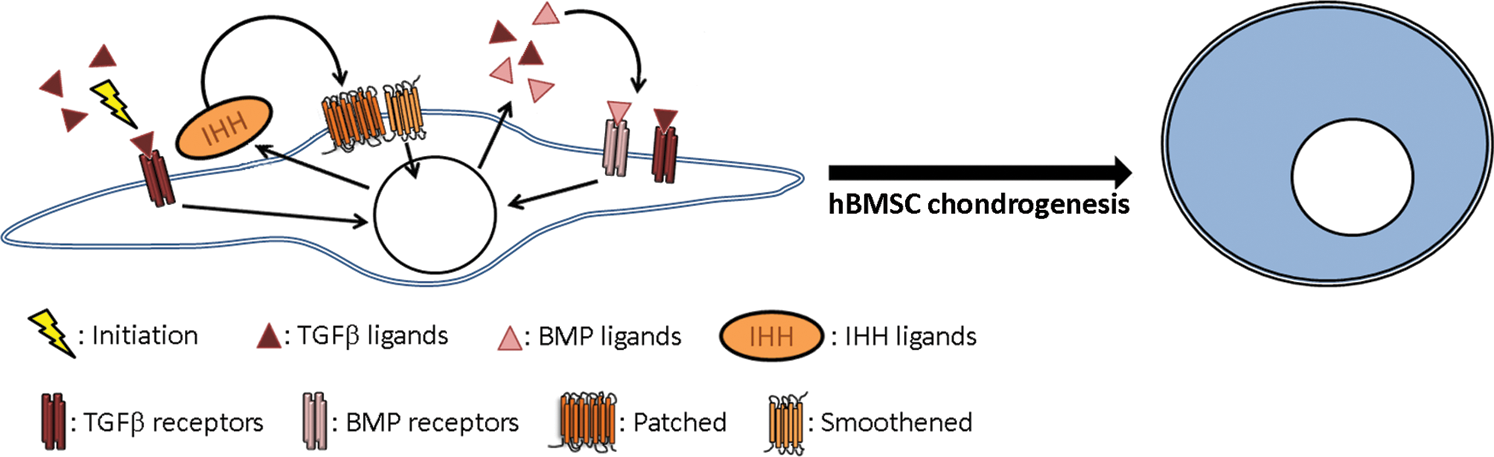
Proposed mechanism driving hBMSC chondrogenesis in response to TGFβ1. Recombinant TGFβ1 stimulation upregulates the expression of IHH, which in turn signals in an autocrine and paracrine manner to mediate the effects of recombinant TGFβ1 and drive hBMSC chondrogenesis. Endogenously produced IHH likewise maintains the endogenous production of TGFβ and BMP ligands, and the cooperative activities of all three signaling pathways together promote optimal chondrogenesis from hBMSCs. Color images available online at
Previous studies have implicated a role for IHH in promoting BMSC chondrogenesis, although IHH has been less well studied than expected. Similar to our findings, Wu et al. blocked Hedgehog signaling with cyclopamine and found that inhibition of Hedgehog signaling significantly reduced rabbit BMSC chondrogenesis [32]. More interestingly, Steinert et al. overexpressed IHH in hBMSCs and, in agreement with our findings, found that IHH-overexpressing hBMSCs were capable of undergoing chondrogenesis in pellet culture without the necessity for exogenous TGFβ stimulation [3]. However, we showed that knockdown of IHH reduced the downstream expression of TGFβ and BMP ligands in response to recombinant TGFβ1 (Fig. 7C), and overexpression of IHH led to a significant upregulation of TGFβ3 and BMP2 expression (Fig. 5D). Thus, in addition to overexpressing IHH, we concomitantly administered pharmacological inhibitors of TGFβ and BMP signaling to rule out the possibility that IHH is driving hBMSC chondrogenesis indirectly by promoting the endogenous production of TGFβ and BMP ligands. Indeed, we found successful induction of chondrogenesis when TGFβ signaling was blocked, but inhibition of both TGFβ and BMP signaling pathways progressively reduced hBMSC chondrogenesis to levels comparable to control pellets (Fig. 5E), suggesting a potentially critical role for endogenous BMP signaling in promoting hBMSC chondrogenesis. However, we found that inhibition of endogenous BMP signaling had no significant effect on TGFβ1-mediated chondrogenesis (Fig. 6). Taken together with our data showing that pharmacological inhibition of Hedgehog signaling blocks hBMSC chondrogenesis, this would suggest that endogenously produced IHH is perhaps more important for driving chondrogenesis than for maintaining endogenous BMP signaling in response to TGFβ1.
Surprisingly, administration of recombinant IHH had no effect on hBMSC chondrogenesis (Supplementary Fig. S1B, C). This is unexpected but may be the reason why IHH has been largely overlooked for its role during hBMSC chondrogenesis. The observed lack of responsiveness to IHH is somewhat difficult to explain, as hBMSCs undergoing chondrogenesis have been shown to express the Hedgehog receptor PTCH1 throughout differentiation [28]. Thus, lack of responsiveness may be due to lack of recombinant protein stability, bioactivity, or diffusivity within the cartilage pellets. IHH undergoes several post-translational modifications before attaining full bioactivity. IHH is synthesized as a 45 kDa precursor but is subsequently cleaved to an active 19 kDa N-terminal fragment that undergoes palmitoylation, which is required for multimerization and leads to about a 30-fold increase in bioactivity [33]. This N-terminal fragment is also modified by attachment of cholesterol, and cholesterol interactions with heparan sulfate proteoglycans facilitate long-range diffusion [34]. Cholesterol addition is also required for targeting to membrane lipid rafts and multimerization. Thus, the absence of several post-translational modifications could account for the lack of recombinant IHH bioactivity.
Similarly, hBMSCs showed no responsiveness to recombinant PTHrP (Supplementary Fig. S1A), despite the fact that activation of signaling components downstream of PTHrP signaling using the pharmacological agent, forskolin, almost completely blocked the chondrogenic differentiation program (Fig. 2B). The lack of responsiveness to PTHrP may be due simply to the absence of its receptor, PTH1R, upon chondrogenic induction, as recent evidence from our lab has found that PTH1R expression is lost upon chondrogenic induction [28]. However, other studies have found that differentiating hBMSCs are indeed responsive to PTHrP, with PTHrP bioactivity depending on the specific isoform administered [35]. Specifically, PTHrP(1–34) was found to enhance hBMSC chondrogenesis while concomitantly repressing hypertrophy. Thus, perhaps the specific isoform of recombinant PTHrP used in this study lacked potent bioactivity or stability.
It is interesting that overexpression of IHH only led to a modest increase in immunostaining for COL2 and COL10 (Fig. 5E), while knockdown of IHH led to a profound blockade of chondrogenesis (Fig. 3C, D). Most notably, overexpression of IHH led to a marginal difference in COL2 immunostaining compared to control pellets when both TGFβ and BMP signaling were blocked (Fig. 5E), despite inducing a significant upregulation in COL2 mRNA levels (Fig. 5D). This may be due to the fact that the efficiency of viral infection of the IHH overexpression plasmid was lower than the efficiency of the shRNA constructs. As a result, the IHH-overexpressing hBMSCs required a longer period of expansion to collect an adequate number of cells for preparation of chondrogenic pellets and thus were naturally closer to senescence at the time of differentiation than the IHH-knockdown cells. Indeed, we found an increase in expression of all senescence markers analyzed in undifferentiated hBMSCs before pellet formation in response to IHH overexpression (Supplementary Fig. S4). However, recombinant TGFβ1 still induced more pronounced chondrogenesis than overexpression of IHH (Fig. 5D, E), demonstrating that IHH-overexpressing hBMSCs were still capable of undergoing chondrogenesis effectively. Thus, it may simply suggest that all three signaling pathways are required for optimal chondrogenesis. Regardless, more intense immunofluorescence staining for IHH (Fig. 5C) correlated positively with the intensity of staining for COL2 and COL10 (Fig. 5E), supporting the notion that endogenously produced IHH is indeed playing a major role in hBMSC chondrogenesis.
From Fig. 7 and previously published findings, it seems plausible that the following sequence of events drives hBMSC chondrogenesis in vitro: first, recombinant TGFβ ligands induce the phosphorylation of SMAD2/3 (Fig. 7B), which then form a heteromeric complex with SMAD4 before translocating into the nucleus and activating the transcription of IHH within 3 h (Fig. 7A). Indeed, previous reports have shown an increase in IHH expression in response to TGFβ stimulation [36,37] and SMAD4 has been shown to bind GTCTAGAC palindromic sequences within the 5′ flanking region of the IHH gene, thus activating its transcription [38 –40]. Concomitantly, SMAD2/3 interacts with SOX9 to increase its transcriptional activity, perhaps by increasing its interaction with CBP/p300 [41], and ERK1/2 is phosphorylated (data not shown), which has been shown to enhance the transcriptional activity of SMAD2/3 [42]. Second, endogenously produced IHH signals in an autocrine and paracrine manner to further upregulate the expression of SOX9. In accord, SOX9 showed a significant increase in expression within 6 h of exposure to TGFβ1 (Fig. 7A), and previous work has identified Hedgehog proteins as positive regulators of SOX9 expression [43,44]. Most notably, Bien-Willner et al. showed that GLI1, a downstream transcription factor of Hedgehog signaling, directly upregulates the expression of SOX9 by binding a cis-acting regulatory element 1.1 Mb upstream of SOX9 [45]. Third, the endogenous expression of TGFβ3 and BMP2 is significantly upregulated within 12 h of exposure to recombinant TGFβ1 (Fig. 7A), which leads to the creation of a positive feedback mechanism that drives the continued phosphorylation of SMAD proteins, activation of IHH transcription, and increase in SOX9 activity and expression that finally induces a significant upregulation in the expression of COL2 and COL10 by 24 h post-TGFβ1 treatment.
While the findings in this study focused on the role that extracellular cues play in driving hBMSC chondrogenesis in vitro, future studies should more thoroughly relate these findings to the intracellular cues driving differentiation. Namely, it would be informative to knockdown components of TGFβ, BMP, and Hedgehog signaling pathways to further elucidate the relative importance of these three signaling pathways for TGFβ-mediated chondrogenesis. Specifically, it would be useful to knockdown the expression of SMAD2/3, SMAD1/5/8, or GLI1/2 and assay their impact on chondrogenesis. Subsequently, immunoprecipitation studies could be carried out to elucidate any cooperative activities between these transcription factors with each other, or conversely with SOX9 and RUNX2, and chromatin immunoprecipitation studies could be carried out to determine which of these transcription factors is interacting with enhancer regions of cartilage- and hypertrophy-related genes. In so doing, these insights would allow us to begin to better control the differentiation process and perhaps even begin to create in vitro environments capable of simultaneously attenuating multiple signaling pathways to drive hBMSC chondrogenesis without hypertrophy.
Overall, we have found that endogenously produced IHH is both necessary and sufficient for the successful execution of hBMSC chondrogenesis, as knockdown of IHH almost completely blocked the induction of chondrogenesis by TGFβ1, and overexpression of IHH was able to induce hBMSC chondrogenesis even when TGFβ signaling was blocked. In addition, knockdown of IHH appears to block the downstream effects of TGFβ1 entirely. Thus, IHH may be a more direct inducer of hBMSC chondrogenesis than TGFβ1. Additionally, the endogenous expression of TGFβ3 and BMP2 are reproducibly attenuated downstream of IHH manipulation, suggesting that IHH maintains endogenous TGFβ and BMP signaling and the cooperative activities of the three pathways together induce the most pronounced chondrogenesis.
Footnotes
Acknowledgments
We thank the University of Wisconsin-Madison Stem Cell and Regenerative Medicine Center for the graduate fellowship supporting A.M.H. and the Emery Bresnick Laboratory for advice regarding plasmid construction.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.