
Abstract
Tumor necrosis factor-α (TNF-α) has multiple effects on proliferation and differentiation of human mesenchymal stem cells. Transforming growth factor-activated kinase-1 (TAK1) mediates the activation of nuclear factor-kappa B (NF-κB), c-Jun N-terminal kinase (JNK), and p38 pathways in response to TNF-α. However, the role of TAK1 in TNF-α-induced effects in human adipose-derived stem cells (hADSCs) and its signaling pathway has not been clearly defined. Therefore, this study was designated to clarify the role of TAK1 in TNF-α-induced actions on proliferation and differentiation of hADSCs and its downstream signaling pathway. Inhibiting TAK1 expression inhibited the TNF-α-induced increase in osteogenic differentiation and basal osteogenic differentiation without affecting the TNF-α-induced effect on proliferation and adipogenic differentiation of hADSCs. A western blot analysis showed that TNF-α treatment induced degradation of IκB, but that TAK1 small interfering RNA (siRNA) transfection did not protect against TNF-α-induced IκB degradation. The transfection of TAK1 siRNA also did not affect TNF-α-induced IκB phosphorylation or ERK1/2 phosphorylation. However, downregulating TAK1 inhibited this TNF-α-induced S536 phosphorylation of the p65 subunit. TNF-α treatment induced p38 phosphorylation, which was inhibited by the transfection of TAK1 siRNA. Adding p38 inhibitor inhibited TNF-α-induced p65 phosphorylation, NF-κB promoter activity, and TNF-α-induced increase in hADSC osteogenic differentiation. These data indicate that TAK1 is involved in the TNF-α-induced activation of p38 kinase, which subsequently phosphorylates the NF-κB p65 subunit, and increases the transactivation potential of p65 and osteogenic differentiation in hADSCs.
Introduction
M
During the inflammation phase, chemokine and proinflammatory cytokines such as interleukins and tumor necrosis factor-α (TNF-α) are released. TNF-α affects the proliferation and differentiation of human MSCs (hMSCs) [4 –6] and the production of biological factors from MSCs [7], and the pretreatment of TNF-α can alter the therapeutic efficacy of transplanted MSCs [8,9]. Therefore, understanding the action mechanisms of TNF-α on MSCs is important for technology development of MSC manipulation for enhancing therapeutic efficacy of their transplantation. TNF-α inhibits adipogenic differentiation in preadipocytes [10,11] and affects osteogenic differentiation and cell proliferation of MSCs. Li et al. showed that TNF-α inhibits osteogenic differentiation of hMSCs by suppressing transcriptional coactivator with PDZ-binding motif (TAZ) expression [4]. In contrast, TNF-α promotes osteogenic differentiation in human bone marrow-derived MSCs [5] and human adipose-derived stem cells (hADSCs) [12,13] by triggering the nuclear factor-kappa B (NF-κB) signaling pathway. The opposing effects of TNF-α on MSC proliferation have also been reported. Bocker et al. [6] and Kim et al. [14] reported that TNF-α increases the invasive and proliferative properties of human bone marrow-derived mesenchymal stem cells (hBMSCs), but Ghali et al. reported conflicting results that TNF-α inhibit the proliferative and apoptotic properties of hBMSCs [15].
Transforming growth factor-activated kinase-1 (TAK1) is a serine/threonine kinase in the mitogen-activated protein kinase kinase kinase (MAPKKK) family. In response to interleukin-1, TNF-α, transforming growth factor-beta1 (TGF-β1), and toll-like receptor agonists, TAK1 mediates activation of the NF-κB, c-Jun N-terminal kinase (JNK), and p38 pathways, and regulates cell survival, differentiation, and inflammatory responses [16]. Therefore, TAK1 may play an important role in MSC proliferation and differentiation. TAK1 interferes with bone morphogenetic protein (BMP)-dependent osteogenic development in murine mesenchymal C2H10T 1/2 cells [17], whereas TAK1 knockout mice display clavicular hypoplasia and hypomineralization of the calvarium [18]. TAK1 mediates TNF-α-induced inhibition of adipogenesis by modulating peroxisome proliferator-activated receptor (PPAR) gamma [19,20].
We have previously demonstrated that TNF-α increases osteogenic differentiation by activating the NF-κB pathway in hADSCs [11] and increases their proliferation [14]. Because TAK1 plays an important role in TNF-α-induced NF-κB activation [21], this study was designed to determine the role of TAK1 in the TNF-α-induced actions of hADSCs.
Materials and Methods
Cell culture
All protocols involving human subjects were approved by the Institutional Review Board of Pusan National University. Superfluous material was collected from four different patients who had abdominoplasty, after informed consent was given by each individual. The patient informations were given in Table 1. The hADSCs were isolated according to methods described previously [22]. The isolated cells were cultured in α-MEM containing 10% fetal bovine serum (FBS). When the monolayer of adherent cells reached 80% confluence, the cells were trypsinized (0.25% trypsin; Sigma, St. Louis, MO), resuspended, and subcultured. Cells between third and fifth passages were used for experiments. We performed duplicate experiments of hADSCs, which were isolated from two different donors (n=4).
BMI, body mass index.
Reagents
Recombinant human TNF-α was purchased from R&D systems (Minneapolis, MN), and SB202190 (p38 MAP kinase inhibitor) was purchased from Sigma.
Small interfering RNA transfection
All transfections were performed using the DharmaFECT transfection reagent according to the manufacturer's instructions (Dharmacon, Lafayette, CO. Small interfering RNA (siRNA) duplex oligo (on-TARGET plus SMART pool; Dharmacon) for TAK1 mRNA or nontargeting duplex oligo (on-TARGET plus siCONTROL; Dharmacon) as a negative control was transfected using the DharmaFECT Transfection Reagent. Cells were harvested after 72 h and subjected to real-time polymerase chain reaction (PCR) and western blot analysis.
Evaluation of cell proliferation
Cells were harvested using 0.025% trypsin and incubated with 4% Trypan Blue solution. The number of viable and nonviable cells was counted using a hemocytometer under light microscopy. Cells failing to exclude the dye were considered nonviable.
Adipogenic, osteogenic, and chondrogenic differentiation
After plating the cells on a 12-well plate (100,000 cells/well), they were grown to confluency. Then adipogenic differentiation was induced by culturing the cells for 10–14 days in the adipogenic medium (10% FBS, 1 μM dexamethasone, 0.5 mM 3-isobutyl-1-methylxanthine, and 200 μM indomethacin in α-MEM) and lipid droplets formation was visualized using the Oil Red O stain as an indicator of intracellular lipid accumulation. To obtain quantitative data, 1 mL of 100% isopropanol was added to the stained culture dish. After 5 min, the absorbance of the extract was assayed by a spectrophotometer at 510 nm.
Osteogenic differentiation was induced by culturing the cells for 2–3 weeks in osteogenic induction medium (10% FBS, 0.1 μM dexamethasone, 10 mM β-glycerophosphate, and 250 μM ascorbic acid in α-MEM) and the level of extracellular matrix calcification was determined by Alizarin Red S staining. To obtain quantitative data, 300 μL of 10% cetylpyridiniumchloride in 10 mM sodium phosphate (pH 7.0) was added to the stained culture dish. After 15 min, the extract was diluted 10-fold with 10% cetylpyridiniumchloride in 10 mM sodium phosphate (pH 7.0), then the absorbance of the diluted extract was assayed by a spectrophotometer at 562 nm.
After extraction, the cells were washed with PBS and added DNAzol reagent (Invitrogen, Gaithersburg, MD) with 10 μL proteinase K solution (20 mg/mL; Sigma). The mixture was incubated at room temperature for 1 h and homogenized with micropipette tip. The homogenate was centrifuged at 10,000 g at 4°C for 10 min. The supernatant was transferred into DNA-binding spin column placed in a 2-mL collection tube (Total DNA Extraction Mini Kit; iNtRON Biotechnology, Inc., Sungnam, Korea). DNA was isolated according to the manufacturer's protocol and the absorbance values were normalized to DNA content.
Chondrogenic differentiation was induced by the micromass culture technique [23]. Briefly, 0.8×104 hADSCs were seeded with growth medium (DMEM/LG, 10% FBS, 1% Penicillin/Streptomycin) in a 24-well plate and the plate was swirled gently. Compact, round cell micromasses formed within 24 h of incubation at 5% CO2 and 37°C. After 48 h, the pellets were transferred to 1 mL chondrogenic media (containing DMEM-HG, 10% FBS, 1% penicillin/streptomycin, 50 μg/mL ascorbate-2-phosphate, ITS premix, and 10 ng/mL TGF-β1). Medium was replaced every 3 days and pellets were cultured for up to 5 weeks.
Quantitation of mineral formation
For the calcium deposition assay, cultures were washed twice with PBS. Mineral was then collected after dissolution with 300 mL of 0.5 N hydrochloric acid at room temperature overnight, and the samples assayed the following day. Incorporation of calcium in the extracellular matrix was quantified using a commercial diagnostic kit (Quantichrom calcium assay kit, DICA-500; Bio Assay Systems, Hayward, CA), in accordance with the manufacturer's instructions. Absorbance was compared with curves prepared using standard solutions of calcium. Calcium deposition was expressed as mM per well of tissue culture 12-well plates.
Real-time PCR
Total RNA was extracted using Trizol (Invitrogen), according to the manufacturer's instructions and reverse transcribed into cDNA with the Reverse Transcriptase M-MLV (Promega Corporation, Madison, WI). Real-time quantitation was based on the LightCycler assay, using the fluorogenic SYBR Green Master Mix for PCR with the LightCycler Instrument (Applied Biosystems, Foster City, CA). β-D-glucuronidase (GUSB) mRNA was amplified as an internal control, because β-actin expression is affected by TNF-α signaling [24]. The primer sequences used in the experiment were as follows: GUSB, 5′-GCGTCCCACCTAGAATCTGC-3′, 5′-CATACGGAGCCCCCTTGT-3′, TAK1, 5′-TACTCGACCACCACTGATAA-3′, 5′-GAGTTGCTCTGTCCTTCATC-3′. For each primer pair, the linearity of detection was confirmed by measuring a dilution curve with cDNA isolated from hADSCs. The data were presented as the relative ratio to GUSB levels of the corresponding samples. Negative and positive controls were included in all experiments.
Western blot analysis
Confluent hADSCs were treated under the appropriate conditions and lysated, after which the protein content of the lysate was determined using a protein assay kit. The proteins were loaded on 10% sodium dodecyl sulfate–polyacrylamide gels, transferred to nitrocellulose membranes (Hybond-ECL; Amersham Pharmacia Biotech, Piscataway, NJ), and probed with monoclonal or polyclonal antibodies [anti-TAK1, anti-Erk1/2, antiphospho (Thr202/Tyr204)-Erk1/2, anti-p38, antiphospho (Thr180/Tyr182)-p38, anti-IκB, antiphospho (Ser32/36)-IkB, anti-NF-κB, antiphospho (Ser536)-NF-kB; Cell Signaling Technology, Danvers, MA]. Immunoreactive bands were detected with anti-rabbit peroxidase-conjugated secondary antibodies and visualized through enhanced chemiluminescence.
Reporter gene assay
All transient transfections were performed using the Lipofectamine Plus Reagent (Invitrogen). The transient transfections were performed using pNF-κB-Luc, pCMV-β-Gal plasmid (Clontech Laboratories, Inc., Palo Alto, CA). The cells were harvested 48 h after transfection in 0.25 M Tris, 2 mM DTT, 2 Mm 1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid, 10% glycerol, and 1% Triton X-100 buffer (lysis buffer), and subsequently assayed for luciferase activity (Luciferase Assay System; Promega Corporation). The β-galactosidase activity was determined using the β-galactosidase Enzyme Assay System (Promega Corporation). Luciferase activity was normalized by β-galactosidase activity. Transfections were conducted in duplicate, and all experiments were repeated four times.
Statistical analysis
All results were presented as the mean±SEM. Comparisons between groups were analyzed through two-sided t-tests or analysis of variance for experiments with more than two subgroups. Post hoc range tests and pairwise multiple comparisons were conducted using the two-sided t-test with Scheffe adjustments. P values <0.05 were considered statistically significant.
Results
Effect of TAK1 on hADSC proliferation and differentiation
The expression profiles of hADSC surface markers were characterized in previous studies from our laboratory [22,25]. The hADSCs to be used in this experiment retained the potential for osteogenic, adipogenic, and chondrogenic differentiation (Supplementary Fig. S1; Supplementary Data are available online at
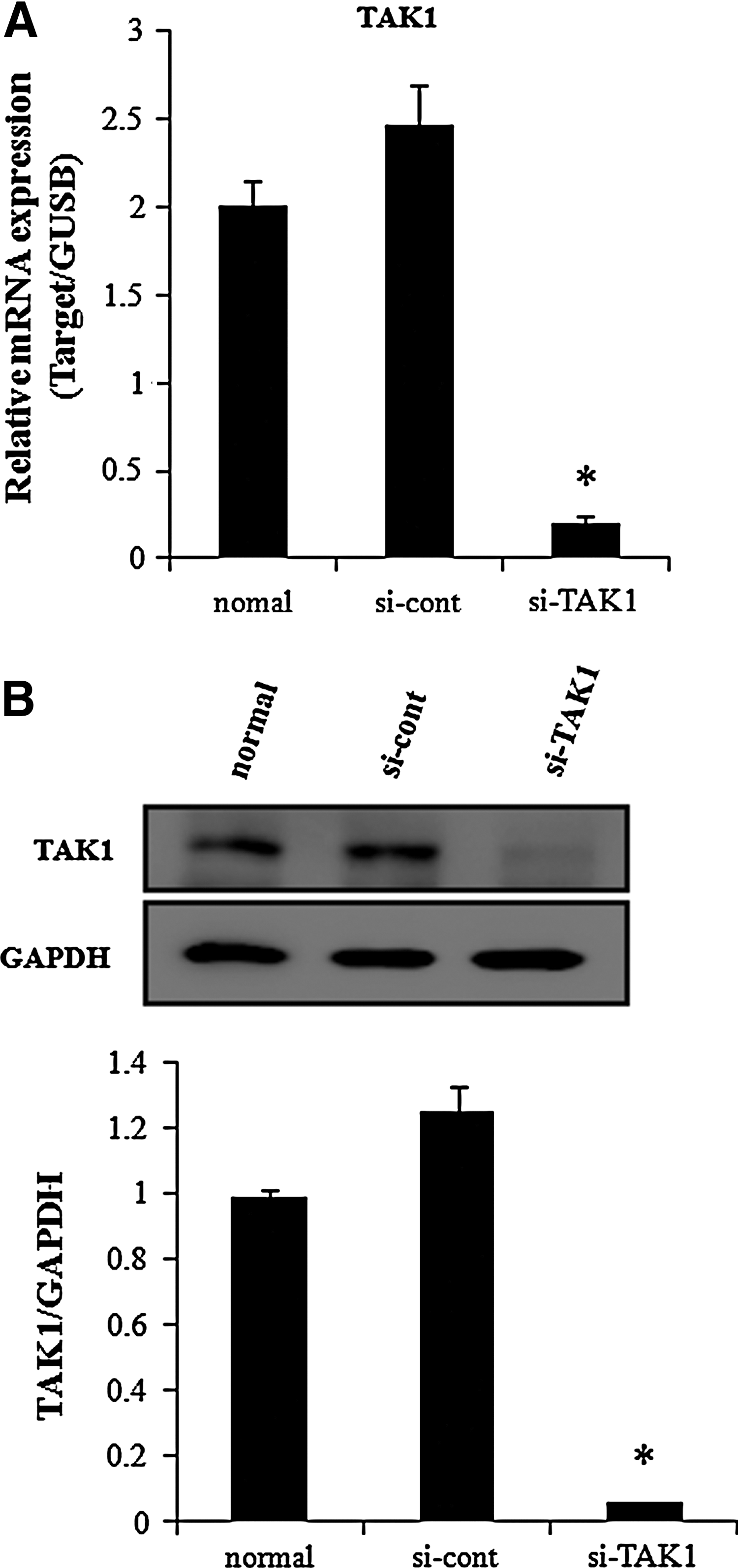
TAK1 downregulation on hADSCs.
TAK1 plays an important role in TNF-α action. We previously reported that TNF-α increases cell proliferation and osteogenic differentiation [12], while inhibiting adipogenic differentiation [11]. Inhibiting TAK1 expression by siRNA transfection inhibited NF-κB promoter activity, which increased following TNF-α treatment (Fig. 2A). We then determined whether downregulating TAK1 expression affected TNF-α actions in hADSCs. TAK1 siRNA transfection did not affect the basal or TNF-α-induced increase in hADSC proliferation (Fig. 2B). Transfection of control siRNA resulted in similar proliferation and NF-κB promoter activities compared with those in naive hADSCs (Supplementary Fig S3A, B). hADSCs were treated with 10 ng/mL TNF-α to assess the effects on adipogenic and osteogenic differentiation and determine whether inhibiting TAK1 expression affected the TNF-α-induced response during hADSC differentiation. Inhibiting TAK1 expression did not affect basal adipogenic differentiation or TNF-α-induced inhibition (Fig. 3A). In contrast, inhibiting TAK1 expression inhibited the TNF-α-induced increase in osteogenic differentiation and the basal osteogenic differentiation (Fig. 3B, C).

Effects of TAK1 downregulation on TNF-α-induced NF-κB activation and proliferation in hADSCs.
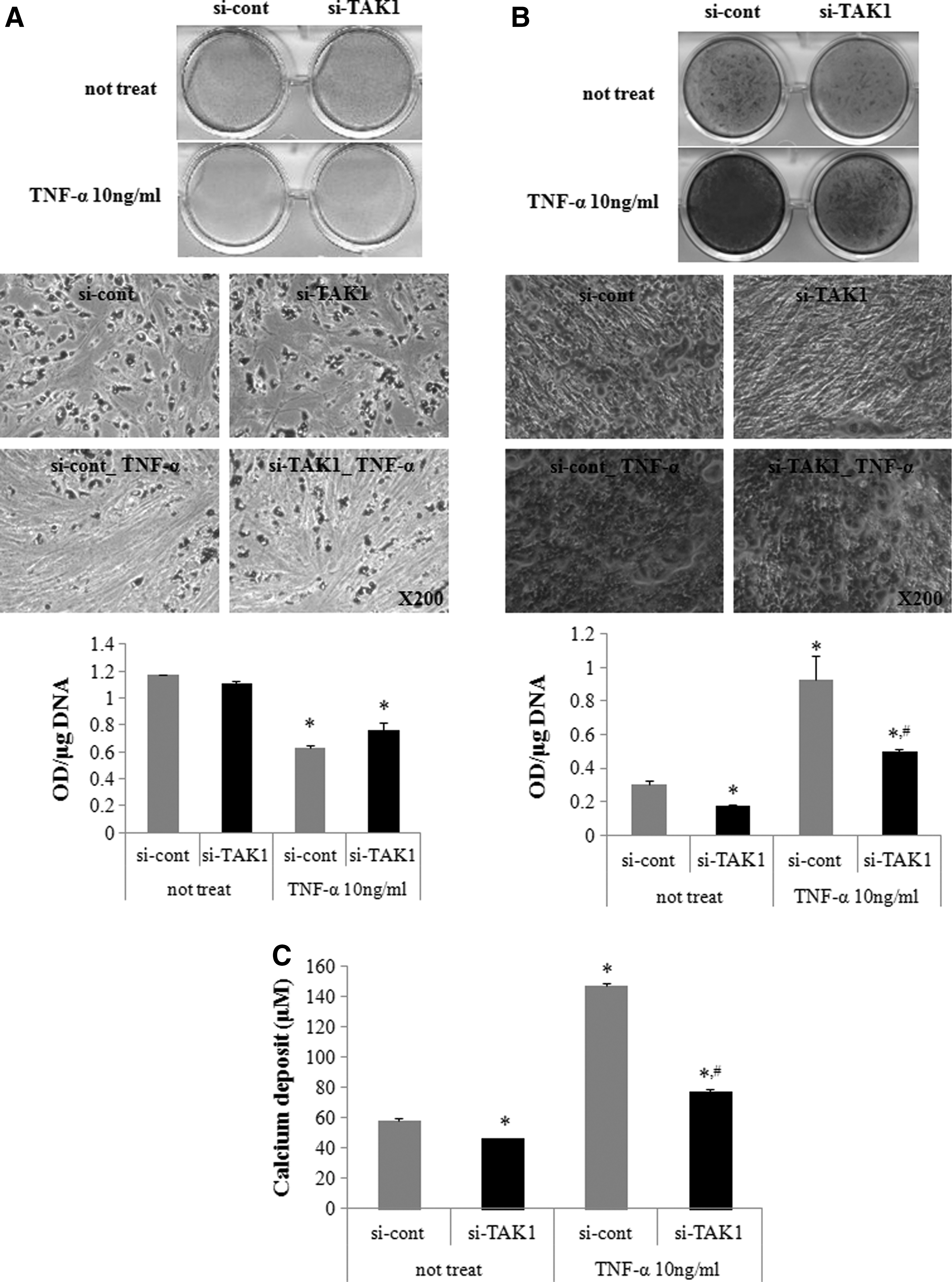
Effects of TAK1 downregulation on TNF-α-induced adipogenic and osteogenic differentiation in hADSCs.
Molecular mechanisms of TAK1-mediated TNF-α actions in hADSCs
We next determined the effect of TAK1 siRNA on the protein levels of Erk1/2 activation, IκB, and IκB phosphorylation, to assess whether TAK1 induced this molecular pathway. Western blot analysis showed that TNF-α treatment induced IκB degradation, whereas TAK1 siRNA transfection did not protect against TNF-α-induced degradation. TAK1 siRNA transfection did not affect TNF-α-induced IκB phosphorylation or ERK1/2 phosphorylation (Fig. 4A, B).
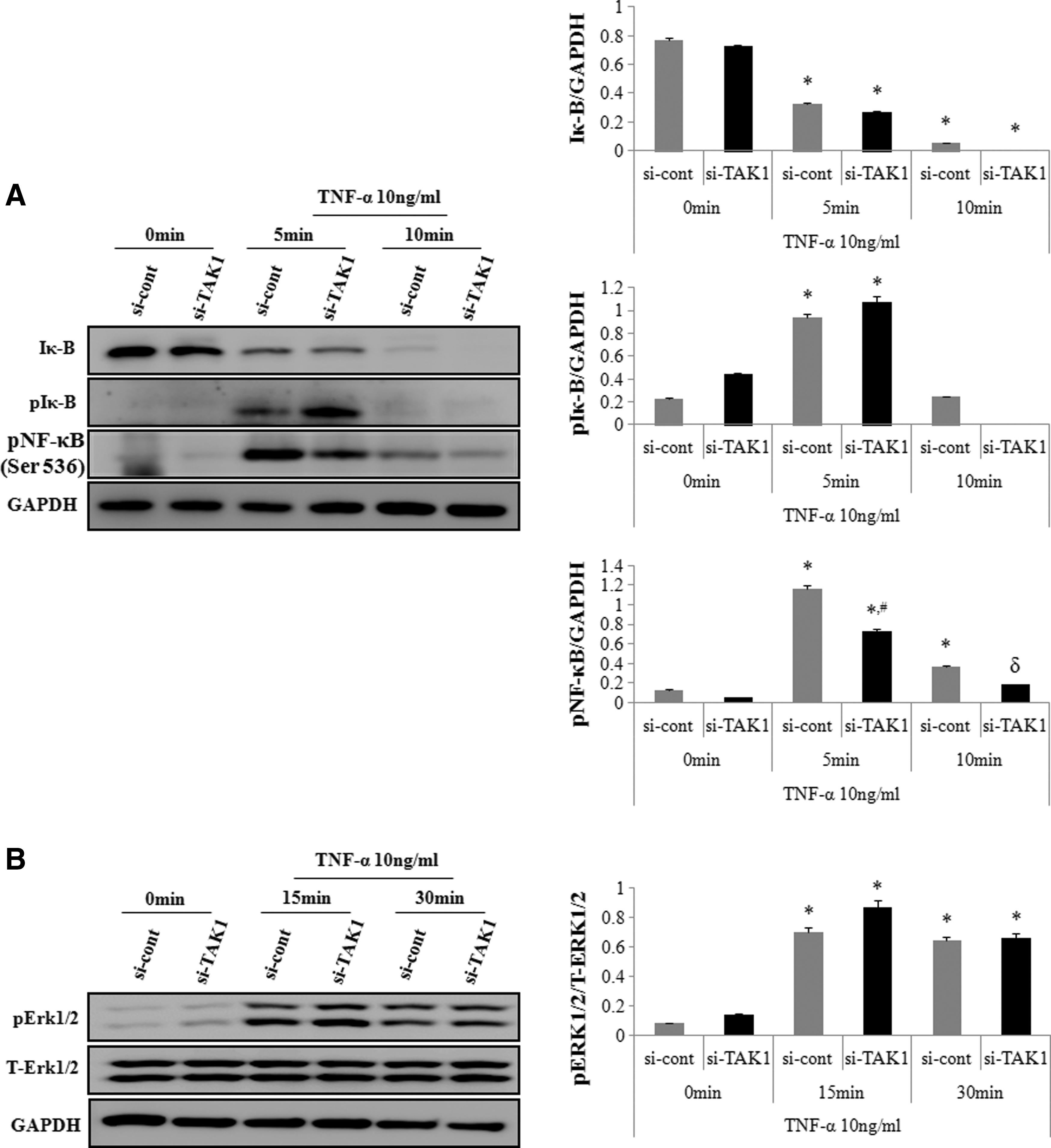
Effects of TAK1 downregulation on TNF-α-induced IκB, NF-κB and ERK phosphorylation in hADSCs.
We then determined the effect of TAK1 siRNA on TNF-α-induced S536 phosphorylation of the NF-κB p65 subunit. Western blot analysis showed that TNF-α treatment increased p65 phosphorylation at 5 and 10 min after TNF-α treatment, which was inhibited by downregulating TAK1 with its specific siRNA (Fig. 4A).
Western blot analysis showed that TNF-α treatment induced p38 phosphorylation in hADSCs. Therefore, the effect of TAK1 siRNA on TNF-α-induced p38 activation was examined. TAK1 siRNA transfection inhibited TNF-α-induced p38 activation (Fig. 5A). Then, we determined the effect of inhibiting p38 on TNF-α-induced p65 phosphorylation and NF-κB promoter activity. Adding 10 μM SB202190, a p38 inhibitor, inhibited TNF-α-induced p65 phosphorylation and NF-κB promoter activity, as determined by luciferase activity (Fig. 5A, B).
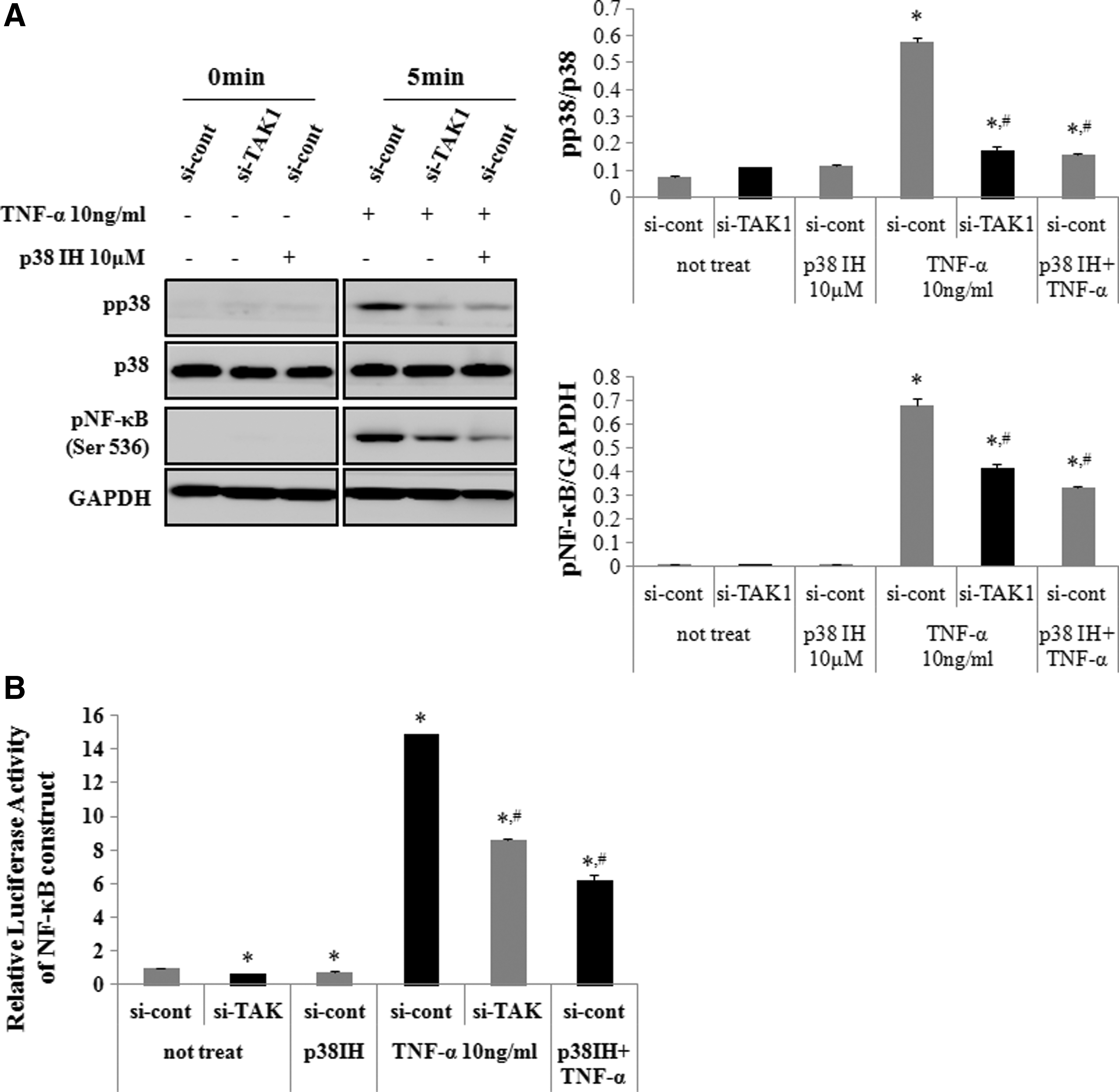
Effects of TAK1 downregulation on TNF-α-induced p38 phosphorylation and NF-κB activation in hADSCs.
Finally, the p38 inhibitor was used to study its effects on the TNF-α-induced increase in osteogenic differentiation. Adding 10 μM SB202190 blocked the TNF-α-induced increase in hADSC osteogenic differentiation (Fig. 6A, B).

Effect of p38 inhibition on TNF-α-induced osteogenic differentiation in hADSCs.
Discussion
The data in this study shows that downregulating TAK1 expression, achieved by siRNA technology, inhibited the TNF-α-induced activation of NF-κB, as determined by the luciferase assay, and that downregulating TAK1 also inhibited the TNF-α-induced increase in hADSC osteogenic differentiation, without affecting TNF-α-induced inhibition of adipogenic differentiation. These results indicate that TAK1 is an important component of the TNF-α-mediated NF-κB activation pathway.
TAK1 plays an important role in the classical NF-κB pathway, although TNF-α did not affect the expression of TAK1 in hADSCs. Binding of TNF-α to the tumor necrosis factor receptor leads to the recruitment of proteins to the receptor complex, including TRADD, TRAF2, TRAF5, and RIP1. The TRAF proteins promote K63-linked polyubiquitination of RIP1 and autopolyubiquitination of the TRAF proteins themselves. Polyubiquitinated RIP1 recruits the TAK1 kinase to the complex, resulting in the activation of IKK. Following activation, IKK phosphorylates the IκB proteins and targets these inhibitors for proteasome degradation. Subsequently, NF-κB enters the nucleus to activate a large array of downstream target genes, including proinflammatory cytokines and negative regulators of the NF-κB pathway, such as A20, CYLD, and IκBα. Therefore, downregulating TAK1 by siRNA inhibits IκB phosphorylation and degradation [21,26]. However, we found that downregulating TAK1 did not affect TNF-α-induced degradation and phosphorylation of IκB, despite that downregulating TAK1 inhibited NF-κB luciferase activity.
Activated TAK1 activates p38 mitogen-activated protein kinase and JNK, besides its association with IKK. Several studies have shown that p38 kinase stimulates the NF-κB transactivation potential. Inhibiting p38 kinase diminishes phosphorylation of the NF-κB p65 subunit transactivation domain, as shown in microglia [27] and in NIH3T3 cells [28], which results in inhibiting the NF-κB transactivation potential. In this study, downregulating TAK1 inhibited the induction of s536 phosphorylation of p65 and the p38 kinase activation, which was usually observed following TNF-α treatment. Furthermore, inhibiting p38 kinase blocked the TNF-α-induced increase in p65 phosphorylation and NF-κB promoter activity. These results indicate that the role of TAK1 in TNF-α-induced activation of NF-κB is mediated by increased p65 phosphorylation and subsequent stimulation of the p65 transactivation potential and not by IκB degradation and phosphorylation.
Activation of p38 by BMP2 in MC3T3-E1 cells [29], which is a noncanonical Wnt-4 signaling pathway in hMSCs [30] and by parathyroid hormone in osteoblasts (MC3T3-E1 cells) [31] is involved in osteogenic differentiation. Greenblatt et al. demonstrated that deleting p38 results in reduced bone mass secondary to defective osteoblast differentiation and that TAK1 is a critical upstream activator of p38 in osteoblasts [18]. The role of p38 kinase on increased hADSC osteogenic differentiation induced by short-term TNF-α exposure has been reported [13]. However, those authors did not investigate how TNF-α-induced activation of p38 kinase increases osteogenic differentiation. The data from our study demonstrate that downregulating TAK1 and inhibiting p38 kinase blocked the TNF-α-induced increase in hADSC osteogenic differentiation. Therefore, the TNF-α-induced increase in hADSC osteogenic differentiation is mediated by TAK1-mediated p38 kinase activation and subsequent enhancement of the NF-κB p65 subunit transactivation potential.
TAK1 plays an important role in TNF-α-induced proliferation of endometrial cells [32]. However, we found that downregulating TAK1 failed to inhibit the TNF-α-induced increase in hADSC proliferation or to activate ERK. In this study, downregulating TAK1 expression did not affect TNF-α-induced ERK activation. Liu et al. reported that the inhibiting TAK1 induced activation of MAPK in Xenopus ectoderm and mesoderm through the modulation of p38 kinase [33]. However, it has been reported that BMPR signaling was impaired and phosphorylation of Smad1/5/8 and p38/Jnk/Erk MAP kinases were reduced in TAK1-deficient chondrocytes [34].
This discrepancy may have resulted from differences in cell types used in the experiment. Downregulating TAK1 did not affect TNF-α-induced inhibition of adipogenic differentiation, indicating that the action of TNF-α on adipogenic differentiation is not mediated by activating NF-κB. Instead, TNF-α-induced inhibition of adipogenic differentiation is mediated by multiple mechanisms, including inhibited PPAR gamma expression [10,35].
In conclusion, our results indicate that TAK1 plays an important role in TNF-α-induced activation of NF-κB and osteogenic differentiation by activating p38 kinase and subsequently phosphorylating the NF-κB p65 subunit in hADSCs.
Footnotes
Acknowledgments
This research was supported by ICT & Future Planning, the National Research Foundation of Korea (NRF), the Ministry of Science, (NRF-2012M3A9B4028558), and by the Korea Healthcare Technology R&D Project, Ministry of Health & Welfare (A101150).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.