
Abstract
Human cord blood (CB) hematopoietic stem cell (HSC) transplants demonstrate delayed early neutrophil and platelet recovery and delayed longer term immune reconstitution compared to bone marrow and mobilized peripheral blood transplants. Despite advances in enhancing early neutrophil engraftment, platelet recovery after CB transplantation is not significantly altered when compared to contemporaneous controls. Recent studies have identified a platelet-biased murine HSC subset, maintained by thrombopoietin (TPO), which has enhanced capacity for short- and long-term platelet reconstitution, can self-renew, and can give rise to myeloid- and lymphoid-biased HSCs. In previous studies, we have shown that transplantation of human CB CD34+ cells precultured in TPO as a single graft accelerates early platelet recovery as well as yielding long-term repopulation in immune-deficient mice. In this study, using a double CB murine transplant model, we investigated whether TPO cultured human CB CD34+ cells have a competitive advantage or disadvantage over untreated human CB CD34+ cells in terms of (1) short-term and longer term platelet recovery and (2) longer term hematological recovery. Our studies demonstrate that the TPO treated graft shows accelerated early platelet recovery without impairing the platelet engraftment of untreated CD34+ cells. Notably, this was followed by a dominant contribution to platelet production through the untreated CD34+ cell graft over the intermediate to longer term. Furthermore, although the contribution of the TPO treated graft to long-term hematological engraftment was reduced, the TPO treated and untreated grafts both contributed significantly to long-term chimerism in vivo.
Introduction
C
The restricted number of CD34+ HSPCs in one CB unit, however, is often a hurdle to treat adults. To overcome this limitation, adults and large children are generally considered for double CB (dCB) transplantation [18 –21], with both improved nonrelapse mortality (NRM) and increased relapse-free survival reported using this dCB approach [18 –21]. The relapse mediated advantages of dCB transplants are still partly offset by NRM as a result of increased infectious complications and bleeding caused by delayed neutrophil and platelet recovery when compared to matched related or unrelated mobilized PB and BM grafts [20]. One solution to this delayed hematological engraftment is to expand the numbers of HSPCs in the graft leading to long-term repopulating cells most preferably in combination with the expansion of neutrophil and platelet progenitors to enable early hematological repopulation as well. Unfortunately most ex vivo manipulations seem to be associated with the loss of long-term repopulation and/or the skewing of progenitors toward neutrophil differentiation.
Although substantially higher CD34+ cell numbers have been generated by ex vivo culture with cytokines and small molecules or the homing/engraftment of these cells has been enhanced with small molecules in the attempts reported so far [22 –32], the cotransplantation of an unmanipulated CB unit with an ex vivo expanded CB unit or part of a unit has been regarded as a sensible precaution as the long-term repopulating hematopoietic stem cells (HSCs) often originate from the nonexpanded CB unit. Importantly, the first cautious attempts to expand one of the grafts before dCB transplantation have significantly reduced the time to neutrophil recovery [25 –28,32], although platelet recovery has remained substantially delayed when compared to current rather than historical transplant outcomes [33].
Thrombopoietin (TPO) is known to be critical for both HSC maintenance and platelet production [34 –40]. Sanjuan-Pla et al. [41] have recently identified, in the mouse, a TPO-dependent platelet-biased HSC expressing Sca-1, c-kit, CD150, and von Willebrand factor (vWF), which exists at the apex of the hematopoietic hierarchy and which not only generates platelets over the short and longer term, but also can give rise to both myeloid- and lymphoid-biased HSCs. This corresponding TPO dependent platelet-biased subset has not been identified in the human, principally because in the mouse a vWF-eGFP reporter is used. However, previous studies from our group and others have demonstrated that culturing human CB CD34+ cells for 7–9 days with TPO can lead to improved early platelet recovery in immune-deficient mice when compared to transplantation with untreated CB CD34+ cells in the single CB transplant setting, without apparently compromising longer term hematological reconstitution [42 –44]. Since thrombocytopenia is a common complication of both single and dCB HSC transplants, it has been proposed from these studies that TPO treatment ex vivo of one or part of one CB unit in a dCB transplant setting might be especially useful in preventing delays in platelet recovery posttransplant and hence in improving the overall patient survival [1,45]. Although purified and ex vivo manipulated CD34+ are most likely to be combined with an unmanipulated CB unit in the transplant setting, the influence of ex vivo manipulation on hematological reconstitution should be compared first for a fixed number of CD34+ cells in surrogate models of dCB transplantation ahead of assessing other immunological and matching dependent effects of the graft on repopulation and final chimerism.
In this article, we, therefore, used a dCB transplant model in NSG [NOD/SCID/IL non-obese diabetic/severe combined immunodeficient/Interleukin-2Rγ(null)] mice and isolated CD34+ cells from three different CB donor pairs to first determine if TPO treated CD34+ HSPCs from one CB unit when combined with untreated CD34+ cells from another CB unit could both improve and sustain platelet recovery when compared with a dCB transplant of untreated CD34+ cells of both donors. Second, we assessed if untreated CB CD34+ cells from one donor had a competitive advantage or disadvantage when cotransplanted with the same number of TPO treated versus untreated CB CD34+ cells from a second donor in terms of short or longer term platelet recovery and longer term hematological reconstitution. Interestingly, the TPO cultured CD34+ cells were responsible for improved early platelet recovery and contributed substantially to longer term platelet recovery in the dCB transplant setting, although the dominant contribution to platelet recovery in the intermediate to longer term arose from the untreated CD34+ cells. Furthermore, although there was less contribution from the TPO-treated CD34+ cell graft, long-term hematological cell reconstitution was dependent on both the TPO treated and the untreated grafts. Finally, the hematopoietic potential of the untreated CB cell units was not negatively influenced by the cotransplantation of TPO treated cells.
Materials and Methods
Umbilical CB collection
CB was drawn from the umbilical vein at birth at >36 weeks gestation after written informed consent from the mother at peripheral hospitals in the Netherlands according to Netcord–FACT standards and with ethical permission from the medical ethics board of the Leiden University Medical Center (LUMC; Leiden, The Netherlands). Blood was collected by gravity drainage into the Macopharma collection bags containing 21 mL citrate phosphate dextrose adenine-1 (Macopharma). The blood was stored at 4°C and processed within 48 h of collection.
CD34+ cell purification
Mononuclear cells were isolated from CB using a sterile Ficoll density gradient (1.077 g/cm3; Pharmacy LUMC). The CD34+ cell fraction was isolated from the mononuclear cell fraction by double CD34+ cell selection using immunomagnetic beads (Miltenyi Biotec GmbH). The purity of the isolated CD34+ cell fraction was verified by flow cytometry (Beckman Coulter) using CD45-FITC and CD34-PE antibodies (Beckman Coulter) and the ISHAGE protocol [46]. Cells were cryopreserved in 10% (v/v) dimethyl sulfoxide in 4% (w/v) human serum albumin; (Pharmacy LUMC) in Iscove's modified Dulbecco's medium (IMDM) and stored at −150°C until use.
Cultures
CD34+ cells were cultured as previously described [44] in 24-well sterile tissue culture plates, at a concentration of 1×105 cells/mL, in IMDM (Gibco) supplemented with 20% (v/v) AB heparinized plasma (Sanquin Blood Supply Foundation), 0.5 mg/mL human transferrin saturated with FeCl3.H2O (Sigma), 0.34% (v/v) human serum albumin [20% (w/v) stock; Cealb® CLB], 1% (v/v) penicillin/streptomycin (PenStrep; Bio-Whittaker), 0.05 mM β-mercaptoethanol (Sigma), and 50 ng/mL mpl-ligand (Nplate) or TPO (a kind gift from KIRIN Brewery Ltd.). At day 7, the medium was refreshed by demi-dilution, with a medium containing 50 ng/mL TPO or Nplate. At day 9, the cells were split into two new wells and diluted 1:1 with medium without TPO and harvested on day 10. The total viable cell number was counted using flow cytometry and the degree of expansion calculated. The composition of the cultured cells was analyzed by flow cytometry using mouse anti-human CD45, CD61, CD34, CD3, and CD56 antibodies (all from Beckman Coulter). It should be noted that previous comparisons of TPO with Nplate in parallel cultures of CD34+ cells from five donors gave similar expansion rates and percentages of the predominant populations during culture. However, because of accessibility to reagents, TPO was changed to Nplate in the last experiment. Cells were analyzed for HLA epitope expression before and after expansion with custom-made, in-house human HLA epitope specific monoclonal antibodies [47 –50].
Transplantation in NSG mice
Female 5–6 week old NSG mice (Charles River) were kept in microisolator cages in laminar flow racks in the LUMC animal facilities. The animal ethics committee of the LUMC approved all animal experiments. NSG mice received 2.85 Gy total body irradiation 24 h before transplantation. Mice were transplanted i.v. with cells (Fig. 1) using four mice per group. Three dCB transplantation experiments were performed with one CB donor pair for each experiment, each experiment consisted of three groups of four mice. Mice received one of the following combinations of cells in their transplants (Fig. 1): (group 1) the first donor CB TPO treated CD34+ cells (X1) and the second donor CB untreated CD34+ cells (N2); (group 2) the second donor CB TPO treated CD34+ cells (X2) and the first donor CB untreated CD34+ cells (N1); and (group 3) both CB donor cells infused as untreated CD34+ cells (N1 and N2). For the analysis of the data, all mice that received one TPO treated graft and one untreated graft (groups 1 and 2) were compared with the mice that received two untreated grafts (group 3). The number of cells that each of the mice received are described in the Supplementary Data (Supplementary Data are available online at
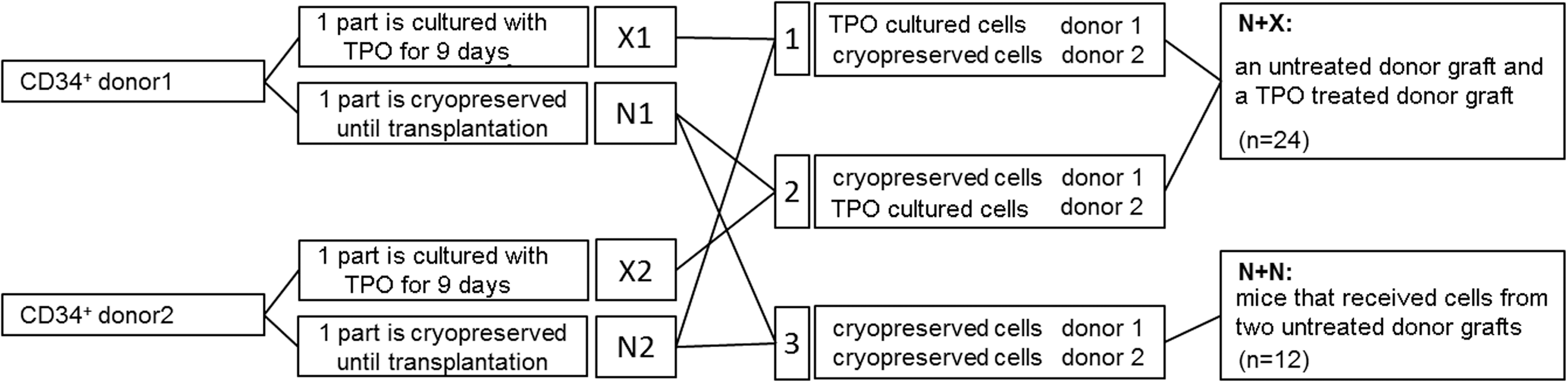
Schematic representation of the experimental strategy. Isolated CD34+ cells from six human cord blood (CB) donors were divided into three donor pairs. The cells from each donor were then split into two parts and cryopreserved. One part was cultured with thrombopoietin (TPO) for 9 days, the other part was thawed before transplantation. After TPO based culture, cells were combined with untreated cells from the other donor of the donor pair creating six groups with TPO treated cells and untreated cells (N+X group), and the untreated cells of both donors of the donor pairs were combined creating three groups with untreated cells from two donors. Cells from each group were transplanted in four NSG [NOD/SCID/IL non-obese diabetic/severe combined immunodeficient/Interleukin-2Rγ(null)] mice.
Analysis of PB after transplantation
Blood was collected from the tail vein twice weekly during the first 3 weeks after transplantation and once weekly, thereafter. Blood collection and human platelet measurements were performed as described previously [47,48,51]. Briefly, human platelets were stained with noncross reactive mouse anti-human CD41-PE and human CD45 cells were stained with mouse anti-human CD45-PC7 (both Beckman Coulter). Cell concentrations of >1×103 cells/mL PB are considered to be reliable. To determine donor origin HLA epitope-specific monoclonal antibodies conjugated with Alexa 488 or PE-Dy647 were used [45,46]. Erythrocytes were lysed with IOTest3 lysing solution (Beckman Coulter) for 10 min at room temperature. All cells were fixed with a 0.1% (w/v) formaldehyde solution (Pharmacy LUMC). Flow-Count™ fluorospheres (Beckman Coulter) were added to the cells to enable the measurement of the absolute number of circulating human platelets. Analysis was performed by flow cytometry (FC500; Beckman Coulter) using the CXP software.
Analysis of the BM, spleen, and blood after 20 weeks
Twenty weeks after transplantation, mice were sacrificed and the BM recovered from the femurs by flushing with IMDM containing 1% (v/v) PenStrep (Life Technologies) and 10 U/mL Na-heparin (Pharmacy LUMC). Single-cell suspensions were prepared from spleens that were washed with IMDM with 1% (v/v) PenStrep (Life Technologies) and 10 U/mL Na-heparin (Pharmacy LUMC) and the blood was collected through cardiac puncture. Evaluation of human cell engraftment and the relative lineage distribution of the engrafted human cells in the BM, spleen, and blood were performed with flow cytometry analysis. Cells were labeled with rat anti-mouse CD45-PE (BD Biosciences), mouse anti-human CD45-FITC, CD33-PE, CD34-PE, CD19-PE, and CD3-ECD (all from Beckman Coulter), and the appropriate isotype controls. Subsequently, erythrocytes were lysed with IO Test3 lysing solution (Beckman Coulter) for 10 min at room temperature and fixed with a 0.1% (w/v) formaldehyde solution. Chimerism was analyzed with HLA specific antibodies [48 –50]. Analysis was performed by flow cytometry (FC500) using the CXP software (Beckman Coulter).
Hematopoietic progenitor cell assays
To investigate whether BM cells harvested 20 weeks after transplantation contained human hematopoietic progenitor cells (HPC), three different in vitro assays were used; HALO assays (Hemogenix, Colorado Springs, CO), myeloid CFU cultures, and cobblestone area-forming cell (CAFC) assays as described previously [51,52] and in Supplementary Data.
Statistics
All statistics were done using the IBM SPSS Statistics (version 20;
Results
Culturing CD34+ cells with TPO
The CD34+ cell purity before TPO culture or transplant was 92%–98%. After culturing the human CB CD34+ cells with TPO, the expression of CD61 (ITGB3, GPIIIa, or beta3 integrin), which forms part of the GPIIb/IIIa (CD41/CD61) complex that binds to vWF, fibronectin, fibrinogen, and vitronectin [40,43,51] was measured. Three broad cell populations were detected: (1) total CD34+ cells (both CD34+CD61+ and CD34+CD61−), (2) CD34−CD61− cells, and (3) CD34−CD61+ cells. As observed previously [43,51], TPO induced differentiation from CB CD34+ cells first to the double negative CD34−CD61− population and then to a strongly expanded CD34−CD61+ population (Supplementary Fig. S1). With the relatively short TPO exposure, we aimed to and succeeded in generating approximately the same number of CD34−CD61− cells as the number of CD34+ cells in the starting population. This double negative population has been shown to be responsible for the earlier platelet recovery in in vivo models [48]. With the expected significant inter-experimental and inter-donor variation, TPO exposure, however, also led to a variable and sometimes higher reduction in absolute CD34+ cell numbers for some donors. However, both CD34+CD61− or CD34+CD61+cells remained present in the graft (Supplementary Fig. S1 and Supplementary Table S1).
Total platelet and leukocyte recovery in the PB of NSG mice receiving TPO-treated and untreated CD34+ cell grafts
At day 4 and onward after transplantation of TPO treated and/or untreated CB cells into NSG mice, human platelets were observed in the PB. A significantly higher number and more rapid increase of human platelets in the PB of mice was seen when mice were transplanted with CD34+ cells where those from one donor were first cultured with TPO (Fig. 2A). Ten days after transplantation, however, a rapid increase in platelet numbers was observed in mice that received two untreated and uncultured grafts (N+N group). With a notable transient peak at 2.5 weeks posttransplantation, the platelet concentration in the N+N mice even exceeded the numbers found in mice that received one TPO treated graft together with one untreated graft (N+X group) for up to 6 weeks. From that time on, the differences, although still present between the two groups, were no longer significant with platelet numbers gradually increasing in both groups up to 18 weeks after transplantation (Fig. 2B).
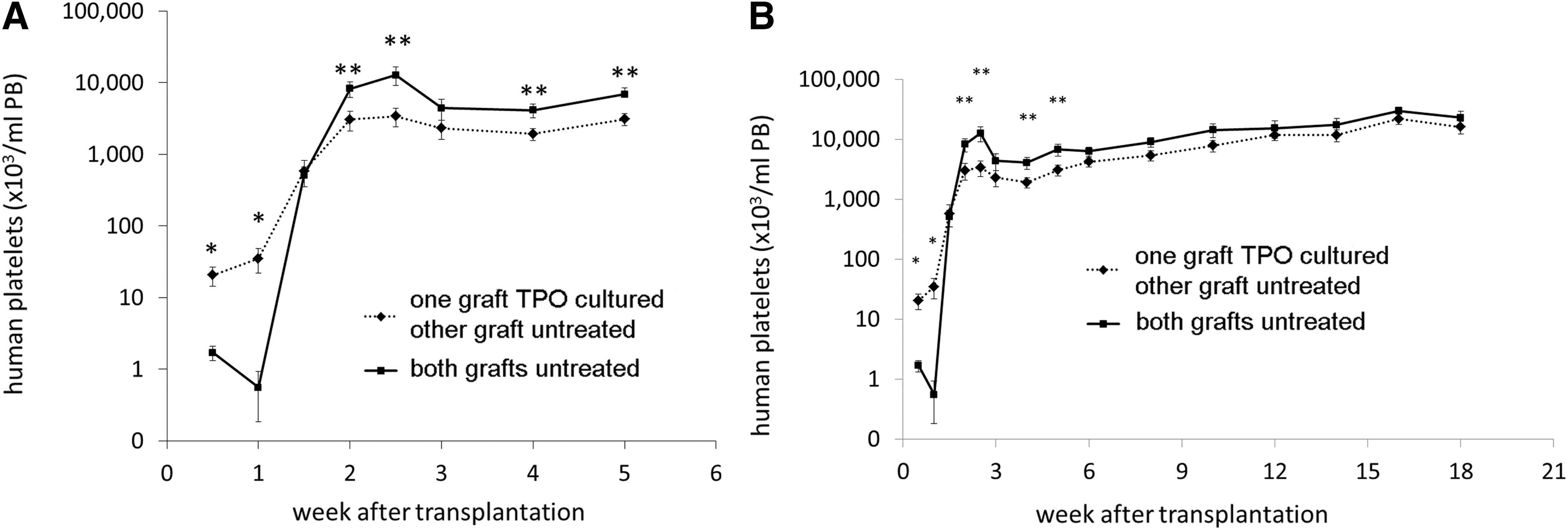
Mean human platelet concentration (plt/μL PB,±SEM) in the peripheral blood (PB) of the mice after transplantation. (♦, dashed line) one of the CB donor grafts was TPO cultured (X), (■, straight line) both CB donor grafts were untreated (N), **P≤0.05, *P≤0.02.
The human CD45+ cell concentrations in PB (signifying total human leukocytes) for the different groups are shown in Fig. 3, with all mice showing a gradual increase in PB human CD45+ cells, which began to plateau for the N+N group at 6 weeks posttransplant and for the N+X group at week 12 posttransplant (Fig. 3). A trend toward a slightly higher total human CD45+ cell recovery was observed in mice from the N+N group between 2 to10 weeks posttransplant, but the difference from the N+X group was only statistically significant at week 6 after transplantation (P≤0.05).

Mean human leukocyte concentration (±SEM) in the PB of the mice after transplantation. (♦, dashed line) one of the CB donor grafts was TPO treated (X), (■, straight line) both CB donor grafts were untreated (N) (*P≤0.05). Leukocyte recovery was slightly faster for the N+N group, but only significantly higher 6 weeks after transplantation.
Overall long-term engraftment in BM, spleen, and PB comparing the TPO treated and untreated grafts with the double untreated grafts
Twenty weeks after transplantation, the mice were sacrificed and their blood, BM, and spleens showed that 72%–89% of the CD45+ cell population was derived from human as opposed to murine leukocytes (Table 1). Additionally, both myeloid and lymphoid lineages were detected. In the lymphoid lineage, CD19+ B cells far outweighed the concentration of CD3+ T cells (Table 1). The percentage of human CD34+ cells based on the total human CD45+ cell fraction in the BM was greater than 0.5% in all mice at 20 weeks, with the variation observed between the 3 experiments being 2.91%±0.42%, 6.39%±1.16%, and 0.80%±1.11% for experiments 1, 2, and 3, respectively. Between the N+N and N+X groups, no significant differences in the percentages of lineage-specific and CD34+ cells in BM or spleens were observed (Table 1).
The number of CD45+ cells in the different hematopoietic organs was approximately similar for the N+N and the N+X group. There were differences between the percentages of the different lineages, but these were not significant.
BM, bone marrow; n.d., not done; TPO, thrombopoietin.
In vitro assays of the engrafted BM cells
Three assays were carried out to assess the human progenitor cell potential of the BM cells 20 weeks posttransplant. We verified that colonies were not mouse derived by performing similar cultures of the BM cell suspension from mice not receiving human dCB transplants and none of these yielded colonies. First, using the HALO assay where the mean concentration of ATP produced is a measure of proliferating cells, no significant difference was observed between human cells derived from the BM from the N+X or N+N groups of mice (Supplementary Fig. S2). Second, to study whether the engrafted human cells were capable of differentiating into different myeloid lineages, CFU assays were carried out with the same marrow samples. In both the N+N and the N+X group, lineage-forming potential was shown (Supplementary Fig. S3) by the generation of colonies of all different myeloid lineages: burst forming unit erythroid colonies (mean=9.1±3.7 colonies for the mice from the N+X group and 9.7±5.1 for the N+N mice), the granulocyte/macrophage colonies (CFU-GM, mean=63.2±10.9 for the mice of the N+X group and 94.0±39.9 for the mice of the N+N group), and the granulocyte/erythrocyte/macrophage/megakaryocyte colonies (CFU-GEMM, 8.8±3.2 for the mice of the N+X group graft and 13.0±9.2 for the mice of the N+N group). Although BM from mice of the N+N group on an average formed more colonies of all different myeloid lineages, none of these differences was significant (P=0.7 for the total number of colonies). Finally, we used the in vitro CAFC assay to show that the harvested BM from the transplanted mice still contained primitive cells, since the CAFC assay is used as a surrogate assay for predicting long-term engraftment potential [49]. Although cells from a representative mouse of each group were capable of generating CAFC colonies in at least two of the five wells (results not shown), less CAFC colonies could be grown from the BM cells of the mice that received mixed transplants. These culture results of all BM cells were followed up by in vivo analysis of donor-specific cell counts both in the PB and in the marrow.
Platelet and leukocyte recovery in the PB comparing the TPO treated and the untreated donor grafts
To analyze how much the TPO treated and the untreated donor grafts contribute to platelet production, PB samples were stained with HLA-specific monoclonal antibodies for epitopes that were present on one and not the other cotransplanted graft. Since the HLA staining of platelet is not absolute, we could analyze the donor contribution in two donor pairs only (n=12 mice). Figure 4A shows that the platelets produced in the first week after transplantation were indeed derived from the CD34+ cells that had been TPO treated. In contrast, when the graft was not TPO treated, platelet production commenced after week 1 only. However, from 2 weeks, the untreated CD34+ cells produced higher platelet numbers and this progressed throughout the remainder of the experiment.
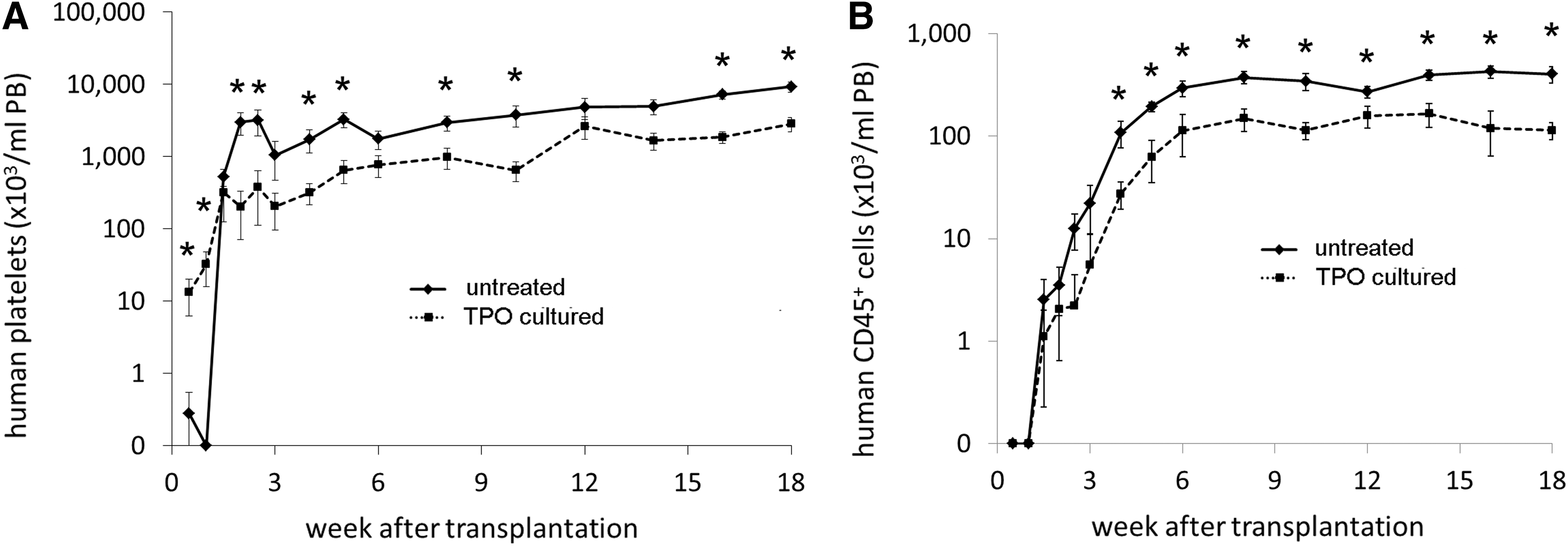
Mean (±SEM) platelet
Total CD45+ leukocyte production from the TPO treated CD34+ cells was significantly impaired. Although detectable CD45+ leukocyte levels were observed after 1.5 weeks from both the TPO cultured unit and the untreated unit, the increase in leukocytes was more rapid when the cells came from a non TPO cultured graft and the total leukocyte production from this untreated graft was higher throughout the experiment (Fig. 4B). Leukocyte production from the TPO cultured graft, however, remained present and constant although at a lower level than leukocytes from the untreated graft.
BM chimerism in mice cotransplanted with one TPO treated and one untreated graft
In agreement with the results in PB, human hematopoiesis (ie, human CD45+ cells) in the BM of mice from the N+X group 20 weeks after transplantation on average demonstrated increased human hematopoiesis from the untreated graft compared to the TPO treated graft (66%±4% vs. 34%±4% mean±SEM). This skewed chimerism in the BM for human CD45+ cells (Table 2) was also observed for CD34+ cells and B lymphoid cells (CD19+), whereas the dominance was less clear for T cells (CD3+ and myeloid cells (CD33+). These combined results indicate that both the untreated graft and the TPO cultured graft can provide long-term reconstitution, but that the initial difference in stem cell numbers between the TPO cultured and untreated grafts is reflected in their eventual chimerism.
In the BM, dominance on average came from the untreated graft.
Platelet and leukocyte recovery in the PB and long-term BM, spleen, and blood chimerism mediated by the untreated donor graft when it is transplanted with a TPO treated or an untreated donor graft
Using the same donor-specific HLA antibodies used for the analysis of the contribution of each donor graft in the N+X group, we next studied if the engraftment efficacy of the untreated CD34+ cells from one donor could be changed by the presence of TPO treated or of untreated cells of another donor. During the experiment, human platelet numbers originating from the untreated donor CD34+ cells were not significantly different regardless of whether they were cotransplanted with TPO cultured cells or untreated CD34+ from another donor (Fig. 5A, n=16). On the contrary, a temporarily higher number of total human CD45+ leukocytes originated from the untreated graft when it was cotransplanted with a TPO cultured graft. This difference became clear at week 2 and was significant at week 4 and 5 after transplantation (Fig. 5B, P≤0.05 for both). The longer term human CD45+ repopulation in the PB was not significantly changed by the manipulation status of the other donor's graft.

Human platelet
Discussion
Thrombocytopenia is a common complication of allogeneic HSC transplantation, with platelet recovery being particularly delayed after CB HSC transplants [1 –3,53]. Transplant patients may also present with secondary failure of platelet recovery [54]. Thrombocytopenia at day+100 and secondary failure of platelet recovery postallogeneic HSC transplant have been associated with poorer survival [45,54]. Current expansion protocols for one CB unit in a dCB transplant setting still do not show substantial improvements in platelet recovery when transplant outcomes are compared with current rather than with historical outcome data [25 –27]. Furthermore, given that their administration can induce reticulin formation in the BM and possibly BM fibrosis, that TPO has differential effects on HSC and platelet precursors [38 –40], and that TPO in the myeloablative setting failed to improve platelet recovery, the use of TPO receptor agonists to promote platelet recovery posttransplant still requires additional safety and efficacy studies [37]. Interestingly, platelet primed or biased HSCs have been identified in mice [41,55,56]. Their phenotypic profiles have not yet been analyzed in a single study from one group and further subset analysis is needed. However, current combined results from three groups suggest that they are c-kithi+ and CD41+, dependent on TPO, capable of generating long-term myeloid- and lymphoid-biased HSCs as well as generating in vitro CD41+CD61+ megakaryocytes, and are responsible for early and longer term platelet recovery [41,55,56]. The therapeutic importance of such specialized HSC subsets in human HSC transplantation remains to be defined.
In this article, we show that TPO treated human CD34+ grafts from one CB donor can successfully be combined with untreated CD34+ cells from another donor to (1) improve short-term platelet engraftment from the TPO treated cell graft and to (2) generate long-term hematological reconstitution in an immune-deficient mouse model of dCB transplantation, whereas (3) the untreated CD34+ HSCs that are cotransplanted as a safety net are not hampered in their engraftment potential by the CD34+ cell manipulation with TPO in the other CB graft. These observations are important in two respects. The first is because delayed platelet engraftment in the weeks following dCB transplantation remains a significant problem [1 –3,53], which has not yet been substantially addressed in the clinical transplant setting. The second is that, if a platelet primed HSC exists in the human as it does in the mouse, we now have developed a validated in vivo dCB transplant model with which (1) to identify and study the efficacy of the putative human TPO dependent platelet-biased HSC, (2) to define its lineage relationships in the context of other identified human HSPC subsets and following their interactions with specific factors, agonists, or antagonists [57,58].
The studies presented in this article build on our previous short-term (6 week) engraftment studies with single CB transplants in immune-deficient mice, where we have shown that TPO treatment can improve early platelet recovery and that this recovery is mostly dependent on the TPO mediated generation of CD34−CD61− cells [44,51]. Importantly, in our current studies, by analyzing engraftment using donor-specific HLA antibodies in the dCB transplant setting, we could further demonstrate that this improved in vivo early platelet recovery (around day 7) was indeed originating from TPO treated CD34+ cell graft, whereas the untreated CD34+ cells formed platelets at later time points (significantly from around week 2) and dominated over the TPO cultured cells.
We limited the number of variables in our experiments because we wanted to specifically investigate if residual CD34+ HSPCs after TPO mediated culture could contribute to long-term hematological reconstitution and if the TPO treated graft could influence the engraftment of untreated CD34+ HSPCs in a dCB setting. For this we cotransplanted untreated CD34+ cells from one donor graft with TPO treated or untreated CD34+ cells from a second donor graft. By using donor-specific HLA antibodies, we could show that, when cotransplanted with an untreated graft, TPO treated CD34+ cells generated fewer CD45+ cells in the PB of NSG mice than the same graft which had not been cultured in TPO. This could be attributed to an absolute loss of a proportion of CD34+ HSPCs, during the TPO based culture because of their redirection toward platelet production. Thus, although its contribution was lower when compared to grafting untreated CB CD34+ cells, we could show that the TPO cultured CD34+ graft provided a stable and substantial contribution to longer term platelet recovery and hematological reconstitution in in vivo dCB transplants. As shown before [51], this indicates that a significant proportion of platelet-biased HSCs might also be maintained in short-term TPO mediated cultures. This finding was corroborated by our CAFC and CFU assays, which are reported to correlate with HSC engraftment. In the BM of all mice, the long-term culture-initiating (CAFC) cells were maintained, but again fewer of these cells were found in the BM from mice that received one TPO cultured CD34+ graft. Similarly, BM cells from mice receiving two nontreated CB CD34+ grafts formed on average a greater number of myeloid colonies than those from mice receiving one untreated and one TPO cultured CB graft; however, in both groups all types of colonies were present. Our combined results, therefore, show a consistent, clearly lower number of HSPCs in the BM of combined untreated and TPO cultured graft (N+X) transplanted mice, which is in agreement with the decreased CD34+ HSPC and possibly platelet-biased HSC content in these combined grafts. Notwithstanding this, there was no difference in the combined hematopoietic repopulation quality between the N+X and N+N group. In our experiments, the percentage of human chimerism in the BM was higher than 80% in both groups reflecting the use of an optimal transplanted CD34+ cell dose in our experiments. However, a smaller number of transplanted untreated CD34+ cells in one graft might not be able to compensate for the TPO induced CD34+ HSPC loss in the cultured graft and might thus eventually compromise the combined engraftment in mice. Another interesting finding is that the untreated CD34+ cells produced more CD45+ leukocytes when cotransplanted with a TPO cultured graft than when cotransplanted with a graft of uncultured CD34+ cells. Whether TPO cultured grafts stimulate the efficacy of unmanipulated CD34+ cells, needs to be confirmed and further investigated. Importantly however, the hematopoietic repopulation capacity of unmanipulated CD34+ cells as safety back up for manipulated CB transplantations is not hampered by cotransplantation of TPO manipulated CD34+ cells.
In conclusion, we have developed an in vivo transplant model with which the relative contribution to short and longer term platelet recovery and longer term hematological reconstitution of and competition between human CB grafts from different donors or lineage-biased HSC subsets from these grafts can be studied. Furthermore, our study approach allows characterization of the effects of exogenous factors on expanding the content of or enhancing the differentiation of such cell subsets before transplant. In this way we can characterize strategies to enhance both short and longer term platelet recovery as well as how to manipulate the HSC and HSPC subset content of such grafts for improved outcomes following clinical CB HSC transplantation.
Footnotes
Acknowledgments
The authors thank Ben Meijer, Jos Lorinser, John Scharenberg, José Milan Rivero, and the staff of the experimental animal facility of the Leiden University Medical Center for technical assistance. This work was funded by grant PPOC 06-030 of the Sanquin Blood Supply Foundation, The Netherlands. S.M.W. and M.v.d.G. would also like to acknowledge support from NHSBT and the National Institute for Health Research (NIHR) under its Program Grants scheme (RP-PG-0310-10003). The views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Author Disclosure Statement
All authors state that there is no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.