
Abstract
Mesenchymal stromal cells (MSCs), as advanced therapy products, must satisfy all the requirements for human use of medicinal products, aiming to maintain the quality and safety of the cells. The MSC manufacturing process for clinical use should comply with the principles of Good Manufacturing Practice (GMP). This ensures that cell preparations are produced and controlled, from the collection and manipulation of raw materials, through the processing of intermediate products, to the quality controls, storage, labeling and packaging, and release. The objective of this document is to provide the minimal quality requirements for the MSC production and its delivery for clinical use, so that the safety of the final cell therapy product will not be compromised. For this purpose, the document evaluates the most important steps of GMP-compliant MSC production: the isolation and expansion process; the validation phase of the process, including all quality controls for the characterization, functionality, potency, and safety of MSCs; and the quality control at the batch release to guarantee the safety of patient infusion.
Introduction
U
For reasons of clarity, complex therapeutic products such as the advanced therapy medicinal products (ATMPs) require a precise legal definition. The Regulation defines ATMPs as the gene therapy medicinal products, the somatic cell therapy medicinal products, and the tissue-engineered products. Each of these products has specific pharmacologic, metabolic, and immunologic activities and the potential for treating a variety of disorders. For these reasons, cellular products for ATMPs, prepared on a routine basis, must meet the same stringent conditions required for drugs before they are placed on the market. In particular, their activity, efficacy, safety, and required dose must be defined. Furthermore, they must be manipulated according to the Good Manufacturing Practice (GMP) [2] and require preclinical testing in Good Laboratory Practice (GLP) and clinical trials conducted in Good Clinical Practice (GCP) before being commercialized [3].
In the last decade, one cell type in particular, the mesenchymal stromal cells (MSCs), has attracted great interest due to the numerous applications proposed for their use [4]. Thanks to their intrinsic properties, MSCs represent an attractive candidate for clinical applications [5 –7].
However, as ATMPs, they must satisfy all the above-mentioned requirements. Therefore, the quality and safety of the expanded MSCs must be maintained throughout their production and quality control cycle, ensuring their safe use in the patient.
The MSC manufacturing process for clinical use should comply with the principles of GMP. This ensures that cell preparations are produced and controlled, from the collection and manipulation of raw materials, through the processing of intermediate products, to the quality controls, storage, labeling and packaging, and release. During the whole production process, critical steps should be known and described. A thorough risk analysis during all phases of production and control ensures a final product with the expected quality, both for patients and for the Regulatory Authorities responsible for controls.
Nevertheless, MSC-based therapy is different from drug-based therapy in which a known chemical entity has known therapeutic effects as well as defined adverse events. For this reason, the simple transfer of the same rules “from drug to cell” may lead to distortions: criteria to establish bioavailability, biodistribution, and excretion profiles, potential adverse effects as well as dosage, potency or shelf-life need to be adapted and stated.
There are several articles that discuss standardization of MSC culture for clinical use. As reported in the review by Ikebe and Suzuki, a total of 47 reports, describing MSC isolation methods, were found in literature, published from January 2007 to 2013 [8]. Today, numerous MSC preparations from academic institutions and private companies are being investigated in nearly 350 clinical trials (>80% of which are phase 1 or 2; 131 have reached their scheduled completion, and 37 have been reported to be placebo controlled). Clinical trials examining the safety and efficacy of MSCs have used both allogeneic (190) and autologous (150) cells [9].
Clinical trials exploring MSC therapy have been driven predominately by US Companies with proprietary allogeneic MSC preparations such as Osiris' (now Mesoblast's) Prochymal. Smaller academic-investigator-driven trials have also been conducted in medical centers and academic institutions in Europe, showing the safety of allogeneic MSC therapy [10,11].
The guideline on human cell-based medicinal products, issued by the European Medicine Agency (EMA) in 2008, is a useful document for stem cell production for marketing authorization application, covering the manufacturing process, quality considerations, process validation, and clinical considerations [12]. Manufacturing and quality control issues are frequently under discussion during the regulatory authorization processes. The recent article by Wuchter summarizes a consensus meeting between researchers, clinicians, and regulatory experts on standard quality requirements for MSC production [13]. However, a practical guideline for standardization of isolation, expansion, and characterization methods is missing. Moreover, quality controls of starting material, intermediate, and final products should also be considered, as MSCs must be produced in compliance with the GMP to ensure reproducibility, efficacy, and safety of the clinical product.
Even if the Regulations, for biological active substances, like GMP Annex 2 [14] can help the ATMP manufacturer to comply with the GMP requirements, the common opinion is that these regulations are still not enough to cover all the Authority requirements for safety, efficacy, and potency related to the ATMPs.
However, a specific step-by-step guideline for MSCs is not available to date.
In this context, the Italian Group of Mesenchymal Stem Cells [Gruppo Italiano Staminali Mesenchimali (GISM)] that aims to promote changes and information among researchers, in particular on clinical application, in this opinion article, proposes the minimal quality requirements for the production and delivery of MSCs for clinical use. This article can be considered as a proposed tentative guideline that provides indications for standardization and optimization of production time and costs according to the GMP requirements, without compromising the safety of the final cell therapy product.
The aim of this document is to clarify the critical control points that must be considered during the process of expansion and release of MSCs as ATMPs. For this purpose, the document will evaluate the most important steps of GMP production for the application of clinical trials with MSCs. These steps include (i) the isolation, identification, and expansion process; (ii) the validation phase of the process, including all quality controls for MSC characterization, functionality, potency, and safety; and (iii) the quality control at the batch release to guarantee the safety of patient infusion. This last step is also applied to products that must be cryopreserved, assuming that the cryopreservation process has been validated during the validation phase. The thawing phase is not covered by this document.
Isolation and Expansion of MSCs
The term “mesenchymal stem cells” initially referred to multilineage progenitor cells isolated and culture expanded from human adult bone marrow (BM), which once in culture display a heterogeneous morphology and are capable of several subpopulations. Horwitz et al. [15] better defined them as “mesenchymal stromal cells.”
Although BM is still the most common source of MSCs, during the last two decades, alternative and more accessible tissue sources of MSCs have been identified. For instance, MSCs have been isolated from tissues, such as fat, deciduous teeth, placenta, and umbilical cord blood, showing comparable features to BM-derived cells [16,17]. In particular, fresh umbilical cord blood (UCB) is the third common source for isolating MSCs for clinical use, but the success rate in isolating and further expanding MSCs depends on the volume of blood collected, the cell content, and the time between collection and processing [18]. Also, MSCs isolated from Wharton's jelly are able to maintain their self-renewal capacity in vitro for a long period of time, showing a higher pluripotency capacity compared to BM-MSCs [19].
MSCs have been isolated from several different placenta regions, although the isolation of cells from this region carries a risk of contamination with maternal decidual cells. Placental MSCs exhibit plastic adherence and fibroblast-like morphology in culture and a phenotype in line to ISCT Guidelines [20]. They are able to undergo multilineage differentiation and even have the capacity to differentiate across germinal boundaries outside their specific lineage [16].
Adipose-derived stem cells, if compared to BM-MSCs, have been found to be genetically and morphologically more stable in culture [21], to have a lower senescence ratio [22], a higher proliferative capacity [21], to be able to retain differentiation potential for a longer period in culture [21], to be a more efficient support to hematopoiesis (both in vitro and in vivo) [23], and to have minor immunophenotypical differences [24]. These in vitro properties have allowed these attractive cell populations to be tested in clinical studies for the treatment of a variety of clinical conditions, including Crohn's disease, end-stage coronary disease, acute myocardial ischemia, femoral head osteonecrosis, calvarial defects, breast reconstruction, and facial lipoatrophy [25].
For the purpose of this article, the intended source of MSCs, for which there are scientifically proven clinical applications, is BM, umbilical cord blood, adipose tissue, and placenta (
All the reagents and materials used during the manufacturing of the cell-based medicinal products should be well defined and controlled, the suppliers should be validated, and all the batches received should be approved by the Quality Control Unit of the manufacturing site, on the basis of specifications and quality control tests.
The cells and tissues used as raw material to produce cell-based medicinal products must comply with the Directive 2004/23/EC, Directive 2006/17/EC, and Directive 2006/86/EC on the donation, procurement, and testing of human tissues and cells [26,27]. In particular, the Annex 1 of the Directive 2006/17/EC defines the selection criteria for donors, and the Annex 2 defines the laboratory test required for donors.
So far, the application of GMP reagent in MSC ex vivo culture has been limited by the fact that only few are commercially available, such as phosphate-buffered saline, cell culture media, and trypsin. The introduction of closed systems for the isolation without density gradient separation, improved the generation of safe clinical protocols. In the case of BM and peripheral blood stem cell collection, blood samples must be taken for testing within 30 days before donation (Table 1).
In case bovine serum is used in the manufacturing of cell-based medicinal products, the viral risk and the risk of transmitting animal spongiform encephalopathy agents must be carefully evaluated according to the EMA Guidance CHMP/BWP/457920/2012 [28]. To comply with the Regulation requirements, the manufacturer of fetal bovine serum (FBS) should provide the serum user with the EDQM certificate of suitability of the fetal calf serum and ensure the safety with regard to the risk of transmitting animal agents through veterinary medicinal products [29].
Several different methods for isolating MSCs have been described. The identification of optimal conditions is important to identify a standard procedure for clinical use. No significant differences among Ficoll, Percoll, or whole BM separation methods have been described in terms of morphology, growth rate at the first passage, cumulative population doubling (CPD), immunophenotype, or differentiative potential. However, direct selection of MSCs from BM cells by adhesion to culture plastic is a more advantageous method compared to MSCs obtained by gradient separation [30] and results in a significantly higher level of HLA-DR [31]. Human platelet lysate (HPL) is being established as a safe and efficient MSC culture supplement for robust MSC cultivation, thus offering certain advantages as a potential FBS substitute [32,33].
However, as the use of human-derived blood materials leads to immunologic and infectious risks, the application of the pathogen inactivation is safe and the inactivated HPL represents a good GMP-compliant alternative to FBS [34]. This is because it reduces manipulation and hematopoietic contamination (cellular growth) and represents a good procedure for MSC expansion for clinical use [35,36]. The gradient separation cannot be applied to those tissues characterized by a solid matrix, such as fat and placenta, for which the first step to isolate MSCs is the enzymatic digestion. After this, the direct selection of MSCs can be achieved by plastic adhesion to the culture surface. Another important parameter to be considered is the cells' seeding density, which has been demonstrated to be important for the expansion of MSCs in culture. Recent studies showed that directly plated BM offers a more advantageous method in terms of fibroblastic colony-forming unit number, minimal manipulation, hematopoietic contamination, and cellular growth, showing a minor hematopoietic contamination at the first passage with whole BM rather than with Percoll or Ficoll [30,35,36]. An initial plating density between 10,000 and 100,000 cells per cm2, using flasks for large-scale cell culture, significantly improves the expansion growth, providing a high number of cells from a small quantity of BM [30].
For the purpose of this article, the validation of the better culture condition, considering the medium, the use of animal-derived supplements, the plastic, and the density and number of passages, should be carried out on at least three representative MSC samples.
Quality Control for Validation of MSC Production
The quality and safety of MSCs must be maintained throughout their production and quality control cycle, ensuring their final use in the patient. According to the International Conference on Harmonization Q2 (ICH Q2) Guidelines [37] and the European (EU) Pharmacopoeia [38], the quality control process should be validated to confirm that the analytical procedure employed for a specific test is suitable for its intended use. The process should follow a validation protocol, also considering instruments, supplies, and reagents, and defining roles and responsibilities of each step [39]. A useful scheme of manufacturing and quality control processes of MSC production as a cell therapy medicinal product is represented by Gálvez et al. [40]. In Fig. 1, two flow charts on validation and production processes are represented. A validation protocol on a minimum of three expansion procedures should be implemented to standardize the best culture condition and to demonstrate the safety and feasibility of the production protocol.
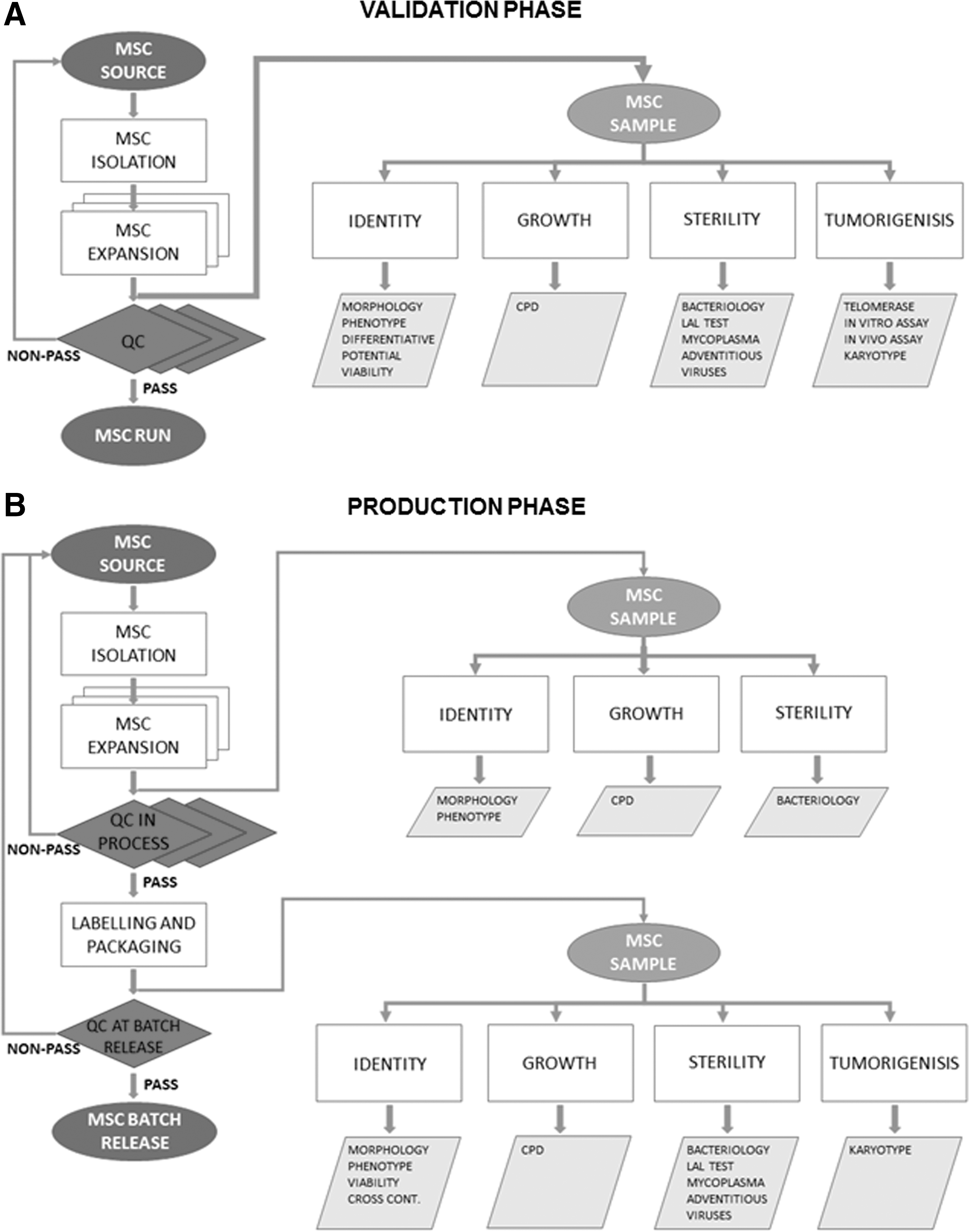
Flow chart representing the workflow and the steps of the validation phase
Results from method validation can be used to judge the quality, reliability, and consistency of analytical results [39].
The elements of the analytical method requiring proof through validation as contained in the ICH Q2A guidelines are specificity, accuracy, precision, repeatability, linearity, and range, as shown in Table 2.
✓ Signifies that this characteristic is normally evaluated.
Identity
The International Society for Cellular Therapy (ISCT) proposed three minimal criteria to identify BM MSCs: the adherence to plastic; the expression of a specific surface antigen (positivity for CD105, CD73, CD90 and negativity for CD45, CD34, CD14 or CD11b, CD79a or CD19, and HLA class II); the ability to differentiate into osteoblasts, adipocytes, and chondroblasts under standard in vitro culture conditions [41]. These criteria can be also applied to MSCs deriving from other tissue sources, although their immunophenotype can be slightly different, above all in the early phases of culture [25]. Plastic-adherent MSCs are typically visible 5–7 days after plating, appear at microscopy as fibroblastoid cells with a heterogeneous shape, also including endothelial-like cells. However, during expansion, the adherent cells become less heterogeneous and fibroblast-like cells become predominant.
MSC surface marker expression may be influenced by the different source of cells and by the method of isolation. Although human MSCs are also positive for several markers, including embryonic stem cell markers, such as Oct-4, Rex-1, and Sox-2 [4] for the purpose of this guideline, the minimal criteria to identify MSCs are the following: positivity for CD105, CD73, CD90 and negativity for CD45, CD34, CD14. In particular, the phenotype of MSCs should at least show positive expression (>90%) for CD105, CD73, CD90, and negative expression (<5%) for CD45, CD34, and CD14.
The capacity to differentiate into osteoblasts, adipocytes, and chondrocytes under standard in vitro differentiating conditions is still the most valid demonstration of MSC multipotency [41]. The evidence of osteogenic differentiation should be demonstrated by alkaline phosphatase activity and calcium deposition staining (Van Kossa staining or Alizarin staining). Adipogenic differentiation should be demonstrated through the morphological appearance of lipid droplets stained by Oil Red O staining. Chondrogenic differentiation can be evaluated by culturing MSCs in a complete commercial chondrogenic medium as cellular aggregates floating freely in suspension culture; after inclusion in paraffin, the pellet could be stained with Alcian Blue to identify the presence of hyaluronic acid and sialomucin.
The multipotent capacity should be demonstrated for all three of the differentiative lineages.
MSCs are a promising tool for cell therapies and also for their immunomodulant proprieties, thanks to their paracrine production of trophic factors together with their broad immunomodulatory and anti-inflammatory functions [42].
The recent article by Menard and Krampera [42,43] suggested some reproducible immunological assays to quantify the immunomodulatory properties of MSCs produced according to GMP. However, these analyses are recommended in the validation (or in preclinical) phase, but not mandatory for the release of MSCs.
Growth characteristics
The cellular expansion growth rate of MSCs should be evaluated by counting the cell at each passage and expressed in terms of CPD using the formula log N/log 2, where N is the cell number of the confluent monolayer divided by the initial number of cells seeded [44].
For clinical safety issues, the expansion should not exceed four passages in an attempt to minimize the administration of senescent cells.
Sterility
For microbiological control, the sterility should be evaluated at each passage on a representative sample of the products, containing cells (EuPh 2.6.27). Due to the small volume of the cell-based medicinal products, the analysis is commonly performed on the supernatant.
- Bacteriology. For microbiological control, the samples could be tested through a rapid microbiology test according to European Pharmacopoeia (EuPh) 2.6.27 [38,45].
- Limulus amebocyte lysate (LAL) test. LAL assay should be used as a quantitative method to detect Gram-derived endotoxin in a solution according to EuPh 2.6.14 [38].
- Mycoplasma. Any mycoplasma contamination could be detected through a semiquantitative polymerase chain reaction. Alternatively, culture methods or the indicator cell culture technique has to be performed. All techniques need to be validated and must be performed according to the current European Pharmacopoeia chapter 2.6.7 [38].
Tumorigenesis
Although tumor formation during the validation process has not been reported in ongoing clinical trials using MSCs [46], tumorigenesis should be tested during validation.
For the purpose of this guideline, the valid methods to exclude the potential tumorigenicity are as follows: - Assessment of telomerase activity, considering that it has been documented that nonmalignant human MSCs display a low/undetectable level of telomerase activity. - Soft agar test, with a commercial cell line as the positive control or with the in vivo test carried out on suitable animals as reported in European Pharmacopoeia 5.2.3 [38].
Karyotype
To demonstrate that the expansion method maintains the genomic stability during the in vitro culture period, the karyotype analysis should be performed. Some studies reported that human MSCs retained chromosomal stability following long-term culture in vitro [46,47]. Tarte et al. showed that MSCs, with or without chromosomal alterations, became senescent, without evidence of transformation either in vitro or in vivo, assuming that karyotyping and fluorescence in situ hybridization (FISH) results are not informative and are thus not adequate controls for the release of MSCs for clinical use [31]. The analysis of Ben-David et al. [48] reveals that MSCs have a 4% probability of acquiring chromosomal abnormalities. However, this study has been argued by the Sensebè's group [49].
Recently, Capelli's group performed a large study, by conventional karyotype analysis, on 92 clinical-grade BM-derived MSCs showing the presence of only spontaneous nonclonal anomalies, assuming that the lack of clonal aberrations or the presence of nonclonal anomalies on 10% of the analyzed metaphases should be set as the release criteria for MSCs distribution [50]. This is on line with the Cell Products Working Party, in which it has been proposed that a karyotype or FISH analysis should be necessary, at batch release, only in cases in which recurrent chromosomal anomalies are found [51].
The demonstration of the absence of genetic instability by karyotype analysis should be assessed by molecular [52] or conventional methods (G banding). At least 20 metaphases should be analyzed, in the presence of aberrations, 20 more metaphases are requested, as defined by the General Guidelines and Quality Assurance for Cytogenetics [46,53,54]. The karyotyping analysis would be sufficient as a release test with the exclusion of two identical abnormal metaphases on 20 metaphases analyzed (10%) [51], comparing the final batch to the donor/patient karyotype status.
Quality Control In-process for MSCs
For identity, morphology, immunophenotype, and growth characteristics, MSCs can be evaluated as described for the validation phase.
For microbiological in-process control, the sterility should be evaluated on cells and on supernatant samples by rapid microbiology methods (EuPh 2.6.27) or by classical methods (EuPh 2.6.1) by a GMP-approved Quality Control laboratory [40].
The quality control analysis and GMP batch release testing are part of the manufacturing process. For this reason, these tests should be carried out in the same GMP facility in a dedicated and validated laboratory or as an outsource in laboratories providing this service in compliance with GMPs.
Quality Control at Batch Release for MSCs
At batch release, MSCs should be analyzed for morphology and immunophenotype, as previously described. In addition, the following tests should be carried out.
Identity
Viability could be evaluated by Trypan Blue exclusion cell counting at microscopy or by Iodure Propidium exclusion with cytofluorimetric analysis. Both the methods should be validated according to ICH Q2 guidelines. The final batch should have a viability >90%.
Cross-contamination evaluation, by the analysis of short tandem repeat DNA profiling to assess the identity of the final product to exclude a cross-contamination, should be assessed only in case it is necessary to share spaces and instruments for the processing of different donors.
Sterility
Bacteriology, LAL test, and mycoplasma should be analyzed as previously described.
For microbiological control, the sterility should be evaluated on cells and on supernatant samples, according to current European Pharmacopoeia by an accredited laboratory.
The rapid microbiology methods applied to ATMP's are common methods to substitute time-consuming testing as Sterility testing and Mycoplasma testing. These rapid tests are described in the European Pharmacopoeia and the methods for validation are also described in details. In particular, the chapter EuPh 2.6.27 Microbiological control of cellular products recommends the rapid method for the release of the ATMPs to evaluate the sterility postinoculation in a shorter time in comparison to the culture-based methods. The Mycoplasma evaluation on ATMP's should be performed according to the chapter EuPh 2.6.7 Mycoplasmas describing both culture methods (Cultural and Indicator cells culture method) and rapid methods as nucleic acid amplification test (NAT) methods qualitative and quantitative.
Since the expanded cells are medicinal products, the tools to assess how and which adventitious agents can contaminate the culture should be defined according to the principles of the Quality Risk Management as referred to the ICH Q9 Guideline [55]. In particular, a Failure Mode Effect Analysis tool can be suitable to evaluate the hazardous agents considering the potential source of contamination such as the ancillary material for manufacturing, the adventitious from the manufacturing operators.
In the United States Pharmacopoeia, chapter 1050, Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin [56], an exhaustive approach to assess the viral contamination from cell-based medicinal products is presented and can be useful to use the suggested sources of contamination also evaluating bacterial, endotoxin, mycoplasma, and transmissible spongiform encephalopathies (TSE) agents contamination. In particular, the evaluation of the risk associated to the TSE agents contamination is well described in the EMA Note for guidance on minimizing the risk of transmitting animal spongiform encephalopathy agents through human and veterinary medicinal products (EMA/410/01 rev.3) [29].
The evaluation of infections of extraneous agents must be performed either on cell batches used as the feeder layer or on the final product. One of the following tests must be performed with the aim to detect adventitious viruses: - Inoculation on selected cell lines; - Detection of retroviruses; - Mice inoculation (test in animals); - Chicken embryo inoculation (test in eggs); - Nucleic acid amplification test.
All of the above-reported tests must be performed according to current European Pharmacopoeia (chapters 2.6.16; 5.2.3).
Regarding the final product of MSCs, due to the small available amount of cells, only the inoculation on selected cell lines and the retrovirus detection test should be performed.
It must be outlined that animal reagents such as FBS used in cell amplification must be tested with the aim to detect contaminants as reported by the EMA document EMA/CHMP/BWP/457920/2012 [28].
Similarly, reagents of swine origin-like proteolytic enzymes have to be tested by NAT techniques or by inoculation in susceptible cell cultures to detect virus contamination. Mycoplasma detection has to be carried out by the methods previously reported.
A summary panel of required quality controls is shown in Table 3.
According to European Pharmacopoeia.
CPD, cumulative population doubling; LAL, limulus amebocyte lysate,
Batch control sample
A sizable aliquot of the final batch should be cryopreserved as a backup to further analyze the cells in case of a clinical need.
Conclusion
MSC production according to GMP requirements is an important issue that has emerged in recent years. Currently, there is no consensus on quality standards for the production of MSC for clinical application. In this article, the GISM Working Group has identified the minimal quality criteria for MSCs at different production stages.
As the recent work of a group of German researchers pointed out the manufacturing and quality control issues [13], we hope that our work could be a significant first step forward toward the awareness of the Regulatory Authority that a wider agreement on this matter is needed.
Footnotes
Acknowledgments
The authors are grateful to Ms. Alexandra Dyer for editorial assistance.
This opinion article reflects the consensus viewpoint of the authors and scientists participating in the GISM Working Group. GISM Working Group includes the following individual investigators: Biagi E, Del Bue M, Frigerio S, Lisini D, Marazzi M, Mareschi K, Nava S, Parolini O, Riccobon A, Romagnoli L, Viganò M.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.