
Abstract
Although chronic wounds are common and continue to be a major cause of morbidity and mortality, treatments for these conditions are lacking and often ineffective. A large body of evidence exists demonstrating the therapeutic potential of mesenchymal stem cells (MSCs) for repair and regeneration of damaged tissue, including acceleration of cutaneous wound healing. However, the exact mechanisms of wound healing mediated by MSCs are unclear. In this study, we examined the role of MSC exosomes in wound healing. We found that MSC exosomes ranged from 30 to 100-nm in diameter and internalization of MSC exosomes resulted in a dose-dependent enhancement of proliferation and migration of fibroblasts derived from normal donors and chronic wound patients. Uptake of MSC exosomes by human umbilical vein endothelial cells also resulted in dose-dependent increases of tube formation by endothelial cells. MSC exosomes were found to activate several signaling pathways important in wound healing (Akt, ERK, and STAT3) and induce the expression of a number of growth factors [hepatocyte growth factor (HGF), insulin-like growth factor-1 (IGF1), nerve growth factor (NGF), and stromal-derived growth factor-1 (SDF1)]. These findings represent a promising opportunity to gain insight into how MSCs may mediate wound healing.
Introduction
N
The wound healing response requires a complex cascade of molecular and cellular events including cellular migration, proliferation, angiogenesis, extracellular matrix deposition, and tissue remodeling [3 –5]. In contrast, chronic nonhealing wounds exhibit decreased production of growth factors and chemokines [6], reduced angiogenesis [7], decreased proliferation and reduced migration of fibroblasts [8,9], and an impaired inflammatory response [10].
Mesenchymal stem cells (MSCs) are self renewing multipotent stem cells derived from the bone marrow stroma and other tissues, which can differentiate into various lineages including bone, cartilage, and fat [11]. In addition to their multipotent potential, MSCs have an extensive ex vivo expansion capacity and can regulate immune and inflammatory processes, making these cells attractive for the treatment of numerous disorders [12]. MSCs have been shown to correct delayed wound healing in diabetic mice by promoting epithelialization, and augmenting angiogenesis and granulation tissue formation [13]. Furthermore, studies have shown that application of MSCs to nonhealing wounds can lead to increased angiogenesis and reduced scarring [14].
Interestingly, these results do not support MSCs differentiating to replace damaged tissue. Instead it is believed that MSCs exert their therapeutic effects by secreting soluble or “paracrine” factors that augment endogenous repair and regenerative mechanisms [15 –18]. Also, work in our laboratory has found that MSCs can enhance fibroblast migration, an integral component of the wound healing process, without direct contact, suggesting the importance of paracrine signaling between these cells [19]. However, the exact molecular mechanisms of this correction are not clear.
Exosomes are small membrane-bound vesicles (diameter 30–120 nm), are secreted by a myriad of cells types and found in essentially all biological fluids, and originate from inward budding of late endosomes with resultant multivesicular bodies that are fused with the plasma membrane [20,21]. Furthermore, exosomes can shuttle transcription factors and genetic materials (mRNA and miRNA), implicating their role in cell-to-cell communication and modulating the molecular activities of recipient cells [22,23].
In this study, we hypothesized that MSC-derived exosomes play a significant role in wound healing. To test this, we examined MSC exosomes, including their characterization, their effect on dermal fibroblasts (derived from both normal and chronic wounds) and endothelial cells, and we tried to deduce possible mechanisms that underlay these effects.
Materials and Methods
Cell lines
Human MSCs were isolated and expanded from normal donor bone marrow acquired from AllCells LLC (Emeryville, CA,
Diabetic wound patient fibroblasts were isolated from the wound edge and collected under a University of Miami IRB approved protocol (HSRO 20080299). Experiments were repeated from diabetic wound fibroblasts. Fibroblasts were collected from a 59-year-old male with uncontrolled diabetes who had a nonhealing ulcer of >2 years duration that had not healed despite standard of care and advanced wound care treatments. Normal adult fibroblasts were obtained from Lonza (Walkersville, MD). Cells were grown until 80%–90% confluence was reached and then passaged at a 1:4 to 1:6 ratios into new tissue culture flasks. Dermal fibroblasts were grown in FB media [Dulbecco's Modified Eagle Medium, 5% fetal bovine serum (FBS), and 1% Pen/Strep and 1% glutamine].
Coculture
For all cell labeling experiments, 1×106 cells were resuspended in 1 mL of α-MEM and incubated with 5 μL of 1 mM Vibrant Dil (MSCs) or Vibrant DiO (normal adult fibroblast) solution (Life technologies, Carlsbad, CA) for 20 min at 37°C in the dark followed by two wash steps in medium. Cocultures of labeled MSCs and normal adult fibroblasts were performed in monolayer at 50:50 ratios in fibronectin-coated four-well Nunc Lab-Tek II Chamber Slide system for 24 h in MSC media. Images were captured with an inverted IX81 Olympus microscope (Olympus America, Center Valley, PA) and ORCA-AG Hamamatsu digital camera (Hamamatsu Photonics K.K., Hamamatsu City, Shizuoka Pref., Japan) using red (for Dil) and green (for DiO) fluorescence microscopy.
MSC exosome purification and fluorescent labeling
FBS used in culture media for exosome isolation was precleared by ultracentrifugation in Beckman pollyallomer tubes (Beckman Coulter, Brea, CA) using the Optima L-XP preparative ultracentrifuge (Beckman Coulter) at 100,000 g for 3 h at 4°C as described by others [24]. It was subsequently filtered using a 0.22 μM filter (Corning, Corning, NY) (depleted FBS). BM-MSCs were grown in MSC media containing α-MEM, 20% depleted FBS, 1% penicillin/streptomycin, and 1% glutamine. Low passage (<5) BM-MSCs were grown to 60%–80% confluence in 5-layer 875 cm2 multi-flasks (∼17×106 to 22×106 MSCs per flask) before isolation. Fresh BM-MSC media was layered and collected after 48 to 72 h (conditioned medium).
Exosomes were isolated by the ultracentrifugation protocol described by Théry et al. [25]. Briefly, hMSCs were conditioned in exosome-free medium for 48–72 h. The conditioned medium was transferred to 50-mL centrifuge tubes (Thermo Fisher Scientific, Rockford, IL) and centrifuged at 2,000 g at 4°C for 20 min to remove cells. The supernatant was carefully removed subsequently transferred to new 50-mL centrifuge tubes and centrifuged for 30 min at 10,000 g and 4°C to remove cellular debris. The supernatant was carefully removed and transferred to Beckman Optiseal™ polypropylene tubes and ultracentrifuged at 100,000 g at 4°C for 70 min with the Beckman Ti70 rotor using the Optima L-XP preparative ultracentrifuge. The supernatant was carefully removed and saved (Depleted conditioned medium) and the pellets were resuspended in 1 mL phosphate buffered saline (PBS). The pellets from all tubes were pooled and ultracentrifuged again at 100,000 g at 4°C for 70 min. The pellet was again washed in PBS and a final ultracentrifuge step was performed at 100,000 g at 4°C for 70 min. The final pellet was resuspended in 100 μL PBS. Recovered exosome protein contents were determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific) per manufacturer's protocol. Exosomes were used immediately or stored at −70°C until use. Supernatant remaining after ultracentrifugation was also collected, filtered via a 0.22 μM filter, and stored at −70°C until use (depleted medium). For all experiments, depleted medium was used at a concentration of 10 μg/mL.
PKH26 labeling of exosomes
Exosomes were labeled with PKH26 (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's protocol. Briefly, 7.5 mg of exosomes were resuspended in 1 mL of diluent C (Sigma-Aldrich) and mixed with PKH26 diluted in Diluent C for a final concentration of 2×10−6 M PKH26. The exosome-dye suspension was incubated for 5 min with regular mixing. Excess dye from the labeled exosomes were removed by centrifugation and resuspended in PBS (for a total of three washes) in 100k Amicon Ultra Centrifugal Filters-100k tubes (Millipore, Billerica, MA). A mixture without exosomes was used as a negative control to examine any carryover of PKH26 dye. For the negative control, labeling was performed as described but without exosomes.
Electron microscopy
Exosome pellets were deposited on Formvar carbon-coated electron microscopy (EM) grids and allowed to air dry. They were fixed with 2% gluteraldehyde, counterstained with 4% uranyl acetate, and visualized using the JEOL 1400 transmission electron microscope (JEOL USA, Peabody, MA). All electron microscopy was completed with assistance from the Electron Microscopy Core facility at the University of Miami.
Immunoblotting
MSCs and exosomes were lysed using RIPA containing 1 mM PMSF and protease inhibitors. After lysis, protein concentrations were measured using Pierce BCA protein assay kit. Cellular lysates and exosomal lysates were subjected to SDS-PAGE and transferred to a PVDF membrane (BioRad Laboratories, Hercules, CA). PVDF membranes were incubated with a 1,000-fold diluted primary antibody solution overnight at 4°C. Washed membrane was probed with a horseradish peroxidase-conjugated secondary antibody at a 1:10,000 dilution. Signals were developed using the SuperSignal West Femto Chemiluminescent substrate from Pierce Biotechnology and imaged by Biorad's ChemiDoc system. Membranes were incubated with the following antibodies: CD-9, CD-81, CD-63, Hsp-70 (System Biosciences, Mountain View, CA), Alix, Flotillin-1, GM-130, Beta-Actin, GAPDH, phosphorylated Tyr705 of STAT3, total STAT3, phosphorylated Ser473 of Akt, total Akt, phosphorylated Thr202/204 of ERK1/2, total ERK1/2 (Cell Signaling, Danvers, MA), and Tsg-101 (Novus Biologicals, Littleton, CO).
STAT3 DNA binding
Cellular and exosomal extracts (10 μg) were assayed for STAT3 DNA binding activity using the TransAM kit (Active Motif, Carlsbad, CA) per manufacturer's instructions.
Cell proliferation
Fibroblasts were seeded at a density of 5×103 cells/well in 24-well plates in FB media. After overnight plating, an MTT assay (3-[4.5-Dimethylthiazol-2-yl]-2.5-diphenyltetrazolium bromide; Life technologies) was performed to obtain baseline values (day 0). MTT assay is a colorimetric assay that measures the chemical reduction of MTT into formazan, which is directly proportional to the number of viable cells. Cultures were incubated for 20 min in FB medium with 0.5 mg/mL of MTT. The resulting formazan was then extracted with DMSO and the optical density was measured at 540 nm. After obtaining day 0 values, wells were randomly selected to be treated with MSC exosomes (0.1, 1, and 10 μg/mL), vehicle (PBS), or depleted media (10 μg/mL). After 3 days of treatment, MTT assay was again performed (day 3).
Cell migration
For migration studies, ∼3×105 fibroblasts were seeded per well in six-well plates in FB media and maintained at 37°C and 5% CO2 for at least 24 h to permit cell adhesion and the formation of a confluent monolayer. To inhibit the influence of cell proliferation, cells were treated with fresh serum-free culture medium containing mitomycin at 10 μg/mL. The confluent monolayer was then scored with a 1 mL sterile pipette tip. Culture medium was immediately removed (along with dislodged cells) and replaced with a fresh serum-supplemented culture medium containing MSC exosomes (0.1, 1, and 10 μg/mL), vehicle (PBS), or depleted media (10 μg/mL). Images were taken after the scratch and 16 h post treatment with the IX81 Olympus microscope. Three fields were analyzed per well using ImageJ software (
Quantitative real-time polymerase chain reaction
For gene expression studies, fibroblasts were seeded in six-well plates in FB media and maintained at 37°C and 5% CO2 for at least 24 h to permit cell attachment. After serum starvation overnight, cells were treated with MSC exosomes or vehicle for 24 h. RNA extraction from fibroblasts was performed using Qiagen RNeasy Mini Kit (Germantown, MD) per manufacturer's instructions. Three hundred nanograms of RNA was reverse transcribed to cDNA using iScript Reverse Transcription Supermix for quantitative real-time polymerase chain reaction (qRT-PCR) (BioRad Laboratories). PCR was performed using the Biorad CFX96 Real-Time PCR Detection System (BioRad Laboratories) machine with the SsoAdvanced SYBR Green Supermix (Bio-Rad). Amplification conditions after an initial denaturation step for 90 s at 95°C were 40 cycles of 95°C, 10 s, for denaturation, 55°C, 10 s, for annealing and 72°C, 30 s, for elongation. CFX96 manager software was used to determine threshold cycles. GAPDH was used as the reference gene for calculations. Data were analyzed by the 2ΔΔCT threshold cycle method. Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). Primer sequences are listed in Table 1.
Tube formation assay
Angiogenesis was measured using an endothelial tube formation assay kit (Invitrogen Life Technologies, Grand Island, NY) per manufacturer's instructions. Briefly, human umbilical vein endothelial cells (HUVECs) were plated at a density of 5×104 cells/well in Geltrex coated 24-well tissue culture plates. Cells were treated with MSC exosomes (0.1, 1, and 10 μg/mL), vehicle (PBS), and depleted media (10 μg/mL). Treated cells were incubated for 6 h in 200PRF media supplemented with 0.2% LSGS (limiting medium) at 37°C and 5% CO2. At the end of incubation, HUVECs were stained with Calcein AM fluorescent dye for visualization of tube formation. Fluorescent images were captured with the inverted IX81 Olympus microscope and ORCA-AG Hamamatsu digital camera. Tube length was measured using NIH's Image J software (
Statistical analysis
Pairwise one-tailed Student's t-tests or one-way analysis of variance with Bonferroni's post hoc test were used in this study and the error bars are shown as standard error of the mean. Statistical significance was set at P<0.05.
Results
Formation of a nanotubular network and secretion of microvesicles during coculture of fibroblasts and MSCs
To examine the transfer and determine the identity of possible transmitted cellular components, we used the lipophilic fluorophores, DiI (red fluorescence) and DiO (green fluorescence). These dyes will label plasma membranes, including exosomes, by stably inserting their hydrocarbon chains in to lipid bilayers. Both Dil and DiO are not cytotoxic and are known not to leak. MSCs were labeled with Dil and fibroblasts were labeled with DiO. The two populations were cocultured in a 1:1 ratio for 24 h. Live cell imaging revealed a nanotubular network among DiO-labeled fibroblasts (green) and Dil-labeled MSCs (red) after 24 h of coculture (Fig. 1A, B). In addition, we found that vesicle structures that appear to be consistent with microvesicle (MV) morphology (which have been found range in size from 50 nm to 1,000 nm) were found to be actively shed from both fibroblasts and MSCs. MSC MVs (red) were also found to be internalized by the DiO-labeled fibroblasts (Fig. 1C).
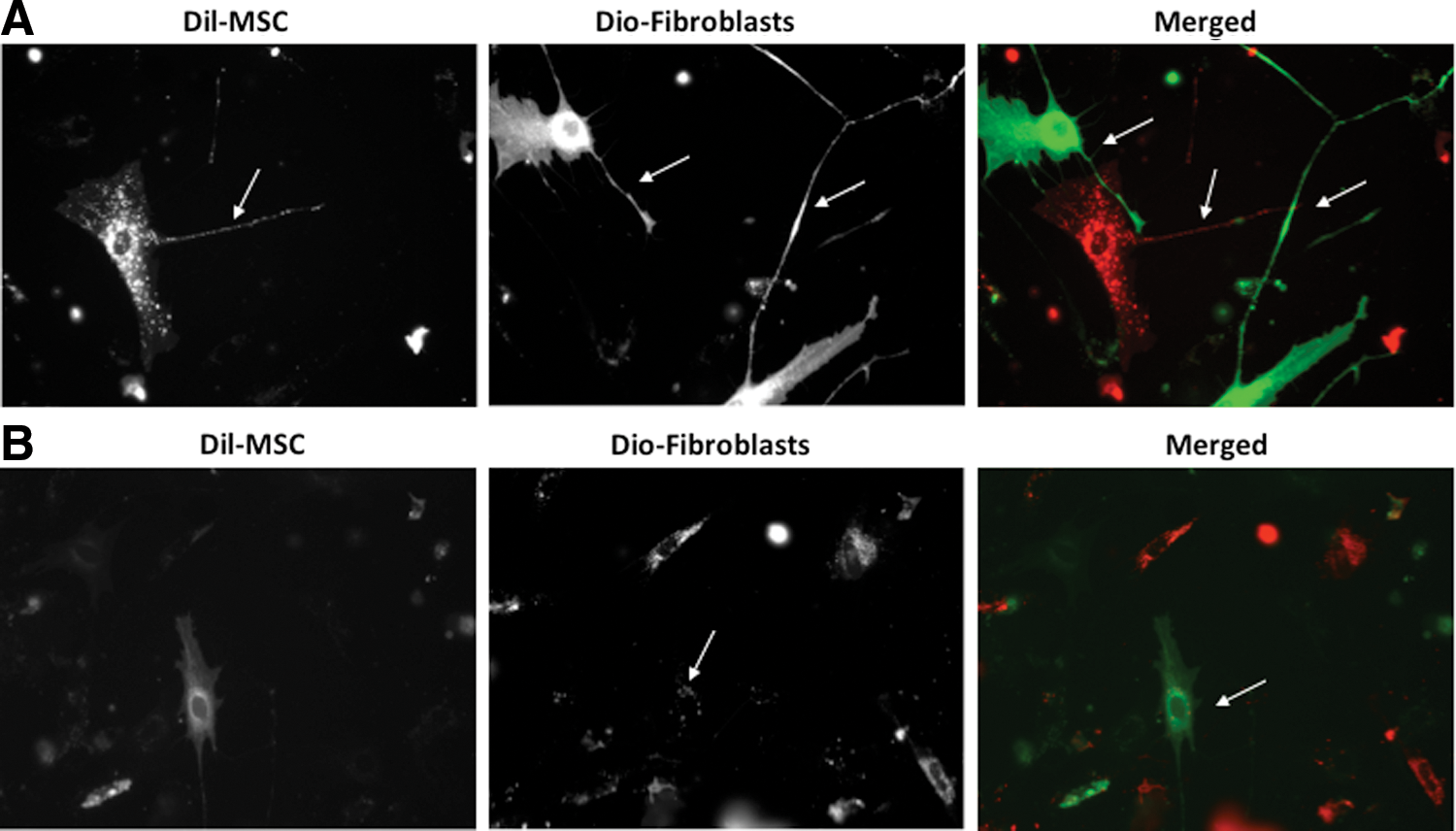
Normal human mesenchymal stem cells (MSCs) and normal human fibroblasts form a nanotubular network and secrete microvesicles in coculture.
Characterization of MSC exosomes
To isolate and further characterize the MVs observed in Fig. 1, we employed an established isolation protocol described by Théry et al. that utilizes differential sedimentation to isolate exosomes, a type of microvescicle that has recently gained attention as being the possible paracrine mediators of MSCs (see Materials and Methods section) [25]. Electron microscopic analysis of the vesicles isolated by ultracentrifugation indicated a cup-shaped morphology with diameters approximately ranging between 30 and 100 nm (Fig. 2A) with an appearance similar to those reported from other cell lines [26 –28]. An extensive biochemical profiling of exosomes was performed by western blot analysis (Fig. 2B) with a panel of antibodies, including those directed against exosomal marker proteins such as the tetraspanins (CD9, CD63, and CD81), proteins involved in multivesicular body biogenesis (Alix and Tsg101) and membrane transport and fusion (flotillin-1) [29]. As expected, CD9, CD81, Alix, TSG101, and flottilin-1 were strongly enriched in exosome preparations compared with cell lysates. We confirmed the presence of CD63 in exosomes. CD63 was, however, also detected in MSC cell lysate, but this was not unexpected as CD63 is expressed by MSCs [30]. Additionally, HSP70, β-actin, and glyceraldehyde-3-phosphate dehydrogenase were found in exosomes, typical of exosomes produced by varied cell types [31]. Further, the cis-golgi matrix protein gm130 was readily detected in cell lysates but was not found in exosomes, indicating that no contaminating cellular debris was present in the exosome preparations.
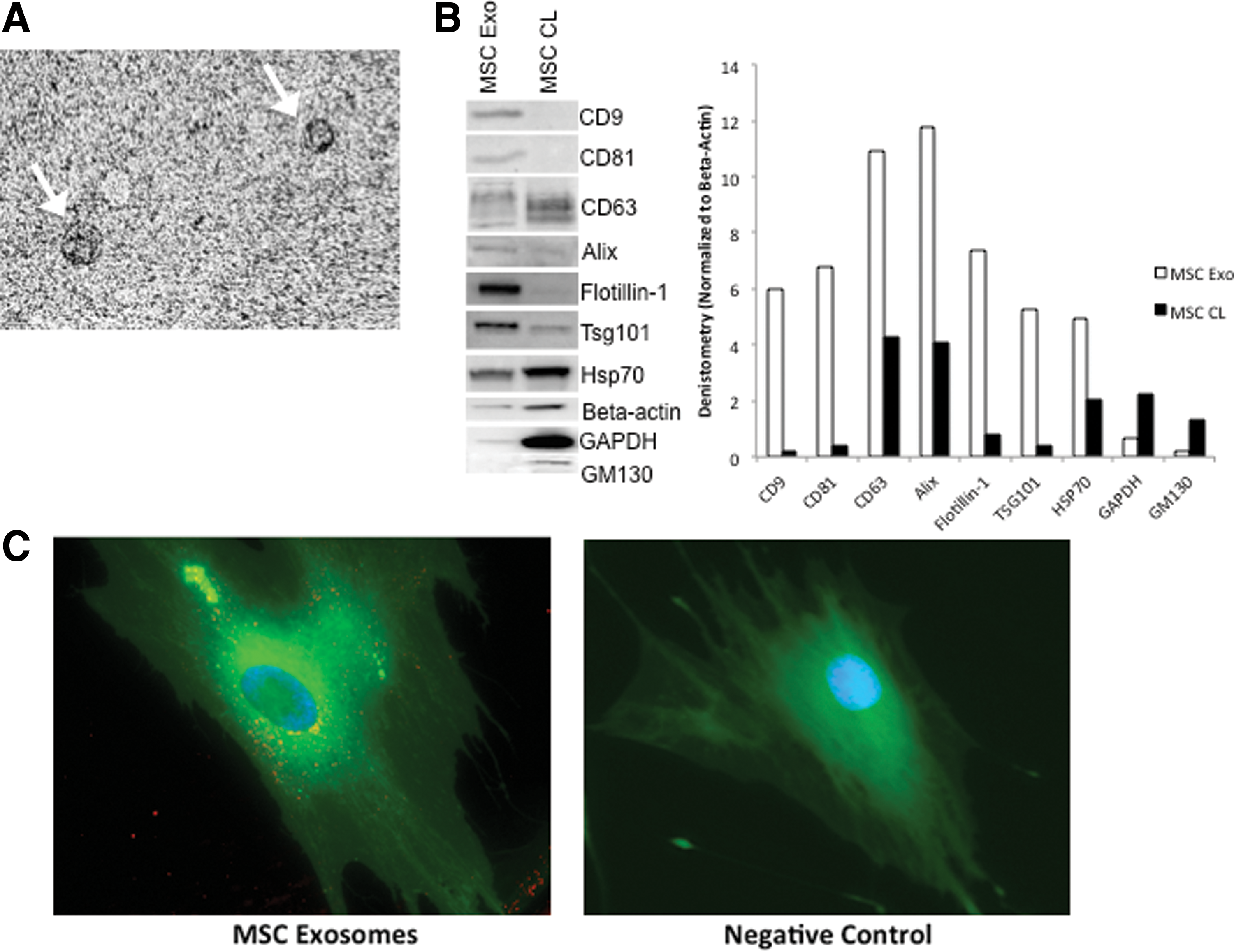
Characterization of MSC exosomes.
We next examined whether MSC-derived exosomes could be transferred to dermal fibroblasts. PKH26 fluorescently labeled exosomes isolated from MSC conditioned medium were cocultured with fibroblast cells overnight. After incubation with exosomes overnight at 37°C, cells were washed thrice and stained with Calcein AM. Live cell imaging revealed PKH26-labeled exosomes to be observed inside the Calcein-labeled fibroblasts, and they were mainly located in the perinuclear region (Fig. 2C), as reported by others [32 –34].
MSC exosomes enhance normal and diabetic wound fibroblast growth
To explore whether MSC exosomes could induce the growth of fibroblasts, both normal adult and diabetic wound fibroblasts were incubated with vehicle (PBS), depleted medium (10 μg/mL), and MSC exosomes at three different concentrations (0.1, 1.0, and 10 μg/mL) for 72 h. Normal adult fibroblast growth, as measured by the MTT assay, was found to be significantly increased and in a dose-dependent manner as compared to vehicle and depleted medium at day 3 (Fig. 3A).

MSC exosomes enhance growth of normal and diabetic wound fibroblasts.
The growth rate of diabetic wound fibroblasts was found to be significantly diminished compared with normal adult fibroblasts (Fig. 3B). A similar dose dependence response was noted when diabetic wound fibroblasts were treated with MSC-derived exosomes; however, it appeared that only the higher concentration of exosomes (10 μg/mL) was significant, compared to the vehicle and depleted medium, in inducing growth. This could be indicative of a blunted ability to respond to growth signals beyond that observed with growth factors.
MSC exosomes enhance normal and diabetic wound fibroblast migration
We next studied MSC exosomes in their ability to induce dermal fibroblast migration during the wound-healing process by using the scratch assay method, an in vitro procedure used to study cell migration [35]. Normal dermal fibroblasts and diabetic wound fibroblasts were plated to confluence (at 3×105 cells/well in a six-well plate). After creation of the scratch, cells were treated with vehicle, depleted medium (10 μg/mL), and MSC exosomes (0.1, 1.0, and 10 μg/mL) for 16 h. The migration rate of normal adult fibroblasts after MSC exosome was found to be significantly increased at all doses as compared to vehicle and depleted medium (Fig. 4A, B). Further, a dose-response was noted with the 10-μg/mL dose demonstrating the greatest migration rate. Similar to normal adult fibroblasts, the migration rate of diabetic wound fibroblasts was significantly increased with MSC exosomes compared with vehicle and depleted medium (Fig. 4C). Interestingly, the maximal effect of MSC exosomes on the migration of diabetic wound fibroblasts was reached at 1 μg/mL, reflecting an enhanced sensitivity of diabetic wound fibroblasts. Hence, MSC exosomes were able to enhance the migration rate of normal and diabetic wound fibroblasts.
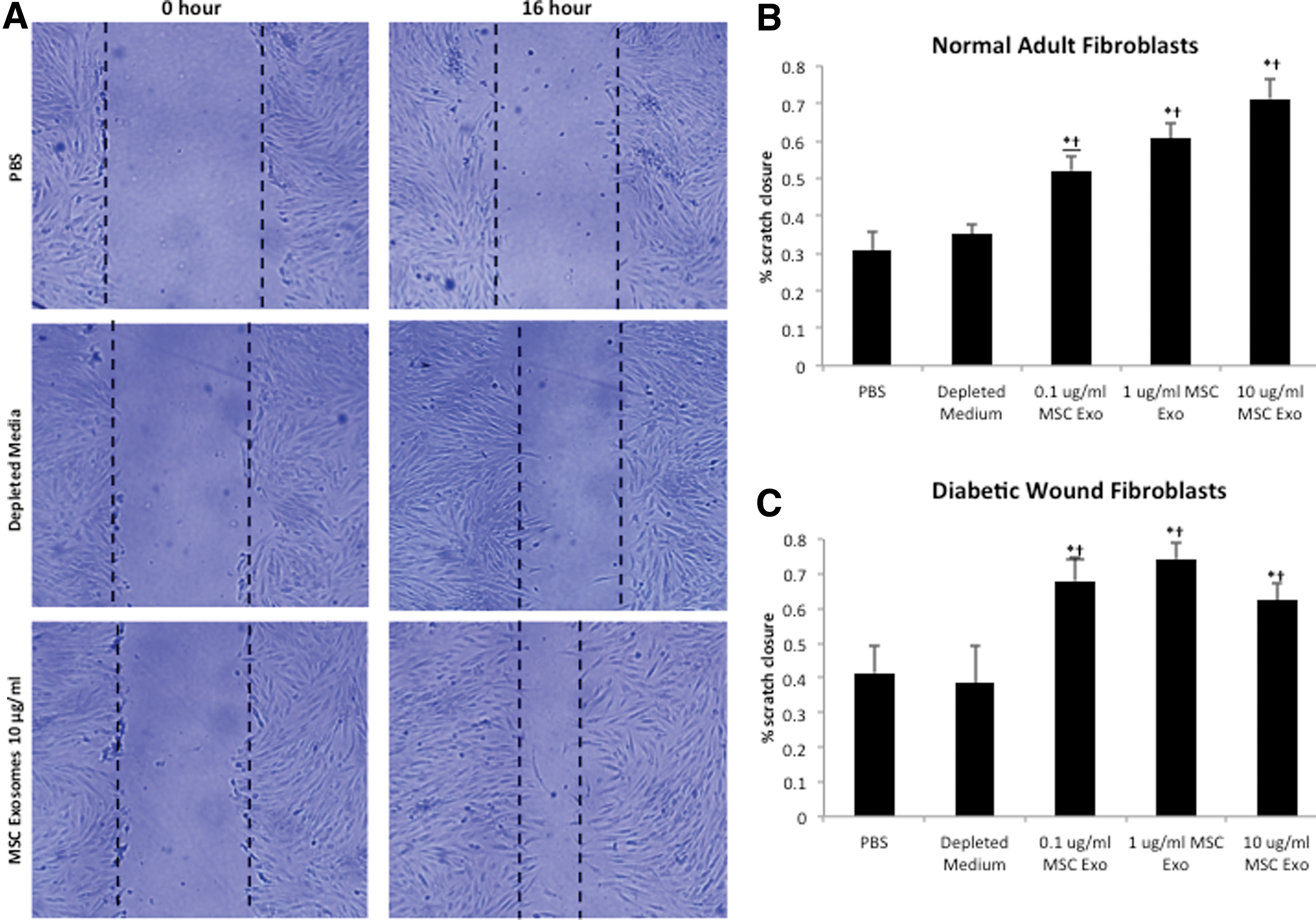
MSC exosomes enhance the migration of normal and diabetic wound fibroblasts. Fibroblasts were plated to 100% confluency, treated with 10 μg/mL mitomycin for 2 h to inhibit the influence of proliferation, and mechanically “wounded” by scraping with a 1,000 μL Fisher-brand pipette tip. Cell monolayers were washed with phosphate buffered saline (PBS), imaged and subsequently treated with different concentrations of MSC exosomes (0.1, 1, or 10 μg/mL), vehicle or depleted conditioned medium.
Endothelial cells internalize MSC exosomes during tube formation
Since endothelial cells play an important role in neovascularization and the wound healing process, we next determined the effects of MSC exosomes on the behavior of cultured HUVECs. We first determined whether MSC exosomes could be uptaken by HUVECs. PKH26 fluorescently labeled MSC exosomes were incubated with HUVECs cultured on matrigel for 6 h under tube formation conditions (2% v/v LSGS-supplemented Medium 200PRF). After incubation with MSC exosomes, cells were washed thrice with PBS and stained with Calcein AM. Numerous small, granular PKH26-labeled MSC exosomes (red) were noted within the tubular network of HUVECs (green) (Fig. 5A). Hence, endothelial cells can internalize up MSC exosomes.

MSC exosomes enhance angiogenesis in vitro.
MSC exosomes induce vascular tube formation in vitro
We next hypothesized that MSC exosomes could induce tubular differentiation of HUVECs in vitro. HUVECs plated on Geltrex in limiting medium (0.2% v/v LSGS-supplemented Medium 200PRF) with increasing concentrations of MSC exosomes formed an extensive tubular network in a dose-dependent manner (Fig. 5B, C). Furthermore, MSC exosomes were found to form greater tube formation compared with controls (vehicle and depleted medium) at all dosages. Depleted medium had a small, yet statistically significant, ability to induce tubular formation compared with vehicle.
MSC exosomes activate several intracellular signaling pathways and induce expression of cell cycle genes and growth factors
We next characterized the effects of MSC exosomes on intracellular signaling in normal dermal fibroblasts by examining the activation of AKT, ERK 1/2, and STAT3. These signaling pathways are known to be play critical roles in wound healing, including roles in cell proliferation, migration, and angiogenesis [36 –38]. Normal adult fibroblasts were serum starved overnight and subsequently exposed to vehicle, depleted medium, or MSC exosomes (10 or 20 μg/mL) for 2 h. Figure 6A (with densitometry shown in Fig. 6B) shows significant activation of Akt (phosphorylation of Ser473) and STAT3 (phosphorylation of Thr705) along with enhanced signaling of ERK 1/2 (phosphorylation of Thr202/204) after MSC exosome treatment.
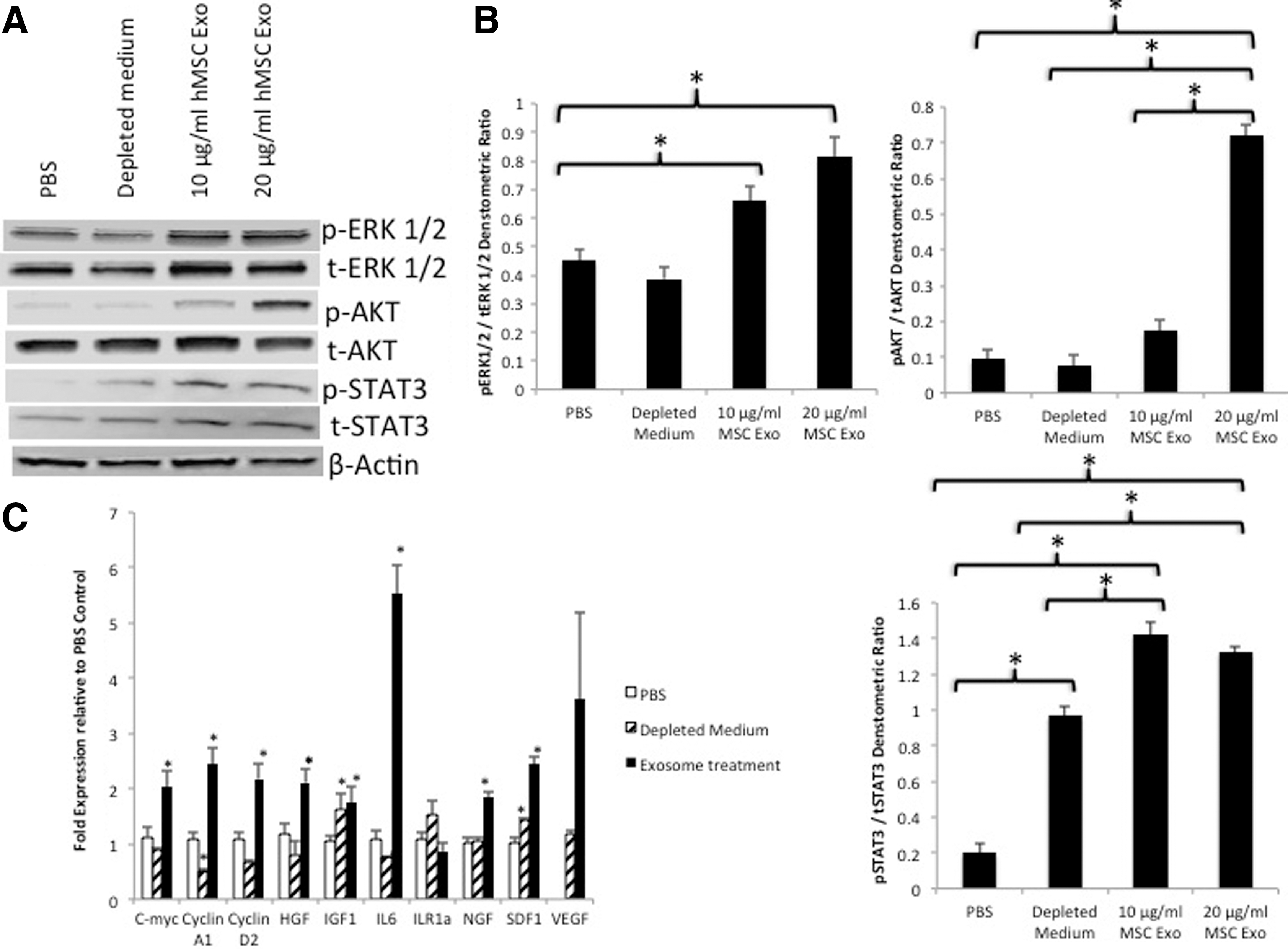
MSC exosomes activate trophic signaling pathways and gene expression.
As the activation of these signaling pathways, particularly STAT3, is known to induce the expression of a number of genes, including those involved in cell cycle progression and growth factor production, we used qRT-PCR to quantify expression of cell cycle, growth factor, and cytokine genes. After serum starvation overnight, cells were treated with MSC exosomes or vehicle for 24 h. Figure 6C reveals there was a significant induction in genes involved in cell cycle progression (c-myc, cyclin A1, and cyclin D2), growth factors [hepatocyte growth factor (HGF), insulin-like growth factor-1 (IGF1), nerve growth factor (NGF), and stromal-derived growth factor-1 (SDF1)] and the cytokine interleukin-6 (IL-6). Although, the induction vascular endothelial growth factor (VEGF) was not statistically significant, there was a trend toward increased expression (P=0.078). There was no difference in the expression of interleukin receptor antagonist-1 between exosome treatment and PBS. In contrast, depleted medium showed only significant increases in IGF1 and SDF1.
MSC exosomes are enriched with STAT3 that is transcriptionally active
STAT3 plays an important role in normal wound healing and response to injury [39]. Furthermore, STAT3 signaling controls a wide array of cellular processes, including cellular proliferation, migration, and angiogenesis by targeting the expression of many genes such as those involved in cell cycle control (c-myc and cyclin) and encoding cytokines and growth factors (IL-6, HGF, and VEGF) [40 –42]. Given that MSC exosomes were able to enhance fibroblast proliferation and migration, enhance tube formation of HUVECs and increase a number of STAT3 genes, we next examined whether MSC exosomes contained STAT3. Figure 7A clearly demonstrate the presence of STAT3 and STAT3 phosphorylated at Thr705 in exosomes isolated from MSCs. Next, we assessed whether exosomal STAT3 had DNA-binding activity utilizing an ELISA-based STAT3–DNA binding assay. Indeed, exosomal STAT3 did demonstrate DNA-binding activity (Fig. 7B).
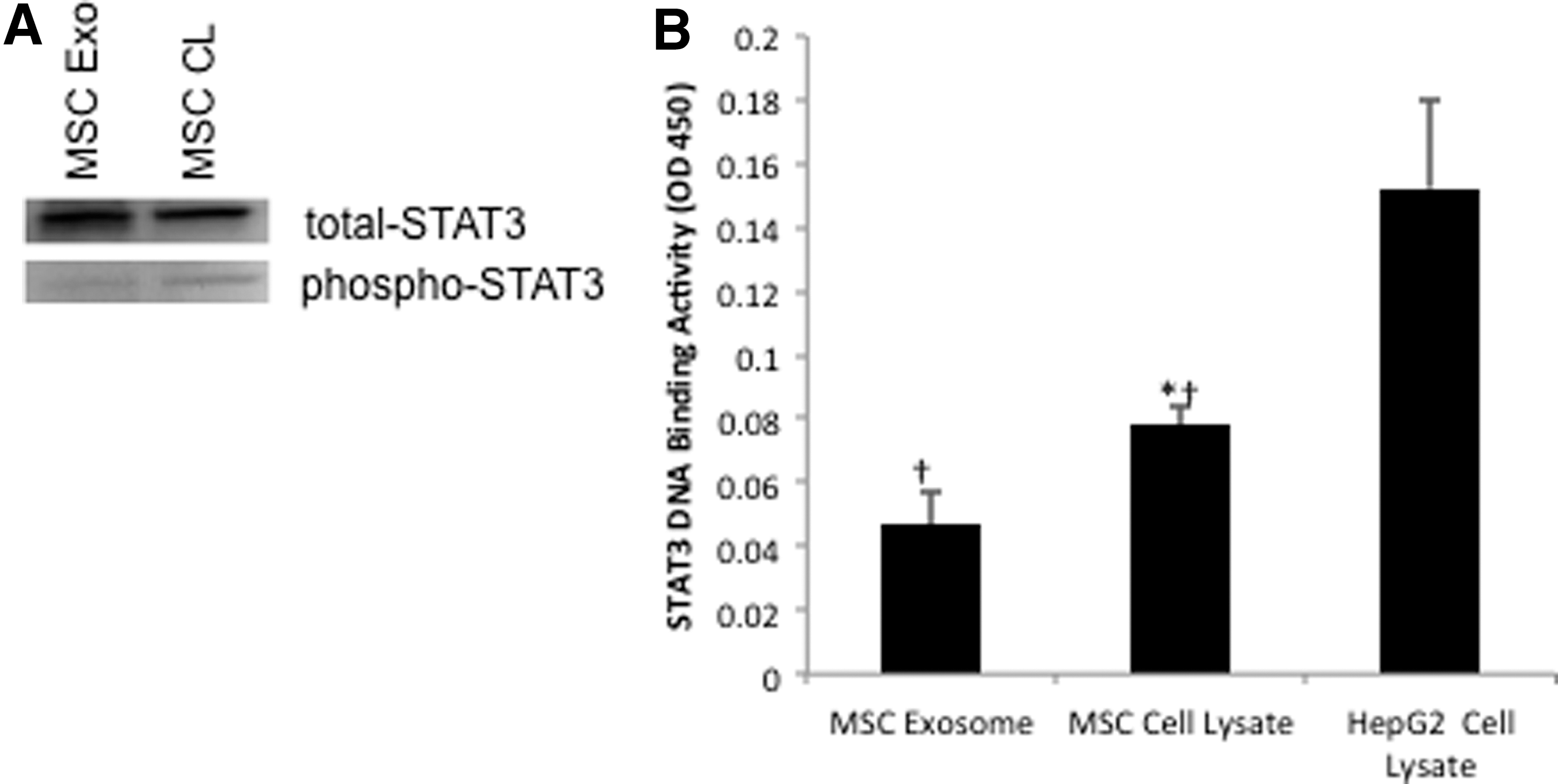
MSC exosomes contain Stat3.
Discussion
Bone marrow-derived MSCs have been shown to be efficacious in the treatment of wounds, including chronic wounds [14,18,19]. Previous work in our laboratory has demonstrated that bone marrow-derived cells play an important role in cutaneous wound healing and that direct application of bone marrow-derived cells to patients with chronic nonhealing wounds leads to wound closure and dermal rebuilding [43 –47].
It was originally thought MSCs homed to injured tissue, differentiated, and replaced damaged tissue, however, subsequent studies have shown that MSC engraftment and differentiation to injured sites are low and transient [48]. Instead, it is believed that MSCs exert their therapeutic effects through secreted paracrine or trophic factors [15 –17]. Experiments utilizing MSC-conditioned media have demonstrated the importance of the paracrine mechanism of BM-MSC-induced wound healing [49 –52]. More recent work in our laboratory has demonstrated that MSCs could enhance fibroblast migration without cell contact in a dose-dependent manner [19].
It has been demonstrated that eukaryotic cells possess highly sophisticated membrane trafficking mechanisms to mediate cellular communication. For example, it has been reported that cells cultured in vitro exchange information by shedding MVs and form tunneling nanotubes [53]. Indeed, we were able to demonstrate that MSCs cocultured with fibroblasts formed ultrafine intracellular structures resembling tunneling nanotubes that connect the cytoplasm of distantly located cells. We also noticed the release and uptake of MVs by MSCs in coculture.
Exosomes, a type of MV are produced by fusion of the multivesicular body with the plasma membrane, and are released from and taken up by most cell types [20 –22]. In our study, we successfully isolated MSCs exosomes, confirmed their smooth spherical shape resembling a lipid bilayer structure and verified the expression of exosomal marker proteins CD9, CD63, CD81, Alix, Tsg101, and flottilin-1. Exosomes have recently gained attention as being the possible paracrine mediators of MSCs in several disease models. Results so far have indicated that these exosomes may be important in reducing myocardial infarction size, protecting against acute tubular injury, promoting functional recovery after stroke, and ameliorating inflammation after Escherichia coli endotoxin-induced acute lung injury [54 –57]. We subsequently hypothesized that MSC exosomes could also mediate the effects of wound healing attributed to MSCs observed in our recent studies.
Dermal fibroblasts provide critical functions during wound healing, including wound contraction, extracellular matrix deposition, and tissue remodeling. Fibroblasts from chronic wounds are defective in their ability to migrate, proliferate, and secrete growth factors [58 –60].
Our experiments found that fluorescently labeled MSC exosomes were uptaken by fibroblasts and that they could enhance the growth and migration of normal and diabetic chronic wound fibroblasts in a dose-dependent fashion. Furthermore, we found that MSC exosomes were uptaken by HUVECs and enhanced endothelial angiogenesis in vitro. This latter finding is in line with several reports demonstrating the angiogenic potential of MSC-conditioned medium [61 –63], and a recent report by Bian et al. that examined the angiogenic effects of MSC exosomes [64].
MSC-conditioned medium includes cytokines such as IL-6, IGF1, VEGF, and basic fibroblast growth factor [65 –67]. Yet, numerous clinical studies of recombinant growth factors used to treat chronic wounds have reported disappointing results [68]. Proteolytic degradation of growth factors and unresponsive growth factor signaling cascades are thought to contribute to an impaired wound healing response [69,70]. Interestingly, consistent with those results, we found that cellular proliferation, migration, and angiogenesis were significantly impaired when the conditioned medium was depleted of exosomes by ultracentrifugation.
Exosomes produced by MSCs would be expected to have several advantages in the wound environment. Their lipid bilayer shell could avert proteolytic degradation and thus more effectively transfer signals to target cells (eg, fibroblasts and endothelial cells). In addition, exosomes contain many potential regulatory components including miRNAs, mRNAs, and proteins, which can be transferred as a type of “physiological lipofection” to recipient cells to modify their characteristics [71]. This ability to transfer complex messages could explain evidence in previously described experiments where cellular extracts were found to induce epigenetic changes in recipient cells. For example, fibroblasts after exposure to lymphatic cell extracts express genes typical for lymphocytes [72] and similarly, hematopoietic stem cells exposed to the extracts from damaged liver cells began to express genes specific for hepatocytes [73]. Exosomes likely accomplish these changes by exerting a direct effect on specific pathways in recipient cells. Dovrat et al. found that β-catenin was secreted in extracellular vesicles and could activate the WNT pathway in recipient cells [74]. MSC exosomes have been found to transfer growth factor receptor mRNA, which was translated to the corresponding protein in renal tubular cells [75].
Also important in wound healing is that MSC exosomes appear to induce changes by activation of growth factor signaling cascades. We have demonstrated that MSC exosomes activate important signaling cascades in target cells, including AKT, STAT3, and ERK. By stimulating these pathways, target cells would increase their expression of a number of growth factors, namely HGF, IL-6, IGF1, NGF, and SDF1. Further, we also postulate that the release of these factors may induce autocrine activation of additional signaling cascades, possibly accounting the activation of additional signaling cascades, including AKT and ERK 1/2, and further activation of STAT3. Activation of these signaling cascades with increased growth factor and growth factor receptor production is particularly relevant, as chronic wounds have attenuated signaling cascades and growth factor expression.
Given STAT3's important role in wound healing, including roles in migration, proliferation, angiogenesis, and growth factor production [37], we examined whether MSC exosomes carry STAT3. To our surprise, we found that MSC exosomes did indeed carry STAT3, which also had DNA-binding activity. Although, elucidating the exact role of exosomal STAT3 requires further study, we speculate it may be in part responsible for transcription of genes involved in our study, including Cyclin D2, c-myc, HGF, VEGF, and IL-6, which are known to be transcriptionally activated by STAT3 [39]. Future work will be required to delineate the exact role of exosomal STAT3 in relation to activation of STAT3 signaling in the target cell.
In conclusion, we found that MSC exosomes could enhance the growth and migration of normal and chronic wound fibroblasts, and induce angiogenesis in vitro. Furthermore, we demonstrate that MSC exosomes contained transcriptionally active STAT3 and MSC exosomes were able to activate AKT, ERK 1/2, and STAT3 and induce the expression of a number of trophic factors. We hypothesize that exosomes derived from MSCs could be used for wound healing as a safe and effective “off the shelf” product and possibly obviate the concerns of MSCs, including transfer of infectious agents, development of unwanted cell types, culture-induced senescence, loss of functional properties, genetic instability, and/or eventual malignant transformation [76].
Footnotes
Acknowledgments
The authors thank Peggy Bates (Electron Microscopy Facility, University of Miami medical school) for technical help.
This work was supported by the National Institute on Aging (R01AG027874) and internal University of Miami funding sources. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.