
Abstract
The primary function of Leydig cells is to secrete testosterone, which is critical in the regulation of male reproduction and development. Low levels of testosterone will lead to male hypogonadism. Stem cell-derived Leydig cell transplantation may be a promising alternative therapy for male hypogonadism. Thus far, others have reported that Leydig-like cells can be derived from mesenchymal stem cells, embryonic stem cells (ESCs), and induced pluripotent stem cells. However, the efficiency of the differentiating Leydig cells remains low, and progress toward generating functional adult Leydig cells (ALCs) is limited. Herein, we describe a robust method of directing differentiation of mouse embryonic stem cells (mESCs) into Leydig-like cells in vitro by overexpression of the transcription factor steroidogenic factor-1 (SF-1) and treatment with a combination of 8-Bromoadenosine-3′,5′-cyclic monophosphate and forskolin. These differentiated cells express mRNA encoding the steroidogenic enzymes and produce progesterone and testosterone. Importantly, when transplanted into male rats that had their original Leydig cells selectively eliminated by ethylene dimethanesulfonate, these in vitro-derived Leydig-like cells further developed into functional ALCs that rescued serum testosterone levels. These data provide evidence that mESCs can be induced to differentiate into Leydig-like cells in vitro, which can develop in the in vivo microenvironment.
Introduction
L
In men, testosterone plays a key role in maintaining muscle bulk, bone growth, and sexual function [2,5]. Low levels of testosterone will lead to a loss of libido, infertility, depression, and fatigue [6]. Male hypogonadism is characterized by low production of testosterone-associated typical symptoms, including mood disturbance, sexual dysfunction, decreased muscle mass and strength, and decreased bone mineral density. Currently, testosterone replacement therapy is required for androgen-deficient males with primary Leydig cell failure. However, most of these patients require therapy for their entire life and are always at risk from certain side effects [7 –10]. Stem cell-derived Leydig cell transplantation may be a promising alternative therapy for male hypogonadism, however, mature Leydig cells are mitotically inactive and the primary immature Leydig cells lose their characteristics during prolonged cultures [1]. Therefore, finding an alternative source of Leydig cells is of paramount interest for both basic research and clinical applications.
The discovery of embryonic stem cells (ESCs) has resulted in an unprecedented opportunity to differentiate tissue-specific cell types that can be used in human disease models, drug screening applications, and patient-specific therapies [11]. While other organs, such as the heart, central nervous system, liver, and pancreas, have benefited from the establishment of differentiation protocols for deriving their functional cell types from ESCs [12], considerably fewer methods have been established to differentiate mouse ESCs (mESCs) into Leydig cells.
Although several studies have attempted to differentiate stem cells, such as mesenchymal stem cells (MSCs) [13,14], ESCs [15 –17] and induced pluripotent stem cells (iPSCs) [18], into steroid-producing cells, the efficiency and purity of the differentiating Leydig cells remain a barrier. These stem cells were induced to differentiate into steroid-producing cells through forced expression of steroidogenic factor-1 (SF-1), which is a transcriptional factor belonging to the nuclear receptor superfamily and is a tissue-specific regulator of the transcription of an array of genes that are involved in reproduction, steroidogenesis, and male sexual differentiation [19,20].
Previous work has reported that SF-1 can initiate a genetic program that enables ESCs, MSCs, and iPSCs to acquire steroidogenic capacity and then produce a variety of steroidal hormones [16,17,21]. However, the steroidogenic capacity of these cells was very limited when treated with retinoic acid (RA) and 8-bromoadenosine-3′,5′-cyclic monophosphate (8-Br-cAMP) in the presence of 20α-hydroxycholesterol as a substrate. Thus, it remains challenging to obtain fully differentiated and functional ALCs in vitro [21]. Additionally, further optimization is clearly needed to establish suitable conditions needed for ESC differentiation into Leydig-like cells in vitro, which will then have the acquired capacity to develop into functional ALCs and produce testosterone in vivo.
In this study, we present a small-molecule-based strategy for the efficient induction of Leydig cells. We found that differentiation toward Leydig-like cells was augmented by 8-Br-cAMP and forskolin (FSK). Transplantation of these Leydig-like cells into an animal model treated with ethylene dimethanesulfonate (EDS) (an alkylating toxicant that selectively eliminates ALCs [22]), develops into functional ALCs after a period of transplantation, which then promote testosterone recovery [22].
Materials and Methods
Cell culture, stable cell transfection, and differentiation
OriCell™Strain C57BL/6 mESCs were obtained from Cyagen (MUBES-01001). The mESCs were maintained with similar conditions as described [23,24]. In brief, cells were cultured on feeder layers of mitomycin-C-treated mouse embryonic fibroblast (MEF) in Knockout™ Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 15% Knockout™ Serum Replacement (Invitrogen), 2 mM GlutaMAXTM-I (Invitrogen), 1% nonessential amino acids (Invitrogen), 0.1 mM 2-mercaptoethanol (Invitrogen), 1% penicillin–streptomycin (Invitrogen), and 1,000 U/mL leukemia inhibitory factor (LIF; Millipore). Cells were passaged at the ratio of 1:6 every 2 days by use of StemPro accutase cell dissociation reagent (Invitrogen), and the culture medium was changed daily. For stable transfection, ESCs were infected with SF-1 lentiviral particles overnight, and subsequent green fluorescence protein (GFP) gene expression was monitored by fluorescence microscopy and flow cytometry.
For differentiation, adherent cells were enzymatically dissociated using the StemPro accutase cell dissociation reagent. For the induction of embryoid body (EB) formation, EBs were formed in hanging drops of 800 cells in 20 μL of culture medium without LIF. After 5 days of culture, EBs were plated on gelatin-coated dishes and cultured in 10% fetal bovine serum (FBS)/DMEM with 8-Br-cAMP (Sigma) or FSK (Sigma) for 8 days of continuous culture.
Plasmid construction
Mouse SF-1 cDNA was amplified from the testis by reverse transcription–polymerase chain reaction (RT-PCR), using forward primer 5′-ACTGAATTCGATATGGACTATTCGTACGACGAGGACCTGG-3′ and reverse primer 5′-TTAGGATCCTCAAGTCTGCTTGGCCTGCAGCATCTCAATGA-3′, cloned into the lentiviral pLVX-EF1α-IRES-ZsGreen1 Vector (Clonetech), and confirmed by sequencing. SF-1 lentiviral particles were packaged into NIH 293T cells following the manufacturer's protocol.
Flow cytometry
Cells were dissociated using the StemPro accutase cell dissociation reagent for 2 min, followed by quenching of trypsin, and then further dissociation in phosphate-buffered saline (PBS) with 10% FBS. The cell suspension was filtered through nylon, and cells were analyzed and sorted by the BD Influx cell sorter.
Animals and treatment
Sprague-Dawley rats (at 8 weeks old) were obtained from the experimental animal center of Sun Yat-Sen University, China. All rats were kept under conditions of controlled temperature (24°C±1°C), relative humidity (50%–60%), and a 12-h light/12-h dark cycle. The standard rodent diet and drinking water were freely accessed by the rats. All surgical procedures and postoperative care were approved by the Institutional Animal Care and Use Committee of Jinan University. Male Sprague-Dawley rats were administered a single i.p. injection of EDS, which was synthesized as previously described [25,26] and dissolved in dimethyl sulfoxide (Sigma–Aldrich) at a dose of 100 mg/kg body weight.
RNA extraction and quantitative RT-PCR
For total RNA preparation, cells were lysed in the RNeasy Lysis buffer (Qiagen) containing 1% β-mercaptoethanol. RNA was isolated using the RNeasy RNA preparation microkit (Qiagen) according to the manufacturer's instructions. One microgram of total RNA was reverse transcribed into cDNA using the Superscript II kit (Invitrogen). The cDNA template was diluted 1:10 and 2 μL of the diluted template was used per 20 μL of the quantitative RT-PCR (qRT-PCR) assay using the Bio-Rad SsoAdvanced SYBR (172-5261). The Bio-Rad CFX connect Real-Time system (Bio-Rad Laboratories) and the Bio-Rad CFX Manager Software (version 2.0) were used to collect the PCR data. Results are presented as linearized values that were normalized to the housekeeping gene GAPDH and the indicated reference value (2−ΔΔCt). The primers are listed in Table 1.
Western blotting
Western blot analysis was conducted as described previously [27,28]. In brief, cells were lysed in 1× RIPA lysis buffer in the presence of a protease inhibitor mixture (Roche)/1% phosphatase inhibitor mixture (Roche). Protein samples were normalized for protein concentration and applied to a 10% SDS-PAGE gel, 40 μg of protein of each was analyzed. For immunoblotting analysis, proteins in the SDS gels were transferred to a polyvinylidene difluoride membrane by an electroblot apparatus. The membranes were blocked with a blocking solution (5% nonfat dry milk protein solution in Tris-buffered saline solution containing 0.5% Tween-20 (TBS-T). The membranes were incubated with primary antibodies in the blocking solution at 4°C overnight, washed with TBS-T thrice (7 min each), and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1.5 h. The membranes were then washed with TBS-T three to five times (7 min each) and subjected to enhanced chemiluminescence detection. The protein expression was normalized to GAPDH.
Cell preparation for in vivo transplantation
Transplantation was performed as previously described [29] with some modifications. Briefly, differentiated cells (grown in 100-mm plates) at day 6 were washed twice with PBS and incubated with the StemPro accutase cell dissociation reagent for 2 min at 37°C. Cells were gently dissociated, resuspended manually, and collected in a 15-mL Falcon tube. Cells were rinsed twice with PBS following centrifugation at 200 g for 3 min. Finally, each pellet was resuspended in PBS for transplantation. The cells were loaded into a 1-mL syringe for injection into the testis of adult Sprague-Dawley male rats that had been treated with EDS. Approximately 1,000,000 cells in a 50 μL volume of PBS were injected into the parenchyma of recipient testes 7 days after the rats received EDS. The control animals for the experimental group were EDS-treated rats that had received a testicular injection of the PBS vehicle. Testes from all animals were examined at 7 and 14 days after transplantation (day 14 and 28 after EDS).
Immunofluorescence and immunohistochemistry
For immunofluorescence experiments, cells were fixed in 4% paraformaldehyde for 15 min and washed thrice in PBS. Cells were blocked in a solution of PBS containing 3% bovine serum albumin (BSA; Sigma), 5% horse serum (Invitrogen), and 0.3% Triton X-100 (Sigma) for 30 min at room temperature. The primary and secondary antibodies were diluted in a solution of PBS containing 3% BSA, 1% horse serum, and 0.1% Triton X-100. Primary antibodies were incubated overnight at 4°C followed by incubation with secondary antibodies for 2 h at room temperature. Nuclei were stained with DAPI (Invitrogen). For histological examination, grafted animals that were previously anesthetized had testis tissues fixed overnight in 4% paraformaldehyde. Immunohistochemistry on paraffin-embedded tissue sections was performed as described. The sections were dewaxed, rehydrated, and treated in a 3% H2O2 methanol solution for 20 min to eliminate the endogenous peroxidase activity. Then, sections were placed in a 70% formic acid solution for 20 min for antigen retrieval. After being rinsed in PBS, sections were incubated in the blocking solution for 1 h at 37°C. Primary antibodies (mouse HSD3B and HSD17B polyclonal antibody, 1:200; Biorbyt) were diluted in the blocking solution and incubated overnight at 4°C. Sections were rinsed thrice in PBS. The secondary antibody, HRP goat anti-mouse IgG (1:200), was used to incubate the sections for 1 h at 37°C. After 3,3-diaminobenzidine and hematoxylin staining, the sections were dehydrated, cleared in xylene, and covered with neutral balsam. Sections from all experimental groups were photographed with a microscope (IX71; Olympus). Nuclei were then stained with DAPI (Invitrogen).
Assay of progesterone and testosterone concentration
Concentrations of progesterone and testosterone in the medium and serum were measured with I125-progesterone Coat-A-Count RIA kits and I125-testoterone Coat-A-Count RIA kits (Beijing North Institute of Biological Technology).
Statistical analyses
All experiments were performed at least thrice, and data are expressed as mean±one standard deviation about the mean. Statistical analyses were performed with an unpaired Student's t-test or one-way ANOVA for more than two groups. The alpha value level for testing statistical significance was P<0.05 (two tailed).
Results
The characteristics of the ES-SF-1 cell line
Even though the molecular mechanisms responsible for specific Leydig cell fate are still uncertain, prior studies have identified the SF-1 transcription factor as an early and essential regulator of Leydig cell specification and differentiation [21]. Several studies have reported that SF-1 could direct stem cells, such as MSCs [13,14], ESCs [15 –17], and iPSCs [18] to differentiate into steroid-producing cells. To direct mESCs into PLCs, we generated an SF-1+ESC line (ESC-SF-1). Fluorescence-activated cell sorting (FACS) and RT-PCR analyzed the characteristics of the ESC-SF-1.
GFP expression, reflecting SF-1 expression in the ES-SF-1 cell line, was analyzed by flow cytometry and the GFP-positive cells were sorted; we obtained over 60% of GFP-positive cells and these GFP-positive cells were allowed to differentiate after FACS. When grown on MEF feeder cells, they organize as spherical colonies, which are transilluminated under phase-polarized light, consequently obscuring the morphology of individual cells (Fig. 1B). RT-PCR confirmed that the expression of undifferentiated mESC genes (eg, oct4, sox2, and Nanog) does not significantly change compared to wild-type mESCs (Fig. 1C). To test whether ESC-SF-1 could form an EB, we cultured ESC-SF-1 in a medium without LIF. We found that almost all of the EBs were GFP positive (Fig. 1D). These results suggested that the ESC-SF-1 had the same stem cell gene expression. Moreover, genetic manipulations did not affect the pluripotent state of the ESCs.

The characteristics of the embryonic stem cells- steroidogenic factor-1 (ESC-SF-1).
Efficient directed mESCs into Leydig-like cells in vitro
In earlier studies, ESC overexpression of SF-1 was shown to produce progesterone after treatment with 8-Br-cAMP, all-trans-RA, and substrate20α-hydroxycholesterol [15,17]. To determine whether ESC-SF-1 lines have the potential of driving the differentiation toward a Leydig lineage, we applied a previously described method to induce differentiation into Leydig cells by using 8-Br-cAMP (a membrane-permeable cAMP analog) [16]. After EB formation during 5 days of suspension culture, the EBs were next plated on gelatin-coated dishes and cultured for an additional 8 days.
We detected the mRNA expression of Leydig cell steroidogenic genes throughout the culture period. The result revealed the expression of mRNA encoding the steroidogenic enzymes, such as steroidogenic acute regulatory protein (StAR), cytochrome P450-containing enzyme (CYP11A1), 3β-hydroxysteroiddehydrogenase (HSD3B), cytochrome P450 17A1 (CYP17A1), and steroid 21-monooxygenase (CYP21A1), whose translation products are required for the synthesis of gonadal steroid hormones were increased as compared to the mock treatment. In addition, the expression of insulin-like factor 3 (INSL-3), a specific marker of Leydig cell differentiation and function [30], was markedly increased on day 2, and the expression level does not significantly change during the next 6 days. Low levels of mRNA expression of these steroidogenic enzymes were also detected in wild mESCs treated by 8-Br-cAMP. Western blot and the levels of progesterone and testosterone further confirmed that 8-Br-cAMP promotes ESC-SF-1 lines to differentiate toward the Leydig lineage. However, the steroidogenic capacity of these cells was very limited when treated with 8-Br-cAMP.
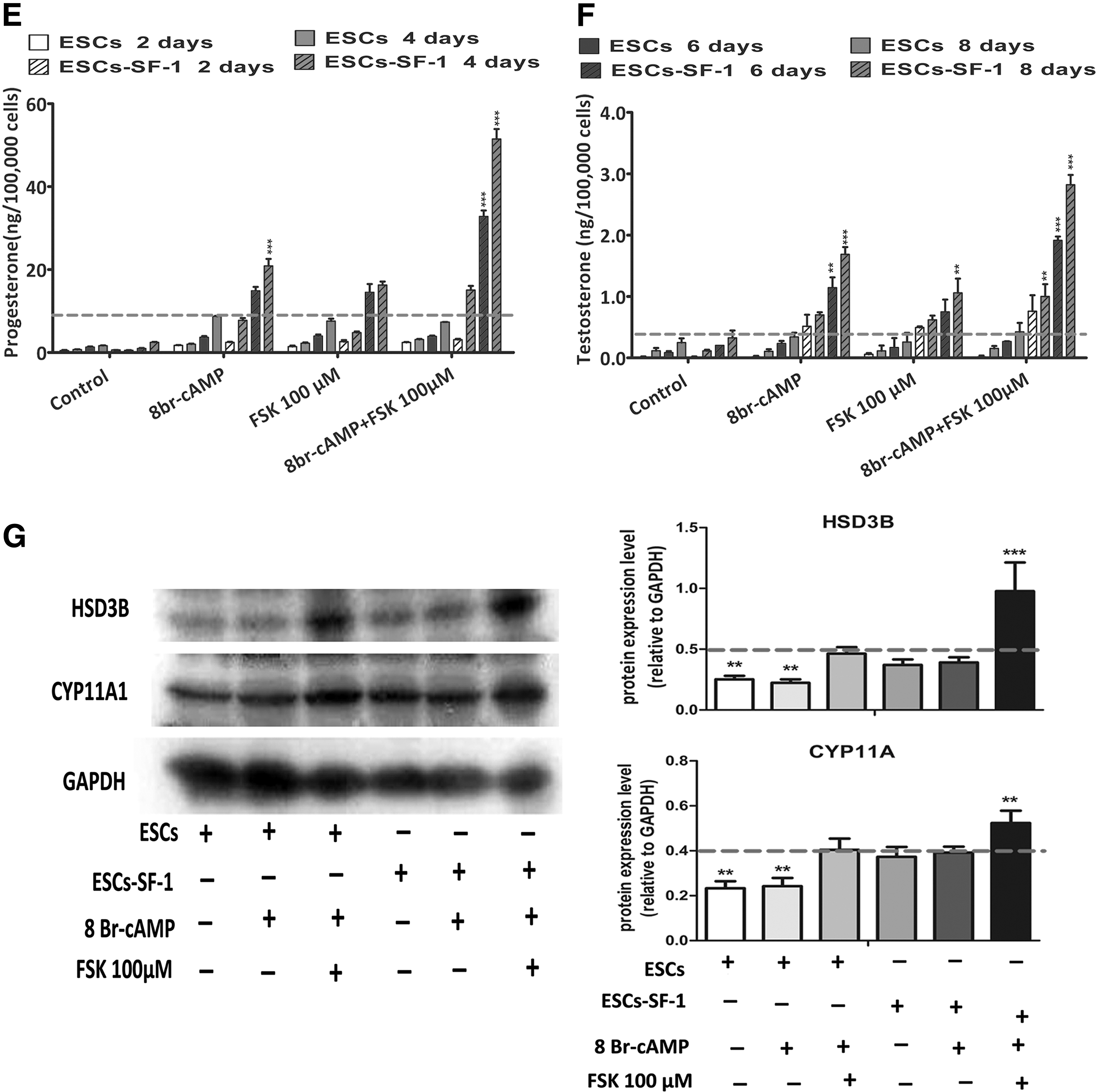
To enhance the steroidogenic cell differentiation of ESC-SF-1, two small molecules, 8-Br-cAMP and FSK, were jointly added to the medium. We found that the expression of mRNA encoding the steroidogenic enzymes StAR, CYP11A1, HSD3B, CYP17A1, CYP21A1, and INSL-3 was robustly upregulated. These steroidogenic gene expression levels were induced more than twofold in cotreatment with 8-Br-cAMP and FSK compared to the 8-Br-cAMP alone. The expression of steroidogenic enzyme genes was significantly higher in the ESC-SF-1 group compared to ESCs differentiated in different culture conditions (Fig. 2B). Protein expression of CYP11A1 and HSD3B and the levels of progesterone and testosterone were also significantly upregulated in the condition of ESC-SF-1 treated with 8-Br-cAMP and FSK (Fig. 3E–G).
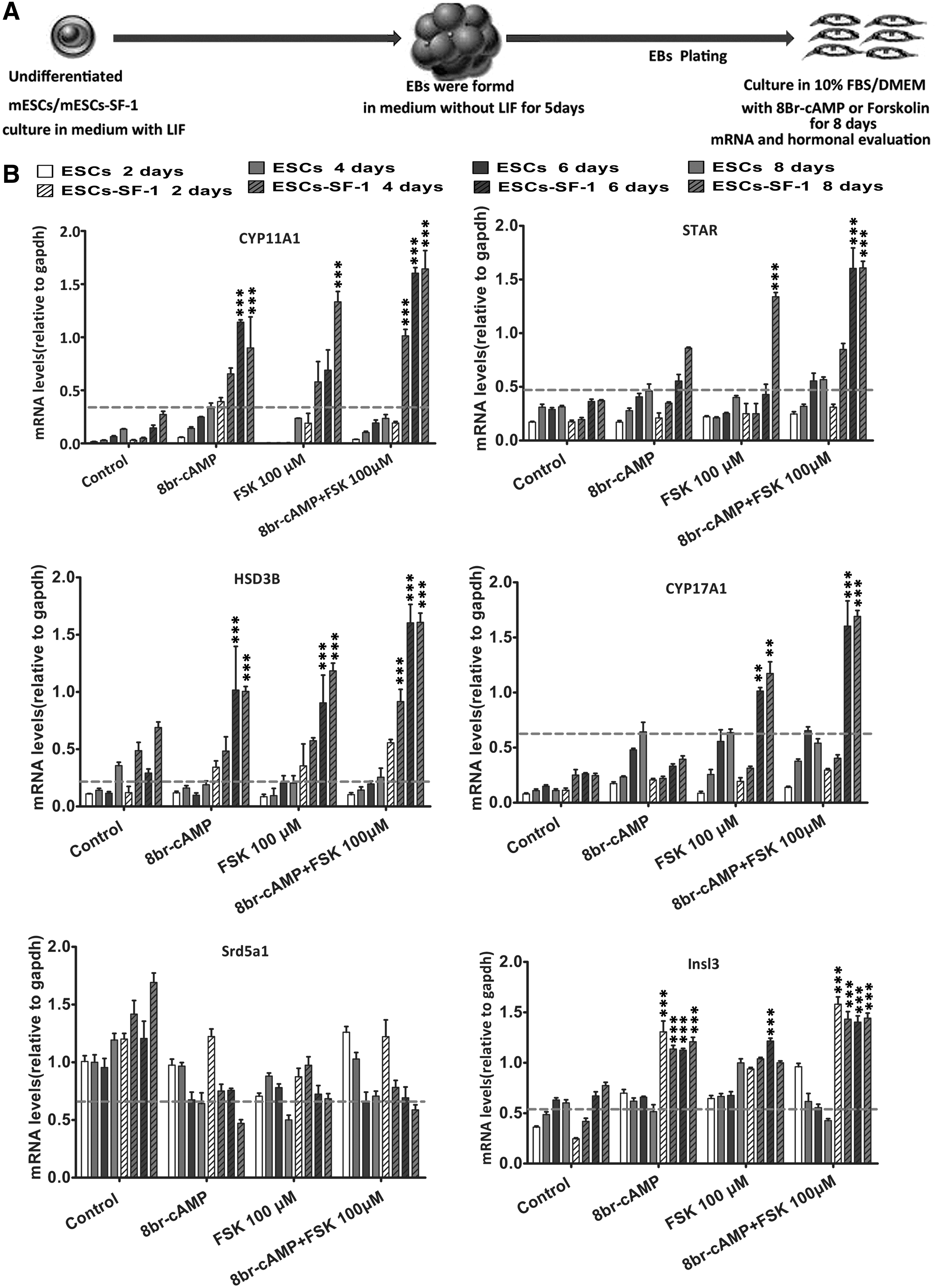
Gene expression analysis of the differentiation of Leydig-like cells from ESCs and ESC-SF-1.
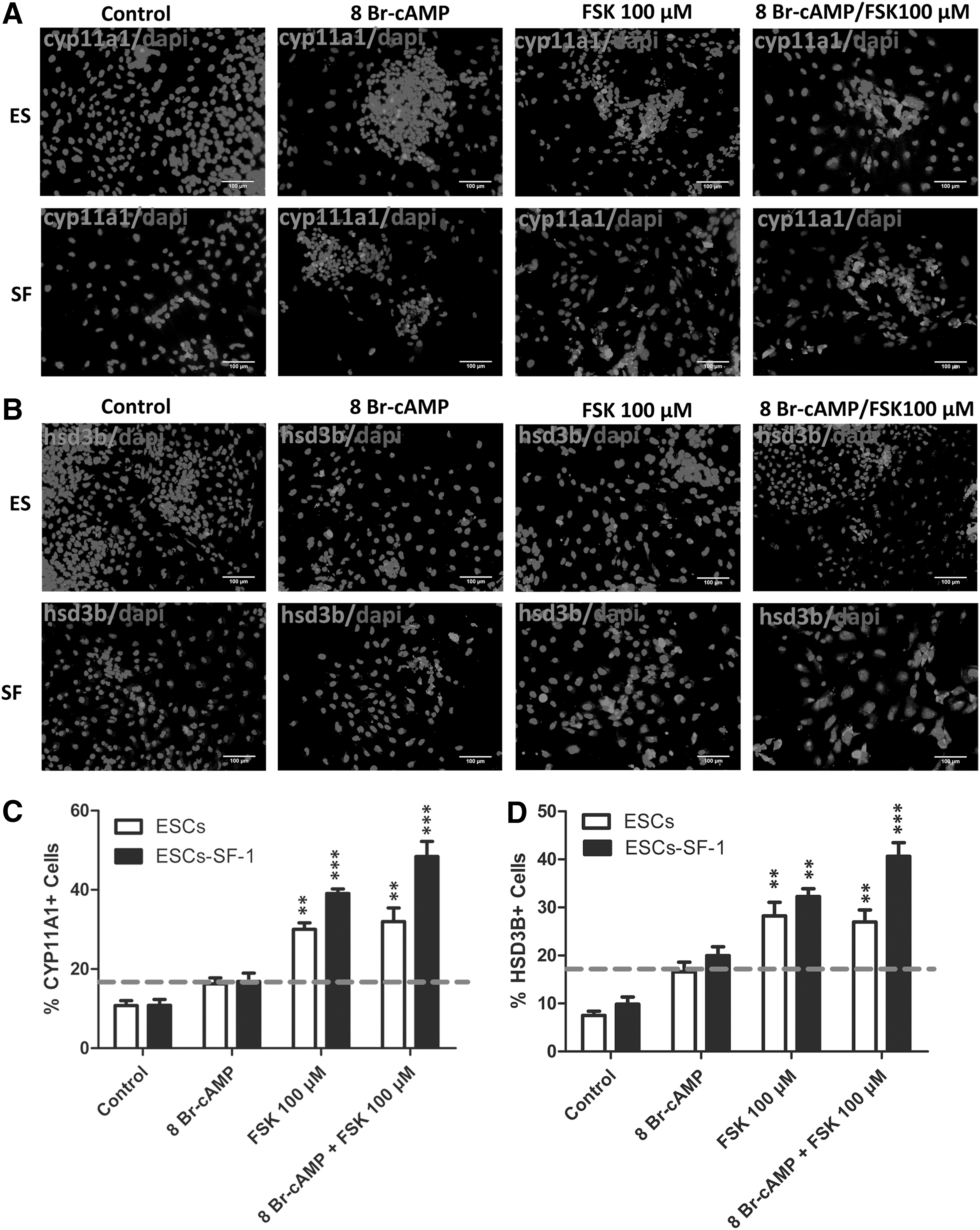
8-Br-cAMP and forskolin (FSK) efficiently induce ESCs and ESC-SF-1 to differentiate into Leydig-like cells.
To test the differentiation efficacy of mESCs and mESC-SF-1 treated with 8-Br-cAMP and FSK, we detected the expression level of CYP11A1 and HSD3B after cell culture for 8 days. The results showed that the percentage of CYP11A1- and HSD3B-positive cells was higher in mESC-SF-1 lines than mESCs. mESC-SF-1 treatment with 8-Br-cAMP alone, at a concentration of 1 mM, induced CYP11A1 and HSD3B expression in 15.60%±5.56% and 19.34%±7.95% of cells (Fig. 3A–D). When mESC-SF-1 cells were treated with FSK, at a concentration of 100 μM, 39.51%±11.6% and 31.40%±6.41% of cells expressed CYP11A1 and HSD3B. The combination of FSK and 8-Br-cAMP increased the percentage of CYP11A1-positive cells to 49.10%±17.53% and HSD3B-positive cells to 38.46%±14.15% of the total population. This percentage was significantly higher compared to the cells treated with 8-Br-cAMP or FSK alone.
Additionally, the measurement of hormones secreted into the medium in the course of 8 days of treatment with 8-Br-cAMP and FSK revealed that the progesterone and testosterone level started to upregulate strongly at day 4 compared to the 8-Br-cAMP alone. These observations suggest that the combination of 8-Br-cAMP and FSK significantly augmented ESC-SF-1differentiation into PLCs compared with 8-Br-cAMP alone.
Derived Leydig-like cells develop into ALCs in vivo
Although differentiated cells in our experiment express key Leydig cell steroidogenic enzyme genes such as StAR, CYP11A1, HSD3B, CYP17A1, CYP21A1, and INSL-3, specific marker genes of ALCs like HSD17B and LHCGR were not detected. This is not surprising since the development of ALCs is regulated by a different hormone. Furthermore, the hormone-regulating system is complex. To assess whether generated Leydig-like cells have the capacity to differentiate into mature Leydig cells in vivo, we transplanted these cells into the testis of adult Sprague-Dawley male rats treated with EDS. On day 7 and 14 after transplantation, the testes were removed for analysis. Hematoxylin and eosin staining showed the localization of the transplanted tissue in the testis of the host rat (Fig. 4B).

ESCs and ESC-SF-1-derived Leydig-like cells survive and integrate, after transplantation in adult rat testes that were depleted of adult Leydig cells (ALCs) by ethylene dimethanesulfonate (EDS).
In order to detect whether ESCs and ESC-SF-1-derived Leydig-like cells possess the characteristic potential of migrating into interstitial regions of the testis, the testis sections at 7 and 14 days after transplantation were histologically evaluated. We found very few cells migrated into the interstitial regions of the testis at post-transplantation day 7 (Fig. 4C). Remarkably, HSD3B+/GFP+ cells were observed migrating into interstitial regions of the testis and formed clusters at postnatal day 14, which demonstrated ESCs and ESC-SF-1-derived Leydig-like cells, which were successfully integrated into the host niche (Fig. 4C). However, the model group that received PBS control injections lacked fluorescence. Moreover, rats that were grafted with ESC-SF-1-derived cells resulted in a significant increase in the percentage of GFP+ cells as compared with the ESC grafts. Coordinately, the expression of key Leydig cell gene HSD3B (Fig. 5A) and mature Leydig cell marker HSD17B (Fig. 5B) was robustly increased in ESC-SF-1-derived cell grafts at day 14 after transplantation.
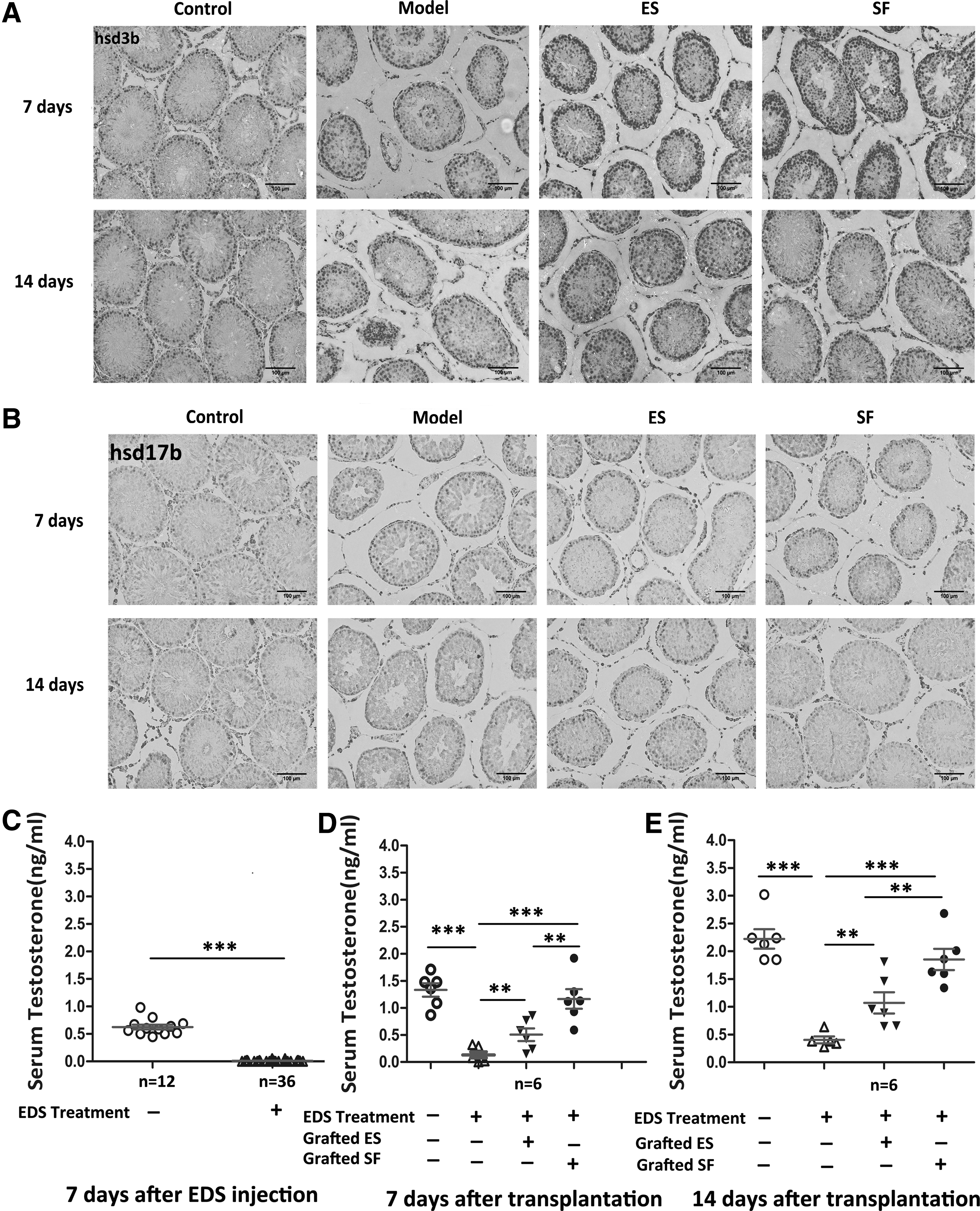
Rescue of experimentally induced EDS by transplantation of ESC-derived Leydig cells.
To confirm whether ESCs and ESC-SF-1-derived cells have the potential to develop into functional ALCs in vivo, we next evaluated the ability of these cells to restore testosterone in EDS-treated rats. The serum testosterone level measured in male Sprague-Dawley rat at 7 days after EDS injection showed a severe downregulation at the time (Fig. 5C). When ESCs and ESC-SF-1-derived cells were transplanted, serum testosterone levels were significantly increased in both the ESC and ESC-SF-1 group as compared with the model group at day 7 after transplantation (Fig. 5D). Serum testosterone levels of rats grafted with ESC-SF-1 cells showed a substantial increase, with a complete rescue of testosterone being evident at day 14 (Fig. 5E). Taken together, these data demonstrated that ESCs and ESC-SF-1-derived Leydig-like cells acquired the characteristics of mature Leydig cells. Additionally, they had the potential capacity to compensate for the lack of serum testosterone when transplanted into rats that were selectively depleted in ALCs.
Discussion
In this study, we report a novel protocol which, to the best of our knowledge, is the first to demonstrate that FSK promotes mESC-SF-1 differentiation into Leydig-like cells in vitro. Importantly, our data demonstrate that transplantation of ESCs and ESC-SF-1-derived Leydig-like cells into Leydig cell-depleted rats has the potential capacity to compensate for the lack of serum testosterone.
Although in earlier studies, ESC overexpression of SF-1 was shown to produce progesterone after treatment with 8-Br-cAMP and all-trans-RA, the steroidogenic cells expressed mRNA encoding adrenal cortical or gonad-specific steroidogenic enzymes, such as CYP17A1, CYP21A2, and CYP11B1 and produced steroid hormones such as progesterone, corticosterone, and cortisol. However, testosterone could not be tested, which means that the earlier inducing method could not acquire fully differentiated and functional Leydig cells [31 –35]. Prior studies demonstrated that mesenchymal progenitors of Leydig cells are present in the testicular interstitium, and that these mesenchymal progenitors are gradually replaced by mature Leydig cells that are thought to differentiate from PLCs during the prepubertal period [3,36 –38]. Based on the above theoretical research observations, we developed a two-step differentiation protocol for differentiating mESCs into Leydig-like cells in vitro, which further developed into mature Leydig cells in vivo.
We first examined whether our mESC-SF-1 could differentiate into Leydig-like cells through EB formation by 8-Br-cAMP treatment. By 2 days of development, marker genes of Leydig cells, such as StAR, CYP11A1, HSD3B, CYP17A1, INSL-3, and testosterone in the medium, were detected. These results are in line with an earlier report [16]. However, the yield of Leydig-like cells after differentiation by 8-Br-cAMP treatment is relatively low and the secreted testosterone is very limited. To further develop a simple protocol for improving the efficient derivation of ESC-SF-1 to Leydig-like cells, we established a method to improve the efficiency of differentiation by a combination of 8-Br-cAMP and FSK. FSK is a mimic of an adrenocorticotropic hormone mode of action. It has been previously reported that FSK dramatically increased SF-1 protein and mRNA levels by repressing DAX-1 protein and mRNA expression [39]. The clinical study also observed the role of FSK in significantly increasing lean muscle mass, bone mass, and testosterone in overweight and obese men [40].
In the presence of FSK, we found that SF-1, StAR, and CYP11A1 mRNA expression significantly increased, and that this effect was dose dependent in ESC-SF-1 (data not shown). These results are consistent with earlier reports [41 –43]. Erik Willems et al. suggested that FSK acted on the expression of SF-1 mRNA at the transcriptional level, and this was determined to be possible because FSK-induced enhanced expression of SF-1 mRNA was almost completely abolished by actinomycin D [39]. Furthermore, multiple SF-1 binding sites are present in the CYP11A1 promoter [44] and the StAR promoter [45].
SF-1 is known to positively regulate CYP11A1, and StAR expression [46 –49] mediates cAMP responsiveness. The combination of 8-Br-cAMP and FSK can significantly improve the efficient derivation of ESC-SF-1 to Leydig-like cells, which is due mainly to improved SF-1 transcriptional levels by the action of FSK. Subsequently, enhanced expression of steroidogenic enzymes promotes the process of ESC and ESC-SF-1 differentiation toward Leydig-like cells.
Thus, it can be seen that FSK treatment is the most critical element in the induction of ESCs and ESC-SF-1 differentiation by direct stimulation of gene transcription of steroidogenic enzymes. However, 8-Br-cAMP treatment induces cellular differentiation by activation of cyclic AMP-dependent protein kinase rather than by direct stimulation of gene transcription of steroidogenic enzymes. Importantly, when Leydig-like cells are transplanted into the testis, these cells have the potential of migration to the testicular interstitium and further develop into functional Leydig cells in vivo, which significantly improve the level of serum testosterone in the experimental animal model group.
However, our data do not clarify the precise mechanisms of FSK-induced ESC-SF-1 differentiation toward Leydig-like cells and do not rule out the possibility of cell fusion between donor Leydig-like cells and recipient testicular Leydig cells or their progenitor cells. Considerable basic research still needs to confirm the long-term safety of transplantation of mESC-SF-1-derived Leydig cells. Additionally, the limitations of our study are that the differentiated cells do not proliferate as well as the undifferentiated cells, making it difficult to obtain large numbers of cells and the differentiated cells are unpurified. To generate ESC-derived Leydig cells at a high efficiency, further studies will be required to address the precise mechanisms of the differentiation of mESCs toward Leydig fate and to identify key molecules that promote Leydig cell induction from ESCs in vitro.
Although further studies are required, the differentiation protocol described here provides an effective method to differentiate mESCs toward Leydig fate in vitro. These results pave the way toward the future refinement and use of ESCs for cell replacement therapy of Leydig cell degeneration and provide a powerful tool for studying the molecular mechanisms of steroidogenic tissue development.
Footnotes
Acknowledgments
The work was supported by grants from the National Natural Science Foundation of China (no. 81070477, 81370704, 31171425, and 31271607) and the project was supported by Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (2012).
Author Disclosure Statement
No competing financial interests exist.