
Abstract
Basic fibroblast growth factor (bFGF) is a crucial factor sustaining human pluripotent stem cells (hPSCs). We designed this study to search the substitutive factors other than bFGF for the maintenance of hPSCs by using human placenta-derived conditioned medium without exogenous bFGF (hPCCM−), containing chemokine (C-X-C motif) receptor 2 (CXCR2) ligands, including interleukin (IL)-8 and growth-related oncogene α (GROα), which were developed on the basis of our previous studies. First, we confirmed that IL-8 and/or GROα play independent roles to preserve the phenotype of hPSCs. Then, we tried CXCR2 blockage of hPSCs in hPCCM− and verified the significant decrease of pluripotency-associated genes expression and the proliferation of hPSCs. Interestingly, CXCR2 suppression of hPSCs in mTeSR™1 containing exogenous bFGF decreased the proliferation of hPSCs while maintaining pluripotency characteristics. Lastly, we found that hPSCs proliferated robustly for more than 35 passages in hPCCM− on a gelatin substratum. Higher CXCR2 expression of hPSCs cultured in hPCCM− than those in mTeSR™1 was observable. Our findings suggest that CXCR2 and its related ligands might be novel factors comparable to bFGF supporting the characteristics of hPSCs and hPCCM− might be useful for the maintenance of hPSCs as well as for the accurate evaluation of CXCR2 role in hPSCs without the confounding influence of exogenous bFGF.
Introduction
S
Materials and Methods
Antibodies and reagents
The antibodies against desmin, alpha-fetoprotein (AFP), FGF2, β-actin, and GATA4 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), and the antibodies against Erk, p-Erk, and neuron-specific class III beta-tubulin (TUJ1) were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Recombinant human interleukin (IL)-8, recombinant human growth-related oncogene α (GROα), anti-IL-8, anti-GROα, and anti-CXCR2 (R&D Systems, Inc., Minneapolis, MN) were used in this study. Recombinant human bFGF, Alexa488, and Alexa594 were obtained from Invitrogen (Carlsbad, CA). The small-molecule inhibitors SB225002 and SB265610 were obtained from Tocris Bioscience (Bristol, United Kingdom). The hESC-qualified Matrigel (BD Biosciences, San Jose, CA) and the mTeSR™1 medium (StemCell Technologies, Inc., Vancouver, BC) were also used in this study. The antibodies against human CXCR2 were obtained from Abcam (Cambridge, United Kingdom). The transfection studies were performed with scrambled small interfering RNA (siRNA) and siCXCR2, both of which were purchased from Santa Cruz Biotechnology.
hESCs induced pluripotent stem cell culture
hPSCs, that is, H1 and H9 cells (listed in the NIH hESC registry under the names WA01 and WA09, respectively), induced pluripotent stem cell (iPSC)-1 (foreskin), and iPSC-2 (IISHi-BM1), were purchased from the WiCell Research Institute (Madison, WI). The hESC line SNUhES3 was obtained from the Seoul National University Hospital (Seoul, South Korea) as previously described [11]. Cells for the control group were cultured on Matrigel-coated dishes in mTeSR™1 (the most widely used feeder-free and serum-free defined culture medium) at 37°C and 5% CO2. Initially, the cells were subcultured with routine passaging once every 5–6 days, using mechanical or enzymatic means (dispase; Worthington Biochemical Corporation, Lakewood, NJ). The cells were washed twice with the medium and plated at a ratio of 1:4. The cells for the experimental groups were cultured in hPCCM in dishes coated with 0.1% gelatin (Sigma-Aldrich Corporation, St. Louis, MO); passaging was performed routinely once every 5–6 days, mechanically or enzymatically, and the cells were plated at ratios in the range 1:6–1:10. Feeder-dependent cultures were established in Dulbecco's modified Eagle medium (DMEM)-F12 (Gibco Life Technologies, Grand Island, NY) supplemented with 20% Knockout™ Serum Replacement (KOSR; Gibco), 0.1 mM β-mercaptoethanol, 1% non-essential amino acid (NEAA) cell culture supplement (Gibco), 1% penicillin–streptomycin (Gibco), and 10 ng/mL bFGF (Invitrogen). All the manipulations and cultivations were performed in a clean germ-free facility at the Korea University Medical Center. The experimental design and procedures using hPSCs of this study were approved by the Institutional Review Board of the Korea University Medical Center (AN12277-003).
Human placenta-derived cells conditioned medium (hPCCM−)
The specific protocol followed was as previously described [11]. Briefly, the placental chorionic plates from healthy women who underwent abortion at 6–8 weeks of gestation were surgically isolated, minced, and incubated in 0.25% trypsin-ethylenedeaminetetraaceticacid (EDTA; Gibco) at 37°C for 30 min. The cells were cultured in DMEM (Gibco) containing 20% fetal bovine serum (Gibco) and 1% penicillin–streptomycin (Gibco) at 37°C, 5% CO2, and 95% humidity. The medium was changed every 2 days. During culture, the cell debris was removed, and the adherent fibroblast-like cells were grown on the tissue culture plates. These fibroblast-like cells derived from the placental chorionic plate (hPC) were cultured till the 10th passage and frozen as stock. Before being frozen as stock, reverse transcriptase-polymerase chain reaction was used to determine whether the hPCs were contaminated with pathogens that commonly infect the placenta (eg, cytomegalovirus, herpes simplex virus types 1 and 2, Chlamydia trachomatis, Chlamydia spp., Mycoplasma genitalium, M. hominis, and Ureaplasmaureaticum). After the ruling out of contamination, cells at 80% confluency were treated with mitomycin-C (Sigma-Aldrich, catalogue no. M4287), harvested, and frozen in CM after 24 h of incubation in DMEM-F12 supplemented with 20% KOSR, 0.1 mM β-mercaptoethanol, 1% NEAA, and 1% penicillin–streptomycin. The supernatants from the cultures were collected every day for 1 week, and the harvested medium was frozen at −80°C.
Human cytokine array
The human cytokine array C Series 1000 (120) (Ray Biotech, Inc., Norcross, GA) was used according to the manufacturer's instructions. Briefly, the cytokine array membranes were incubated overnight at 4°C in equal quantities of conditioned media obtained from basal medium (DMEM-F12+20% KOSR), hPCCM−, and mTeSR™1. After washing with phosphate-buffered saline (PBS), the membranes were incubated in biotin-labeled primary antibodies followed by the addition of 1,000-fold diluted horseradish peroxidase (HRP)-conjugated streptavidin. The membranes were then developed. After developing, the films were scanned, and the images were processed and quantified using the ImageJ software (National Institutes of Health, Bethesda, MD). The signal intensity was normalized to that of internal positive controls for comparison.
Multiplex bead assay
Quantitative measurements of IL-8 and bFGF were performed according to the instructions in the Bio-Plex Pro™ Cytokine, Chemokine, and Growth Factor Assay kit (Bio-Rad Laboratories, Hercules, CA). Fluorescently labeled beads with a spectral range that permitted the discrimination of individual tests within a multiplex suspension were used in this assay. The capture antibodies directed against the desired biomarker were covalently coupled to the beads, and the coupled beads were subsequently treated with the sample containing the biomarker of interest. After a series of washes to remove the unbound protein, a biotinylated detection antibody was added to create a sandwich complex. The final detection complex was formed with the addition of a streptavidin-phycoerythrin conjugate. Phycoerythrin serves as a Bio-Rad Bio-Plex®200 Systems Instrument. The markers were grouped together according to their dilution factors after checking the cross-reactivities of all the analytes. The control samples at the two levels in the dynamic range of the standard curve were run together in quadruplicate for quality control throughout the study.
Enzyme-linked immunosorbent assay
The detection of GROα in hPCCM− was performed using a Human GROα ELISA kit (Ray Biotech, Inc.), as per the instructions in the kit. Briefly, 100 μL of hPCCM− and standards (the recombinant human GROα provided with the kit) were added to a 96-well enzyme-linked immunosorbent assay (ELISA) plate. After washing, the plates were incubated with 100 μL of the biotinylated anti-human GROα antibody for 1 h at room temperature (RT), followed by the addition of HRP-conjugated streptavidin. The reaction was monitored with a tetramethylbenzidine one-step substrate reagent followed by a stop solution. The 96-well ELISA plate was read at 450 nm using a microplate reader. The GROα concentration in the samples was analyzed based on the standard curve prepared in accordance with the instructions provided in the kit.
Immunofluorescence
For immunofluorescence staining, the hPSCs were cultured and fixed in eight-well slide chambers (BD Biosciences) with 4% (w/v) paraformaldehyde, permeabilized with 0.1% (v/v) Triton-X100, and blocked for 1 h with 3% (v/v) normal horse serum (Gibco) in PBS containing 0.1% (v/v) Tween-20 (Sigma-Aldrich). The cells were incubated with the primary antibody overnight at 4°C and with the secondary antibody for 1 h at RT. Between incubations, the cells were washed three to five times with 0.1% (v/v) Tween-20 in PBS buffer. Before mounting, the cells were incubated with 4′,6-diamidino-2-phenylindole (Molecular Probes, Invitrogen) for 5 min in the dark. The cells were preserved in the fluorescence mounting medium (Vector Labs, Burlingame, CA) and observed under a fluorescence microscope (Olympus, Tokyo, Japan). The primary antibodies for TRA-1-60, TRA-1-81, and stage-specific embryonic antigen (SSEA)-4 were purchased from Merck Millipore, Inc. (Billerica, MA). Antibodies against octamer-binding transcription factor (OCT)-4, NANOG, and sex-determining region Y-box 2 (SOX2) were purchased from Cell Signaling Technology, Inc., and the other antibodies were purchased from Santa Cruz Biotechnology.
Quantitative PCR
Quantitative PCR (qPCR) was performed as previously described to compare the expression of the pluripotency markers [8]. Total RNA was isolated from cells using a Qiagen RNeasy kit (Qiagen, Hilden, Germany), and the extracted RNA was quantified using a Nano Drop Spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE). The cDNA was synthesized by adding 2 μg of the total RNA to a 20 μL reaction mixture containing oligo (dT) primers and Superscript II reverse transcriptase (Gibco), according to the manufacturer's instructions. The synthesized cDNA was amplified using a Bio-Rad iCycler iQ system, with the iQ SYBR Green qPCR Master Mix (Bio-Rad Laboratories). The primers for the markers used in the qPCR have been described in Supplementary Table S2 (Supplementary Data are available online at
Detection of alkaline phosphatase activity
The alkaline phosphatase (AP) activity was detected using the ES Cell Characterization kit (Chemicon International, Inc., Temecula, CA), according to the manufacturer's protocol. The stained cells were examined and imaged using an Olympus microscope (IX71; Olympus).
Transfection (siRNA)
The transfection of the hPSCs was performed as per the protocols adopted by Braam et al. [12]. Briefly, to prepare siRNA/lipid solutions for each well of a gelatin-coated 12-well plate, a total of 100 nM of the siRNA was diluted in 75 μL of OPTI-MEM (Invitrogen), and incubated at RT for 5 min. In a separate tube, 1.5 μL of Lipofectamine® 2000 (Invitrogen) was diluted in 75 μL of OPTI-MEMI and incubated for 5 min at RT. The contents of the two tubes were mixed by gentle pipetting, followed by RT incubation for 20 min. The resulting 154.5 μL of the siRNA/Lipofectamine 2000 transfection solution was added dropwise to the plate wells. After 4 h of incubation, 1.5 mL of the hPCCM− medium was added. The siRNA was also purchased from Santa Cruz Biotechnology [control siRNA (sc-37007), siCXCR2 (sc-40026)].
Western blotting
The western blot analysis was performed as previously described [9]. Briefly, the protein concentrations were determined using protease inhibitors. Equal amounts of protein were resolved on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and transferred onto a polyvinylidene fluoride membrane. Next, the blot was blocked and probed overnight with different primary antibodies at 4°C, followed by incubation with HRP-conjugated secondary antibodies for 1 h. The signals were detected using an ECL reagent (Dogen, Korea).
Proliferation assay
The cell growth was calculated based on the CCK-8 assay (Dojindo Laboratories, Kumamoto, Japan). After 24 h of the hPSC transfection with siCXCR2 , 1×104 cells were plated in gelatin-coated 96-well plates. Four wells were selected every 48–144 h. CCK-8 (10 μL) was added to each well and incubated for 3 h. The absorbance of the samples was measured at 450 nm. Each experiment was repeated in triplicate.
Cell viability assay
The cell viability was determined after 1×105 cells seeded in 12-well plates on Matrigel and gelatin were incubated for 48 h using a LIVE/DEAD assay kit, according to the kit manufacturer's instructions (Molecular Probes, Invitrogen). Calcein AM (1 μM) and EthD-1 (2 μM) were added to 10 mL of PBS. After incubation for 30 min, the stained cells were viewed under a fluorescence microscope (Olympus). The live cells produced green fluorescence, whereas the dead cells displayed red fluorescence. All the images were analyzed using the Metamorph software (Molecular Devices, Sunnyvale, CA), to determine the percentage of live and dead cells.
Flow cytometry analysis
Cells were removed from the culture dishes using trypsin-EDTA, dissociated into a single-cell suspension, and resuspended in cold fluorescence-activated cell sorting (FACS) buffer [0.1% (v/v) bovine serum albumin]. These cells were then incubated with the primary fluorescence-conjugated antibodies against SSEA-4, SSEA-1, OCT-4, SOX2 (from R&D Systems, Inc.), Tra-1-60, or Tra-1-81 (both from Merck Millipore, Inc.) for 1 h on ice in the dark, and then washed twice with the cold FACS buffer. The control cells were incubated with the immunoglobulins (IgG and IgM) and then with the secondary antibody as described earlier. The cells were subsequently sorted using an FACS Calibur Flow Cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ), and the data were analyzed with the CellQuest software (Becton, Dickinson and Company).
Embryoid body formation
The hPSCs were transferred to the low-attachment plates and allowed to spontaneously differentiate through embryoid body (EB) formation in the DMEM-F12 medium containing 20% KOSR, 1% NEAA, and 0.1 mM β-mercaptoethanol with medium changes every 2–3 days. After 2 weeks in suspension, the EBs were transferred to gelatin-coated dishes and cultured for 1–2 weeks. They were then fixed and stained with antibodies against the markers for all the three embryonic germ layers (AFP, TUJ1, desmin, and GATA4, respectively), and analyzed as described later for immunofluorescence.
Teratoma formation
The teratoma formation experiments were performed as previously described [13] with subcutaneous implantation of ∼5×105–1×106 cells into young (7 week old) severe combined immunodeficiency (SCID)-beige mice. Four animals were used per cell line (n=4). The teratoma growth was determined by palpation every week, and the mice were sacrificed at 8–9 weeks after the implantation. The teratomas were fixed, and the sections were stained with hematoxylin and eosin. The tissue components of all three embryonic germ layers were detected in the stained sections. All the animal experiments were approved by the ethics committee of the Korea University Medical School (KUIACUC-2013-76) and performed at the specific pathogen-free animal facility at the medical school.
Short tandem repeat genotyping
For the short tandem repeat (STR) analysis, the genomic DNA was extracted from the control cells (Matrigel+mTeSR™1) and experimental cells (gelatin+hPCCM−) at the same passage number using a QIAamp® DNA Micro kit (Qiagen), according to the manufacturer's instructions. The extracted DNA was amplified for 16 different genetic loci using the Promega PowerPlex 16 System kit (Promega, Madison, WI) or the AmpF/STR® Identifiler® PCR Amplification kit (Applied Biosystems, Inc., Carlsbad, CA), and capillary electrophoresis was carried out using an Applied Biosystems® 3130xl Genetic Analyzer (Applied Biosystems).
Karyotype analysis
The karyotyping of the cell lines was carried out using G-banding studies as previously described [11]. Briefly, the hPSCs were cultured on gelatin in hPCCM− supplemented with 0.1 mg/mL of the colcemid solution for 3–4 h, trypsinized, and then incubated in 0.075 M KC1 for 20 min at 37°C. After fixation with a solution of 3:1 methanol/acetic acid, the karyotypes of the hPSCs were determined at the 300-band level of resolution.
Apoptosis analysis
Apoptosis analysis was performed after the transfection of hPSCs with siCXCR2, using the EzWay Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit (Koma Biotech, Inc., Seoul, Korea). Briefly, 1×106 cells/mL were transferred to a 0.5 mL cell suspension (5×105 cells/mL) in a microtube. The medium was removed, and the cells were washed with 0.5 mL of cold PBS. After centrifugation at 1,000 rpm for 5 min at RT, the cells were washed with 0.5 mL of cold 1×binding buffer. Next, 1.25 μL of annexin V-FITC was added, and the cells were incubated for 15 min at RT in the dark. Then, 10 μL of propidium iodide (PI) was added to the cell suspension, and the cells were immediately analyzed using flow cytometry or fluorescence microscopy. The flow cytometry analysis was performed with the FL1 channel for detecting FITC (518 nm), and with the FL2 channel for detecting PI (620 nm).
Statistical analysis
The statistical significance of the differences was determined using the two-tailed Student's t-test for comparing two groups, or by performing the two-way analysis of variance for multiple comparisons, followed by Tukey's test. All experiments were repeated in triplicate. A P-value of >0.05 was considered statistically significant.
Results
Human placenta CM contains many ligands that stimulate CXCR2 but nearly undetectable bFGF levels
We estimated the concentrations of cytokines present in hPCCM− and compared the data with those obtained from mTeSR™1 (a commercialized CM from mouse embryonic fibroblasts using Matrigel). Cytokine array analyses revealed that hPCCM− contains high concentrations of GROα and IL-8, which commonly activate CXCR2. Furthermore, we identified seven CXCR2 ligands (CXCL1, 2, 3, 5, 6, 7, and 8) included in hPCCM−. However, bFGF was nearly undetectable in hPCCM− compared with a similar quantity of DMEM-F12 (Fig. 1A). The levels of major cytokines in hPCCM− were quantitatively measured using multiplexed bead and ELISA, which confirmed the presence of high levels of CXCR2-related ligands and minimal bFGF content (Fig. 1B–D). These findings suggested that CXCR2 ligands might play a role in the maintenance of hPSC pluripotency and proliferation, so we developed a culture system using hPCCM− to evaluate CXCR2 effects on hPSCs [hESCs (H1) and human foreskin iPSC-1] and eliminate the influence of exogenous bFGF. We found out that hPSCs cultured in hPCCM− can attach to gelatin in a manner similar to their attachment to Matrigel (Fig. 5E), and this attachment is not facilitated by IL-8 or GROα (data not shown). This finding has not been previously reported. Accordingly, we developed a new feeder-free culture system for hPSCs in hPCCM− on a gelatin substratum for this study. Matrigel was not employed, because it contains bFGF [14].
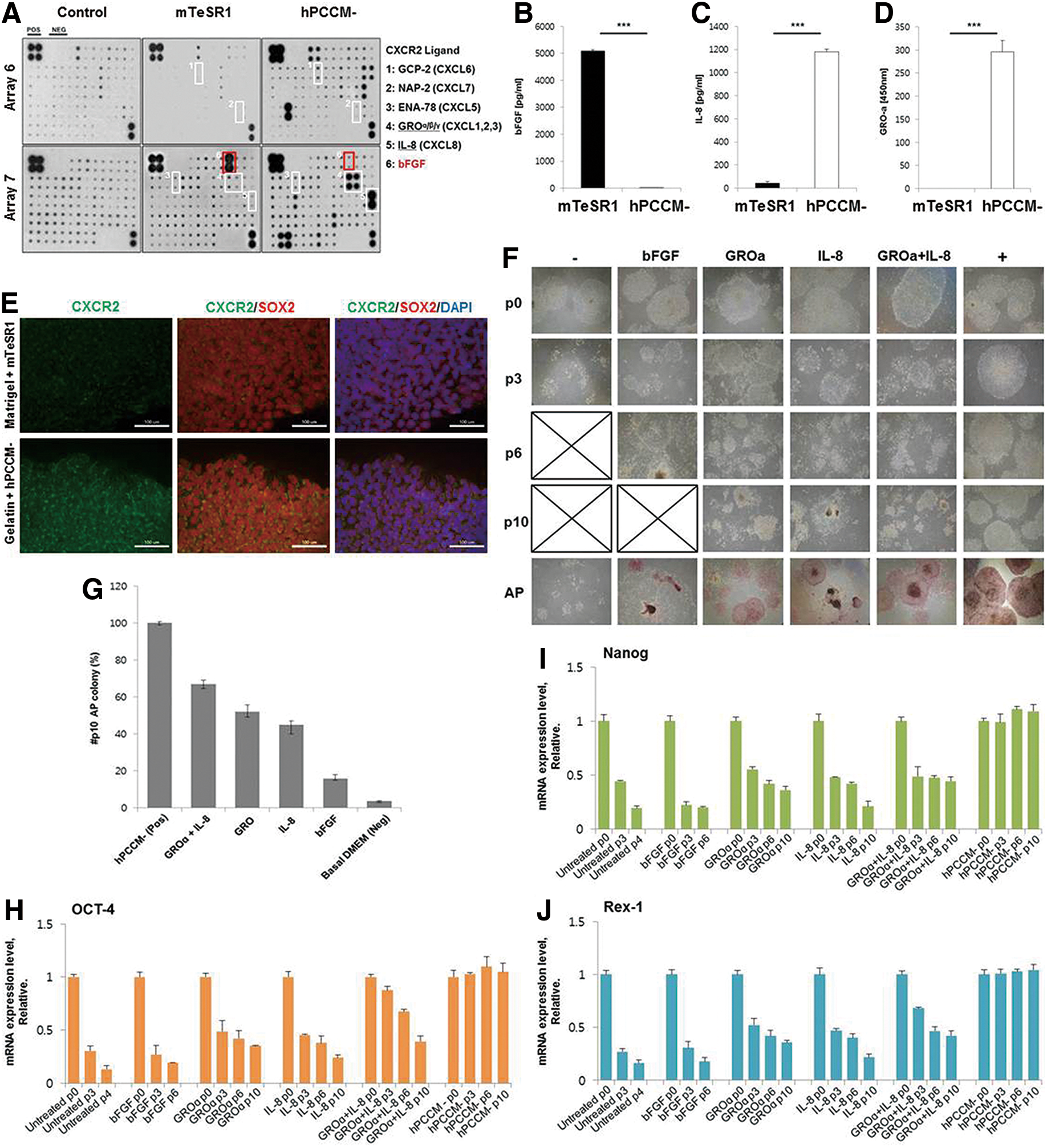
hPCCM− contains CXCR2 ligands propagating hPSCs without bFGF supplementation.
IL-8 and/or GROα support the maintenance of hPSC characteristics and proliferation
Next, we investigated the direct effects of IL-8 and/or GROα on hPSC propagation and compared them with the effects of bFGF. Since IL-8 and GROα do not facilitate the attachment of hPSCs, we initially cultured hPSCs in hPCCM− on gelatin for 24–48 h to enhance their attachment. None of the experiments included treatment with factors that aid in cell attachment such as Rho-associated kinase inhibitor (Y-27632). Subsequently, we cultured hPSCs in a basal medium (DMEM-F12+20% KOSR) without any additives (basal medium group), medium containing IL-8 and/or GROα (CXCR2 ligand group), medium with bFGF only (bFGF group), or hPCCM− (hPCCM− group). Cells were cultured for 3–4 days and subcultured thereafter. hPSCs survived only for 3 and 6 passages in the basal medium and bFGF groups, respectively. In the CXCR2 ligand group, the hPSCs survived for more than 10 passages (10–15 passages) (Fig. 1F). In the hPCCM− group, hPSCs were propagated for more than 35 passages. After 10 passages, hPSCs of the CXCR2 ligand group exhibited pluripotency to an extent comparable to those in the hPCCM− group, as estimated by measuring AP activity and by qPCR. In the CXCR2 ligand group, the expression levels of pluripotency-related genes were higher in the IL-8+GROα combination than in groups treated with either IL-8 or GROα individually (Fig. 1G–J). Using the hPCCM− culture system, we suspected that IL-8 and GROα play a direct role in the maintenance of hPSC characteristics and that this role is novel and greater than that of bFGF. However, the inability of IL-8 and GROα to sustain hPSCs for more than 15 passages might suggest that these ligands have limited effects on hPSCs in hPCCM−. Therefore, we evaluated the contribution of CXCR2, the common receptor for IL-8 and GROα, toward the maintenance of hPSC characteristics and proliferation.
CXCR2 suppression inhibits the maintenance of hPSC characteristics and proliferation
We observed little to no visible change in hPSCs on directly blocking IL-8 and GROα in hPCCM− with neutralizing antibodies (Fig. 2A, B and Supplementary Fig. S1), suggesting that other ligands of CXCR2 in hPCCM− such as GCP-2 or NAP-2 might stimulate hPSCs (Fig. 1A). It is not physically possible to block all CXCR2 ligands completely in hPCCM−. Hence, we investigated CXCR2 activation and found that it is higher in SOX2-positive hPSCs in hPCCM− than in mTeSR™1 on Matrigel (Fig. 1E). This suggests that the CXCR2 might be associated with the maintenance of hPSC characteristics in hPCCM−. Subsequently, we blocked CXCR2 in hPSCs using neutralizing antibodies or low doses of the specific antagonists SB225002 or SB265610 for 2 weeks or 20 days, respectively (Fig. 2C, D and Supplementary Fig. S2). CXCR2 blocking impeded hPSC self-renewal, and the proportion of differentiated populations gradually increased compared with the vehicle control group (Fig. 2C, D). Pluripotency-associated gene expression was also markedly decreased (Fig. 2E). In contrast, the expressions of three embryonic germ layer markers, especially NESTIN and trans-acting t-cell-specific transcription factor (GATA3), were greatly increased (Fig. 2F). To investigate these findings more thoroughly, we suppressed CXCR2 in hPSCs by siRNA to examine the function of CXCR2 in accordance with the work by Braamn et al. [12]. CXCR2 expression in hPSCs was suppressed by ∼80% at 72 h after siRNA transfection (Fig. 3A). During this period, qPCR measurements showed a significant decrease in the expression of pluripotency markers and an increase in the expression of three germ layer markers (Fig. 3B–E). However, we did not observe any associated changes in bFGF expression (Fig. 3C). It has been established that phosphorylation of extracellular signal-regulated kinases (ERK) is suppressed when hPSCs lose their stem cell characteristics and that bFGF activates the ERK pathway in hPSCs [15]. To investigate the correlation between CXCR2 suppression and the ERK pathway in hPSCs, we performed western blotting, which revealed a lower ERK phosphorylation level in the CXCR2-suppressed group than that in the group transfected with control siRNAs (Fig. 3F). Moreover, the proliferative ability of hPSCs was markedly decreased during this period (Fig. 3G–I), indicating that CXCR2 influences the ERK pathway in a manner similar to bFGF. In addition, we conducted the experiments described earlier to block and suppress CXCR2 in hPSCs cultured in mTeSR™1 on Matrigel to verify the role of CXCR2 in hPSCs under the influence of exogenous bFGF. Blocking CXCR2 using a neutralizing antibody or specific antagonist showed no significant changes, unlike the results in hPCCM− (Fig. 4A, B). Quantitative analysis of pluripotency marker mRNA levels at each time point indicated slightly decreased (<20%) expression after 20 days of antagonist treatment. In addition, changes in expression of the three germ layer markers were insignificant (Fig. 4C, D). Interestingly, suppression of CXCR2 in hPSCs by siRNA induced the decrease of p-ERK expression and proliferation with a few changes in pluripotency marker expression (Fig. 4E–H). Considering the fact of which mTeSR™1 and Matrigel scarcely contain CXCR2 ligands, the result of siRNA experiment might be more important than that of blocking CXCR2. Therefore, these results lead us to suspect that CXCR2 is mainly associated with the proliferation of hPSCs rather than the maintenance of pluripotency characteristics in the condition of which hPSCs is supported by exogenous bFGF. We also investigated the possibility that CXCR2 suppression might influence apoptosis and the adhesive properties of hPSCs but observed no significant changes in these aspects (Supplementary Figs. S3 and S4). These findings suggest that CXCR2 in hPSCs is not associated with apoptosis but is mainly associated with pluripotency and proliferation. We, therefore, examined the feasibility of the bFGF-independent importance of CXCR2 to sustaining hPSC pluripotency and proliferation using the hPCCM− feeder-free culture system.

Blocking CXCR2 induced the differentiation of hPSCs.

CXCR2 knockdown leads to the loss of pluripotency markers and growth inhibition of hPSCs.
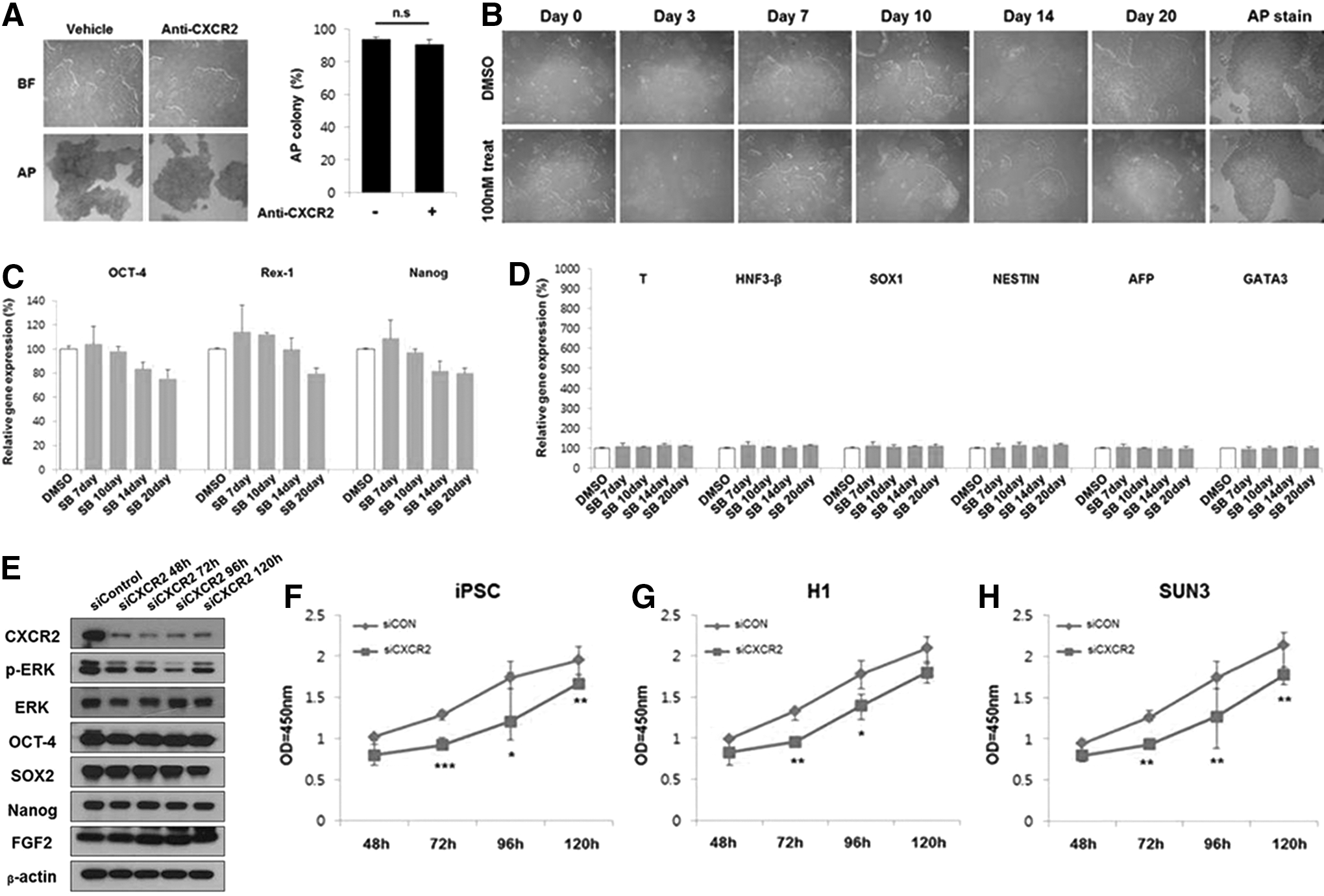
CXCR2 suppression deceases the proliferation of hPSCs in bFGF-containing medium. hPSCs were cultured on Matrigel™ in mTeSR™1.
The hPCCM− culture system can support propagation of various hPSCs
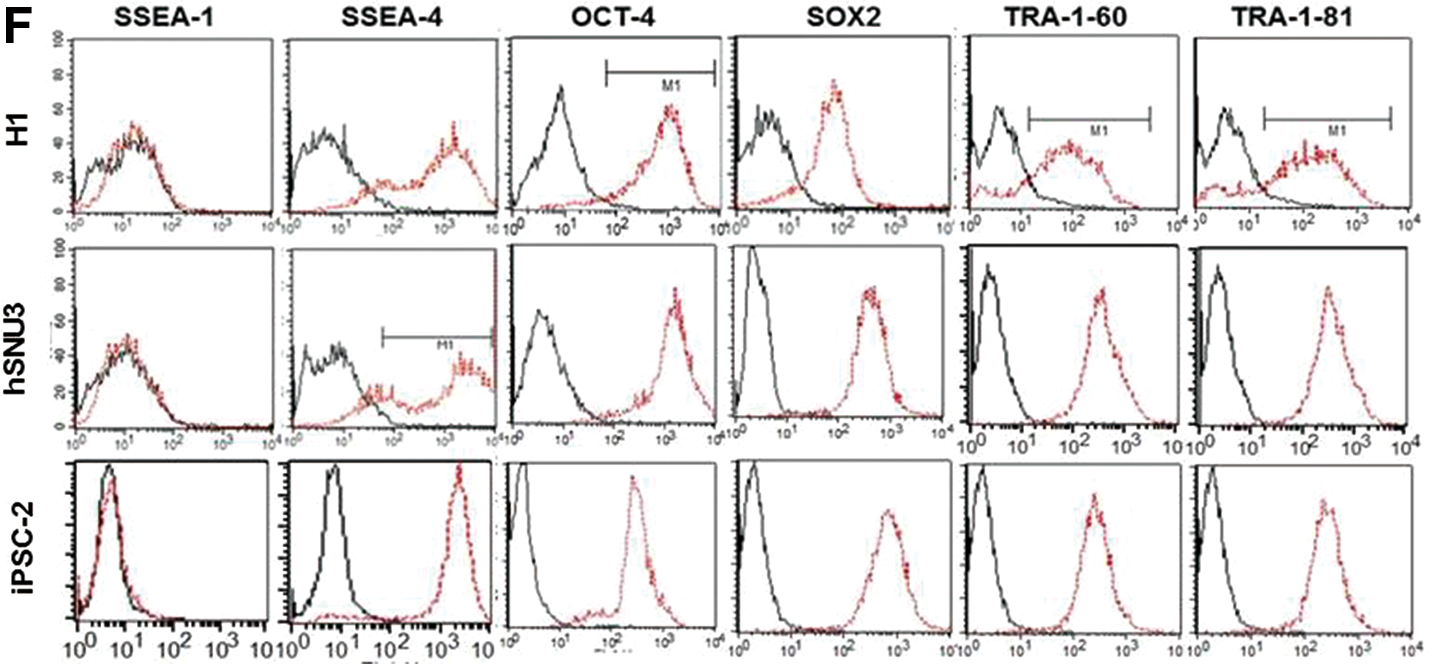
We cultured various hPSC lines (three hESC lines: H1, H9, and SNUhES3; and two human iPSC lines: foreskin iPSC1 and IISHi-BM1) in hPCCM− on gelatin for more than 35 passages (5–6 months) and compared their characteristics with those of hPSCs cultured in mTeSR™1 on Matrigel to assess the feasibility of hPCCM−. The long-term growth of hPSCs in hPCCM− was vigorous and higher than that in mTeSR™1 on Matrigel (Fig. 5A). qPCR and immunofluorescence showed that the hPSCs maintained high expression levels of pluripotency markers (Fig. 5B, C). Moreover, the densitometrically quantified endogenous CXCR2 expression levels of various hPSC lines cultured in hPCCM− were significantly higher than those in mTeSR™1. (Fig. 5D). This finding might also suggest the positive role of CXCR2 ligands in hPCCM− for the proliferation of hPSCs. The attachment of hPSCs cultured in hPCCM− on gelatin was similar to that of hPSCs cultured in mTeSR™1 on Matrigel (Fig. 5E). Flow cytometry analysis confirmed the maintenance of pluripotency markers (SSEA-4, OCT-4, SOX2, TRA-1-60, and TRA-1-81) intracellularly or on the surface of hPSCs after 20 passages in hPCCM− (Fig. 5F). To examine whether hPSCs retained the ability to differentiate, we induced spontaneous differentiation by EB formation and confirmed the capacity to differentiate into three germ layers in vitro (Fig. 6A). After 30 passages, hPSCs were injected subcutaneously into SCID mice, and subsequent teratoma formation was verified in vivo (Fig. 6B). The STR analyses revealed that the original DNA fingerprint was maintained without any changes after more than 30 passages (Fig. 7A, B). Furthermore, after 35 passages, we observed that various hPSCs sustained high AP activity when cultured in hPCCM− on gelatin (Fig. 7C). The karyotypes of the hPSCs were normal after 28 and 31 passages (Fig. 7D, E). This result demonstrates that hPCCM− is a unique CM for supporting the maintenance of hPSC characteristics and proliferation by CXCR2-related ligands on a gelatin substratum.
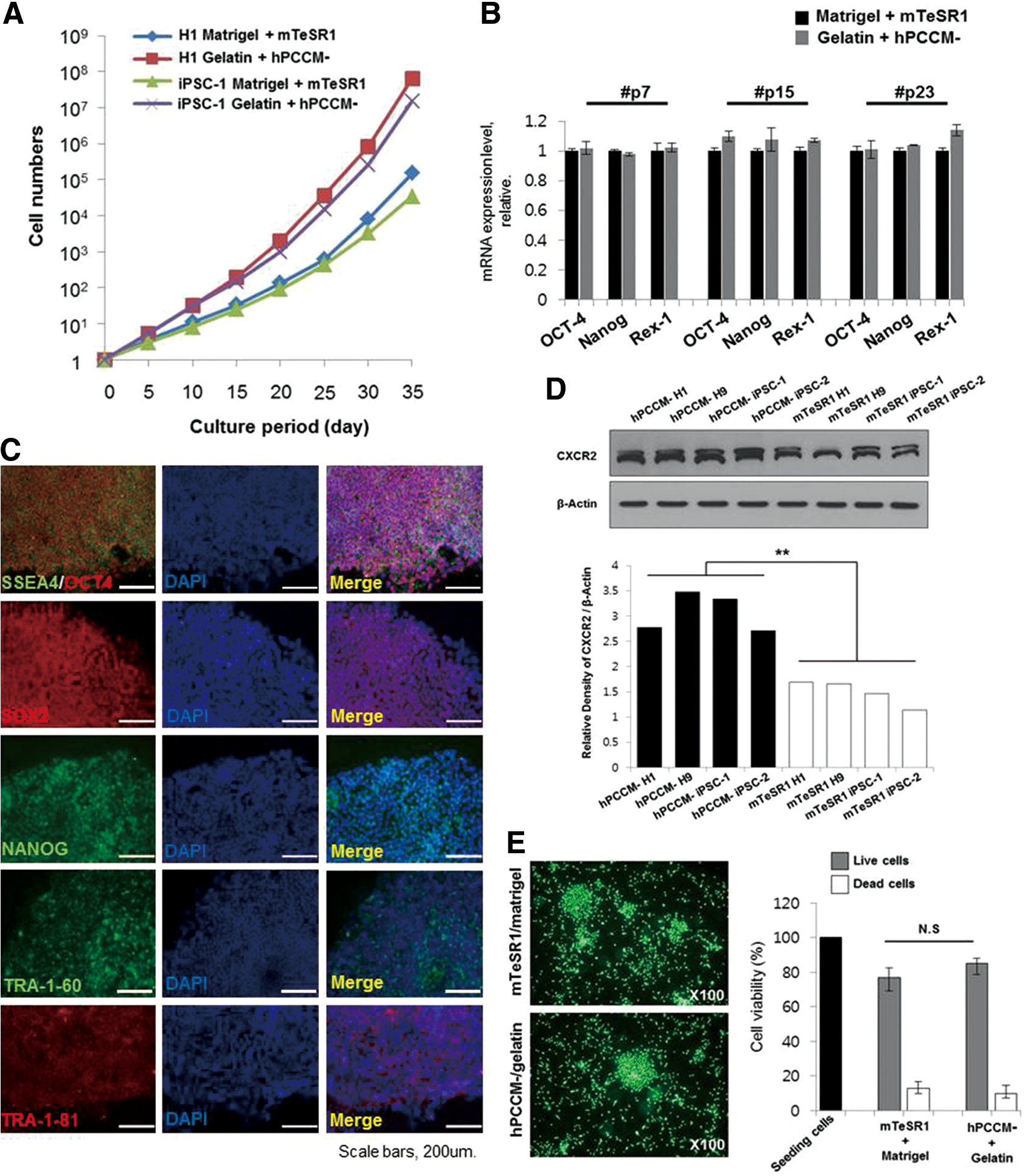
Establishment of conditioned media without bFGF supplements for the characterization of hPSCs.

hPSCs sustain the capability of three germ layer differentiation both in vitro and in vivo.
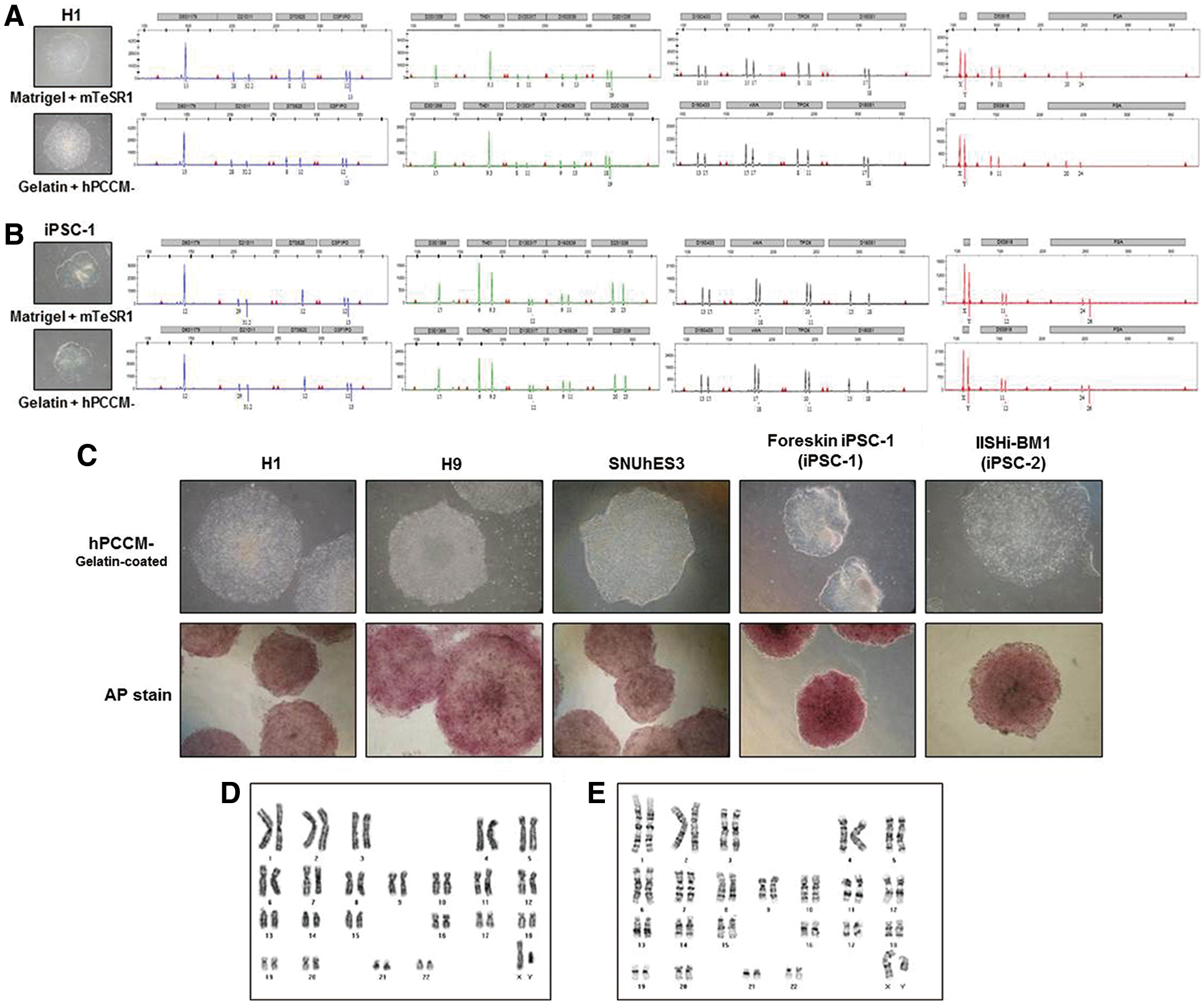
Various hPSC lines maintained long-term self-renewal without chromosomal abnormality in hPCCM− on gelatin.
Discussion
The CXCR2 has diverse biological roles. Normal terminally differentiated senescent cells activate a self-amplifying secretory program in which CXCR2 ligands reinforce growth arrest [16,17]. In cancer cells, however, CXCR2 mediates malignant cell invasion and metastasis [18 –21]. Furthermore, the importance of CXCR2 in cancer stem cells was recently emphasized by Singh et al., who showed that targeting CXCR1/2 significantly reduces breast cancer stem cell activity [22]. This indicates that the role of CXCR2 in cancer cells might be different from that in normal cells. Elucidation of the role of CXCR2 in normal stem cells will aid in determining the cause of this discrepancy because the nature of cancer cells, especially cancer stem cells, is somewhat similar to that of normal stem cells. To study this topic, hPSCs have advantages over human adult stem cells due to the greater homogeneity and the higher degree of experimental reproducibility in hPSCs than in human adult stem cells. In addition, human placenta cells are suspected to offer the best condition for establishment and maintenance of hPSCs because the placenta supports a fetus originating from the inner cell mass.
However, the effect of CXCR2 on stem cells has rarely been studied. Tirotta et al. reported that CXCR2 signaling restricts apoptosis of oligodendrocyte progenitor cells derived from hESCs induced by interferon-γ [23]. This result might suggest that CXCR2 plays a different role in progenitor cells compared with differentiated normal cells. As a single study to evaluate the effects of CXCR2 ligands on hPSCs, Krtolica et al. reported that human placenta fibroblasts secrete high amounts of GROα. They, therefore, developed a GROα-containing medium [Knockout Serum Replacement medium consisting of 100 ng/mL bFGF, 10 mM lactate, 0.5 ng/mL transforming growth factor (TGF)-β, and 10 ng/mL GROα] for the feeder-free culture of hESCs on Matrigel or human serum [24]. However, since the medium developed by Krtolica et al. [24] contained GROα as well as bFGF from Matrigel, the exact role of CXCR2 and its related ligands could not be confirmed. Thus, to date, there has been no study investigating the effects of CXCR2 and its related ligands on hPSCs. Accordingly, our study appears to be the first report to confirm that CXCR2 and its related ligands play an important role in the preservation of hPSC characteristics and proliferation and can substitute for bFGF as an essential factor for the ex vivo proliferation of hPSCs. In addition, this result suggests the possibility that the role of CXCR2 in hPSCs might be somewhat similar to its role in human cancer (stem) cells with regard to proliferation potentiation.
To test this possibility, it is very important to examine the signal transduction pathway after CXCR2 stimulation. In cancer stem cells, Singh et al. reported that IL-8/CXCR1/2 signaling is partly mediated via a human epidermal growth factor receptor 2 (EGFR/HER2)-dependent pathway in patient-derived breast cancer stem cells [22]. We attempted to study the role of CXCR1 and EGFR1/HER2 in hPSCs, but the results were inconclusive (data not shown). In our previous studies, we observed that the ERK pathway is activated in hESCs cultured on human placenta or bone marrow feeders without exogenous bFGF, and we suspected that hESCs could be supported by bFGF synthesized in human feeder cells [8,10]. Considering the results of this study, we hypothesize that CXCR2 and its related ligands are closely related to the ERK pathway of hPSCs in addition to our previous supposition focusing on bFGF. Although we have determined that CXCR2 is related to the ERK pathway, we were unable to obtain positive data identifying the additional mechanisms by which CXCR2 stimulates hPSCs because the mechanism for the maintenance of hPSC characteristics and proliferation is complex, and there is currently little consensus. We also investigated protein kinase B, which is a serine/threonine-specific protein kinase; stress-activated protein kinases/c-Jun N-terminal Kinase, which are known to play a key role in multiple cellular processes; and STAT3 status, which is known to control the migratory response to CXCR2 ligands by direct activation of granulocyte-colony stimulating factor-induced CXCR2 expression in neutrophils, but our findings were insignificant (data not shown) [25 –27]. We are continuing to investigate this question.
Sánchez et al. previously reported that CM from human mesenchymal stem cells secreted sufficient amounts of bFGF for hESC expansion [28]. However, they could not fully eliminate the possibility of exogenous bFGF influence on the hESCs, because Matrigel containing bFGF was used to enhance hPSC attachment. In our study, hPSCs were successfully cultured in hPCCM− on a gelatin substratum, not Matrigel, permitting a xeno-free humanized culture environment without bFGF influence. This is significant because no similar culture systems for hPSCs have been reported. The underlying mechanism by which hPSCs attach to a gelatin substratum remains unclear, and we are continuing to study and clarify the factors facilitating the attachment of hPSCs on gelatin (data not shown). The challenge in verifying the effect of CXCR2 on hPSCs might be bFGF, which is a well-established hPSC-sustaining factor for hPSC propagation, because bFGF can confound the results of CXCR2 research through its own role in maintaining hPSC characteristics and proliferation. This concern was proved in this study showing that the suppression of CXCR2 on hPSCs cultured in mTeSR™1 and Matrigel containing exogenous bFGF decreased the proliferation of hPSCs, but hardly influenced the expression of pluripotency-associated genes. Therefore, the development of a culture system for hPSCs without bFGF might be important. This study shows that a new culture system for hPSCs on a gelatin substratum using hPCCM− containing many ligands that stimulate CXCR2, such as IL-8 and GROα, without exogenous bFGF, insulin growth factor, or TGF-β, which are known to be important for hPSC self-renewal and propagation, is as effective as mTeSR™1 on Matrigel (Fig. 1A and Supplementary Table S1). So, this new culture system might enable the more correct verification of the role of CXCR2 and its related ligands to support hPSCs than the ordinary culture media containing exogenous bFGF.
In conclusion, CXCR2 and its related ligands might be important targets for potential breakthroughs in a new area of stem cell research and for elucidation of the proliferation mechanisms of both hPSCs and cancer (stem) cells. Moreover, our feeder-free humanized PCCM− system for hPSCs on a gelatin substratum might provide a suitable humanized ex vivo environment to evaluate the role of CXCR2 in hPSCs without the confounding influence of exogenous bFGF.
Footnotes
Acknowledgments
The authors thank Gabsang Lee (PhD, assistant professor, Stem Cell Biology, Johns Hopkins University) for his valuable advice and for reviewing the data. They also thank Soon-Cheol Hong (MD, PhD, associate professor, Department of Obstetrics and Gynecology, Medical College Korea University) for providing placental tissue and for his valuable advice. This work was supported in part by grants (R1211902) from the National Research Foundation of Korea (NRF), Republic of Korea.
Author Disclosure Statement
The authors indicate no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.