
Abstract
Embryonic stem cells (ESCs) undergoing neural differentiation form radial arrays of neural stem cells, termed neural rosettes. These structures manifest many of the properties associated with embryonic and adult neurogenesis, including cell polarization, interkinetic nuclear migration (INM), and a gradient of neuronal differentiation. We now identify novel rosette structural features that serve to localize key regulators of neurogenesis. Cells within neural rosettes have specialized basal as well as apical surfaces, based on localization of the extracellular matrix receptor β1 integrin. Apical processes of cells in mature rosettes terminate at the lumen, where adherens junctions are apparent. Primary cilia are randomly distributed in immature rosettes and tightly associated with the neural stem cell's apical domain as rosettes mature. Components of two signaling pathways known to regulate neurogenesis in vivo and in rosettes, Hedgehog and Notch, are apically localized, with the Hedgehog effector Smoothened (Smo) associated with primary cilia and the Notch pathway γ-secretase subunit Presenilin 2 associated with the adherens junction. Increased neuron production upon treatment with the Notch inhibitor DAPT suggests a major role for Notch signaling in maintaining the neural stem cell state, as previously described. A less robust outcome was observed with manipulation of Hedgehog levels, though consistent with a role in neural stem cell survival or proliferation. Inhibition of both pathways resulted in an additive effect. These data support a model by which cells extending a process to the rosette lumen maintain neural stem cell identity whereas release from this association, either through asymmetric cell division or apical abscission, promotes neuronal differentiation.
Introduction
C
In the adult mammalian brain, astrocyte-like neural stem cells persist in both the subventricular zone (SVZ) of the lateral ventricle and the subgranular zone (SGZ) of the dentate gyrus, and these cells retain many of the characteristics of a polarized epithelium [1,3,10]. The apical ends of the neuroepithelial, radial glia cells, and the type B neural stem cells of the adult SVZ are adjacent to the lumen or ventricle, and contain adherens and tight junctions, marked by the localized expression of constituent proteins, including neural cadherin (N-cad) and zona occludens-1 (ZO-1) [11,12]. Polarity complex proteins, including Par3, are also associated with the apical domain and play a role in regulating symmetric versus asymmetric cell divisions in the developing neocortex [13]. The basal ends of the radial glia cells are also specialized. They abut the pial surface and attach to a basal lamina via the extracellular matrix receptor β1 integrin, which localizes to the basal cell surface [14]. The establishment of the apical/basal polarity of neural progenitors is a key event in embryonic and adult neurogenesis.
The apical ends of radial glia and adult SVZ neural stem cells extend a single primary cilium into the ventricle [15 –17]. These nonmotile, microtubule-based structures consist of an axoneme containing nine microtubule doublets arranged in a circle anchored by a basal body base [18]. The primary cilium is a dynamic structure constructed and deconstructed according to environmental conditions. For example, primary cilia are resorbed during mitosis [19] and elongate when cells are deprived of nutrients [20]. The molecular motors kinesin and dynein transport components in and out of the ciliary axoneme [21,22]. The vital role played by this structure is highlighted by the observation that a variety of mutations in cilia components result in ciliopathies in humans, with complex disease phenotypes that may affect the CNS [23 –25]. Recent analysis using conditional gene targeting in the mouse has revealed a role for the ciliary small GTPase Arl13b in establishing correct polarity of radial glial cells during development [26]. Mutations in the gene encoding this protein have been linked to Joubert syndrome in humans, characterized by a number of neurological phenotypes, including hippocampal malformations and enlarged ventricles [26].
Within the neurogenic niche, a number of signal transduction pathways, including Hedgehog and Notch, regulate the proportion of neural stem cells and neurons produced during embryogenesis and adult neurogenesis. Lineage tracing experiments show that the neural stem cells of the SVZ and SGZ are Hedgehog responsive [27]. Loss of Hedgehog signaling leads to a reduction in neurogenesis, while increased levels promote the production of new neurons [28]. A direct role for Hedgehog signaling in maintenance and proliferation of neural stem cells has been demonstrated [29 –33]. High levels of Notch are associated with maintenance of proliferating neural stem cells and inhibition of neuronal differentiation, whereas lower levels are associated with neuronal differentiation [34,35], though evidence suggests that Notch is active at additional steps during neurogenesis [35]. A study in which the Hedgehog response is upregulated in neural stem cells by targeted inactivation of the Hedgehog inhibitor Patched 1 (Ptc1) highlights a cooperative role for SHH and Notch in promoting symmetric versus asymmetric cell division of neocortical progenitors [36].
The action of both of these pathways can be spatially restricted by the subcellular localization of specific pathway components. In vertebrates, SHH and Notch signaling are mediated via primary cilia. The Hedgehog inhibitor Patched1 (Ptc1) is concentrated along the ciliary membrane in the absence of the SHH ligand. In the presence of ligand, the Hedgehog effector Smoothened (Smo) translocates to the ciliary membrane, replacing Ptc1 [37 –39]. Once Smo is localized to the primary cilium, it inhibits Suppressor of Fused (Sufu) from processing Gli proteins into their repressor forms, and active Gli proteins move into the nucleus and initiate transcription of downstream targets [40,41]. The key role played by the ciliary machinery is supported by numerous studies demonstrating that mutations in genes regulating cilia structure and function, including intraflagellar transport proteins IFT-A and IFT-B, also affect SHH signaling [42,43]. The primary cilia that mark the apical end of neural stem cells in the embryonic and adult brain are required for SHH-dependent regulation of progenitor proliferation [15,25,44,45]. Hedgehog signaling through primary cilia has also been implicated in the tangential migration of GABAergic interneuron progenitors from the medial ganglionic eminence into the cortical plate [46].
Though not as extensively examined, recent analysis demonstrates that Notch signaling also relies on nonmotile primary cilia. In differentiating epidermal cells, Notch 3 and Presenilin-2, a part of the γ-secretase complex mediating the proteolytic cleavage of Notch to its active Notch intracellular domain (NICD), localize to primary cilia [47]. Furthermore, cilia mutants display defective Notch signaling [47].
Pluripotent stem cells undergo neural differentiation in vitro in a manner that, in many aspects, is surprisingly reminiscent of neurogenesis in vivo. Using either direct monolayer adherent culture or embryoid body intermediates subsequently plated onto adhesive substrates, neural differentiation of embryonic stem cells (ESCs) is characterized by the organization of cells into epithelial structures called neural rosettes. Within rosettes, cells arrange themselves radially, elongate, and differentiate into radial glia-like intermediates [48 –51]. Several studies have reported the formation of these structures before the emergence of mature neuronal subtypes, suggesting a role for epithelialization in regulating ESC neurogenesis. Neural rosettes bear a striking resemblance to the embryonic neural tube and the developing telencephalon in a number of respects, including expression of the radial glia markers nestin and brain lipid binding protein [52]. Cells within rosettes acquire apical polarity based on the localized expression of adherens and tight junction proteins, including N-cad and ZO-1, as well as components of the polarity complex, in a ring surrounding a central lumen [48,50,53]. In addition, INM occurs, with dividing cell bodies adjacent to the rosette lumen while a spatial gradient of differentiation forms, with mature neurons concentrated at the periphery of the rosette [48,53]. Radial glia-like cells characteristic of both the inner and outer SVZ are present in these neural rosettes [54].
We report here the use of human ESC (hESC)-derived rosette cultures to identify key relationships between structural features and neurogenic potential. We show, based on expression of β1 integrin, that neural progenitors have a specialized basal as well as apical cell surface in vitro. Individual cells extend an apical process that terminates at the adherens junction. Primary cilia become concentrated at the central lumen as rosettes mature, further defining the apical end of differentiating neural stem cells. Components of both the Hedgehog and Notch cascades localize to the apical domain of neural rosette cells, where they act on cells that have a connection to the rosette lumen. Altering levels of Notch signaling changes the balance of neural progenitors and neurons produced in differentiating cultures, with lower levels of activity associated with enhanced neuronal maturation and decreased progenitor proliferation, as previously described [50]. A similar though less robust outcome is observed with altered Hedgehog signaling, and we now establish that inhibition of both pathways has an additive effect. Taken together, these data suggest that the rosette structure facilitates localized response to key signaling molecules that play a role in regulating the balance of neural progenitors to neurons during neurogenesis.
Materials and Methods
hESC culture and neural differentiation
H9, H9 Ubi:RFP, [55], hES3 NKX2.1 GFP/w with NKX2.1:GFP reporter [56], NKX2.1:GFP/Ubi:RFP reporter [55], and an H9-derived cell line with HES5:GFP reporter [57] were routinely cultured on mitomycin C-treated mouse embryonic fibroblasts (MEFs, 2×104 cells/cm2) and passaged mechanically by scoring and picking fragments of colonies as previously described [55]. hESC medium consists of DMEM/F12 (Sigma) supplemented with knockout serum replacement (KSR; Gibco), nonessential amino acids (NEAA; Gibco), L-glutamine (Gibco), penicillin/streptomycin (Gibco), βME (Sigma), and bFGF (CONNStem, 8 ng/μL).
Neural differentiation was promoted as previously described [55], except as an alternative to noggin; the small-molecule BMP inhibitor LDN was used at 100 nM. Rosette cultures were either generated from fresh material at the time points described or alternatively formed from a stock of cells frozen at approximately day 18 of differentiation and thawed onto laminin-coated glass chamber slides. Rosettes reassemble to produce immature rosettes at around day 4 post-thaw and mature rosettes by around day 7 post-thaw.
Hedgehog and DAPT treatment
For Smo localization experiments (Fig. 4), freshly differentiated cells were treated at day 6 with 500 ng/mL of rhSHH [55]. We have previously shown that this concentration of rhSHH alone effectively promotes Hedgehog signaling based on upregulation of the reporter NKX2.1:GFP [55]. For experiments determining the effect of altering Hedgehog and Notch signaling on neurogenesis (Fig. 6), thawed day 18 HES5:GFP differentiated cells were treated with 750 nM of the Hedgehog antagonist Cur 199691 (Genetech) or a combination of 250 ng/mL rhSHH plus 1 μM of the Hedgehog agonist purmorphamine (Calbiochem) [58]. Comparable upregulation of the SHH target NKX 2.1:GFP was achieved in a direct comparison of the effect of the rhSHH/purmorphomine combination versus 500 ng/mL rhSHH alone. The Notch inhibitor DAPT (Tocris) was used at a final concentration of 5 μM. This concentration has been previously used and was effective at inhibiting expression of the Notch reporter HES5:GFP (data not shown).
Immunocytochemistry
Cultured cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 10 min followed by permeabilization with 0.5% PBS-Triton×100. Cells were then blocked for 1 h at room temperature in 2% bovine serum albumin (BSA) 5% or 10% serum/0.1% PBS-Triton X-100 before incubation with primary antibodies in blocking solution overnight at 4°C. Secondary antibody incubations were applied for 1 h at room temperature in blocking solution. The following antibodies were used: MAP2 (Sigma; mouse, 1:1,000), Pax6 (DSHB; mouse, 1:50), Musashi1 (Abcam; rabbit, 1:400), N-cadherin (Sigma; mouse, 1:200), integrin β1 (Abcam; mouse, 1:100), nestin (Chemicon; mouse, 1:1,000), Par3 (Chemicon; rabbit, 1:400), phosphohistone H3 (Sigma; rabbit, 1:400), Presenilin 2 (Abcam; rabbit, 1:200), Arl13b (gift of T. Caspary; 1:1,000 or NeuroMab; mouse 1:1,000), and Notch 3 (Abcam; rabbit, 1:200). All fluorescent secondary antibodies (Alexa Fluor-conjugated; Life Technologies) were used at 1:1,000. Rhodamine-conjugated Phalloidin (Molecular Probes) was used to visualize actin. Cells were counterstained with Hoechst 33342 for 10 min before coverslipping with gelvatol. Slides were imaged using a Nikon Eclipse Ti with NIS Elements software.
Primary cilia quantification
To quantify the location of primary cilia in neural rosettes, chamber slides were scanned while visualizing Hoechst staining and images were taken of rosettes, which were identified by their radially arranged, elongated nuclei. Rosettes were labeled as “mature” if they displayed a defined lumen in the center of the rosette that lacked nuclei. Rosettes were labeled as “immature” if they did not display a defined lumen but contained cells that were radially arranged around a center point. After the Hoechst image was taken and labeled as “mature” or “immature,” an image of the primary cilia was taken and these images were overlaid. The total area of the rosette was calculated in Photoshop by drawing a circle around its periphery. The total area of the rosette was divided by four, and three additional concentric circles were centered inside of the rosette to divide the area into four rings of equal area. Primary cilia within each ring were counted and then divided by the number of nuclei within each ring. Statistical significance was determined using a two-tailed t-test.
Quantification of MAP2 and phosphohistone H3
H9 HES5:GFP neural stem cells frozen at day 21 of differentiation in the absence of agonists or antagonists were thawed. Treatments with Hedgehog agonist and antagonist and DAPT was begun at 4 days after thawing and continued for 7 days, after which cells were fixed (32 days total). Immunocytochemistry for MAP2 or phosphohistone H3 was combined with counterstaining with Hoechst, and four randomly selected fields were photographed. Nuclei and positively stained cells were counted in NIS Elements, and the results were analyzed in Microsoft Excel. Statistical significance was determined using a two-tailed t-test.
Quantification of Smo/primary cilia colocalization
For experiments in which we quantified colocalization of Smo and primary cilia, cultures were treated with 500 ng/mL recombinant human SHH (rhSHH). Smo colocalization at the base of the axoneme was scored only when immunofluorescence labeling for primary cilia (Arl13b) and Smo did not overlap, to ensure that Smo had not yet entered the ciliary membrane. Instances of Smo colocalization with primary cilia along the axoneme were scored when immunofluorescence labeling for Arl13b and Smo were completely overlapping or when Smo was located immediately adjacent to the primary cilia, indicating that Smo was located within the ciliary membrane of the primary cilia, but not necessarily completely overlapping with Arl13b, a component of the ciliary axoneme.
Scanning electron microscopy
A stock of hES3-derived neural progenitors frozen at day 18 were thawed and cultured for 4–7 days. Cells on glass chamber slides were fixed for 10 min in electron microscopy-grade 3.7% formaldehyde and 2.5% glutaraldehyde. Cells were rinsed in PBS and then put through the following ethanol exchange series: 25%, 50%, 70%, 85%, 95%, and 100% EtOH. The ethanol was allowed to slowly evaporate and when dry the chamber slides were sputtered with a thin layer of gold and imaged under SEM.
Results
Monolayer protocol produces neural rosettes in which cells display apical and basal domains
We have recently reported a robust, monolayer-based protocol for neural differentiation of hESCs in which nearly 90% of the cells express the neural stem marker Musashi-1 and very low levels of expression of mesendoderm markers are observed [55]. This approach readily generates relatively synchronous cultures undergoing rosette differentiation, with immature rosettes present by Day 12 and mature rosettes by Day 18. Rosettes formed using this approach resemble the neural tube and developing neocortex in a number of respects (Fig. 1A): the expression of radial glia markers Musashi-1 and nestin (Fig. 1B), apically localized N-cadherin expression (Fig. 1C), INM based on localized expression of the M-phase marker phosphohistone H3 (Fig. 1C), and a gradient of neural differentiation based on the expression of the neuronal marker Hu at the rosette periphery (Fig. 1D). These data indicate that this protocol generates rosettes from hESCs with similar properties to those generated by alternative means [48,59].
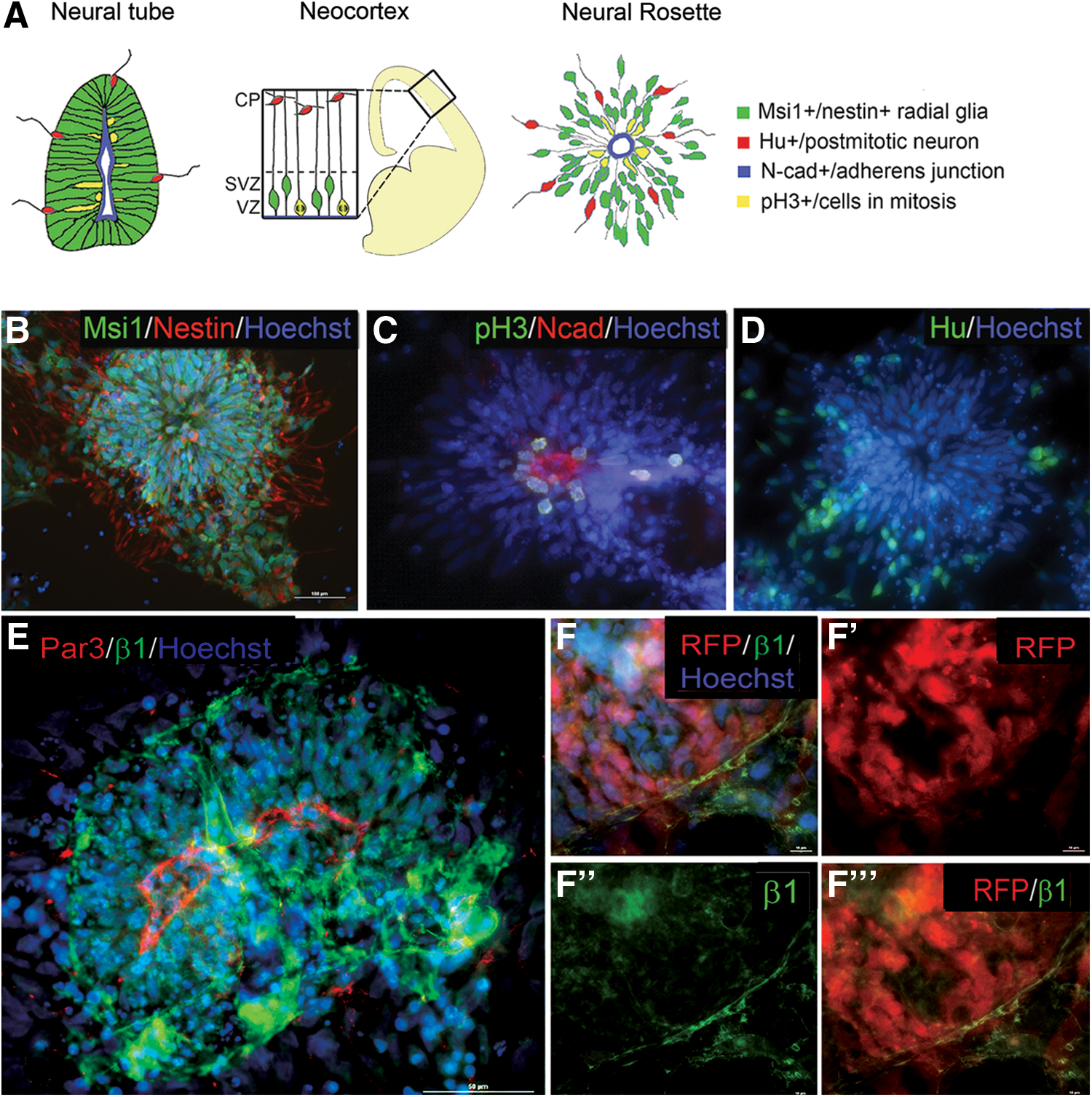
Mature human embryonic stem cell (hESC)-derived rosettes resemble the developing neural tube and neocortex and establish a basal as well as apical domain.
In the developing brain, the basal extension of radial glia terminates at the extracellular matrix-rich pial surface [60]. The basal surface of the cells adjacent to the basal lamina contains enriched levels of the extracellular matrix receptor β1 integrin [61,62]. To determine whether ESC-derived neural stem cells within rosettes have a specialized basal, as well as an apical domain, we examined the expression of β1 integrin. Figure 1E shows that in mature rosettes, a circumferential ring of β1 expression is present several cell diameters away from the lumen. To determine whether this ring of expression corresponds to the basal surface of differentiating cells, we used a human ESC line constitutively expressing RFP throughout the cell body so that the outer edge of the cell and the ring of β1 integrin could be readily visualized. Rosettes were prepared from a mixture of RFP-positive and RFP-negative cells. Figure 1F-F’’’ demonstrates that the line of accumulated β1 corresponds to an apparent outer edge of the rosette, though there are still cells located outside of this ring. These data suggest that a basal, as well as an apical domain, is established by ESCs in rosette culture.
Rosette morphogenesis: apical markers
To examine rosette morphogenesis, we characterized the localization of the polarity complex marker Par3 and the adherens junction protein Ncad at various time points during monolayer differentiation: prerosette (∼6 days differentiation), intermediate rosette (∼12 days of differentiation), and mature rosette (∼18 days of differentiation) (Fig. 2A–C). As previously reported using alternative differentiation protocols, for polarity complex and adherens junction proteins [48,50,53], at the prerosette stage we see both Par3 and Ncad between individual or small groups of cells, in a honeycomb pattern (Fig. 2D, G). Over time, as immature (no defined lumen) and then mature rosettes (defined lumen) form, both proteins become restricted to the apical domain of cells as they elongate and arrange themselves radially, concentrating in a ring around a central lumen (Fig. 2E, F, H, I). Actin colocalizes with Ncad at emerging apical domains, based on Phalloidin localization (Fig. 2G–I). Par3 and Ncad distribution overlap in mature rosettes, based on double immunolocaliztion (Fig. 2J, J’). These data suggest that both adherens junctions and the polarity complex become spatially restricted concurrently; one does not precede the other.
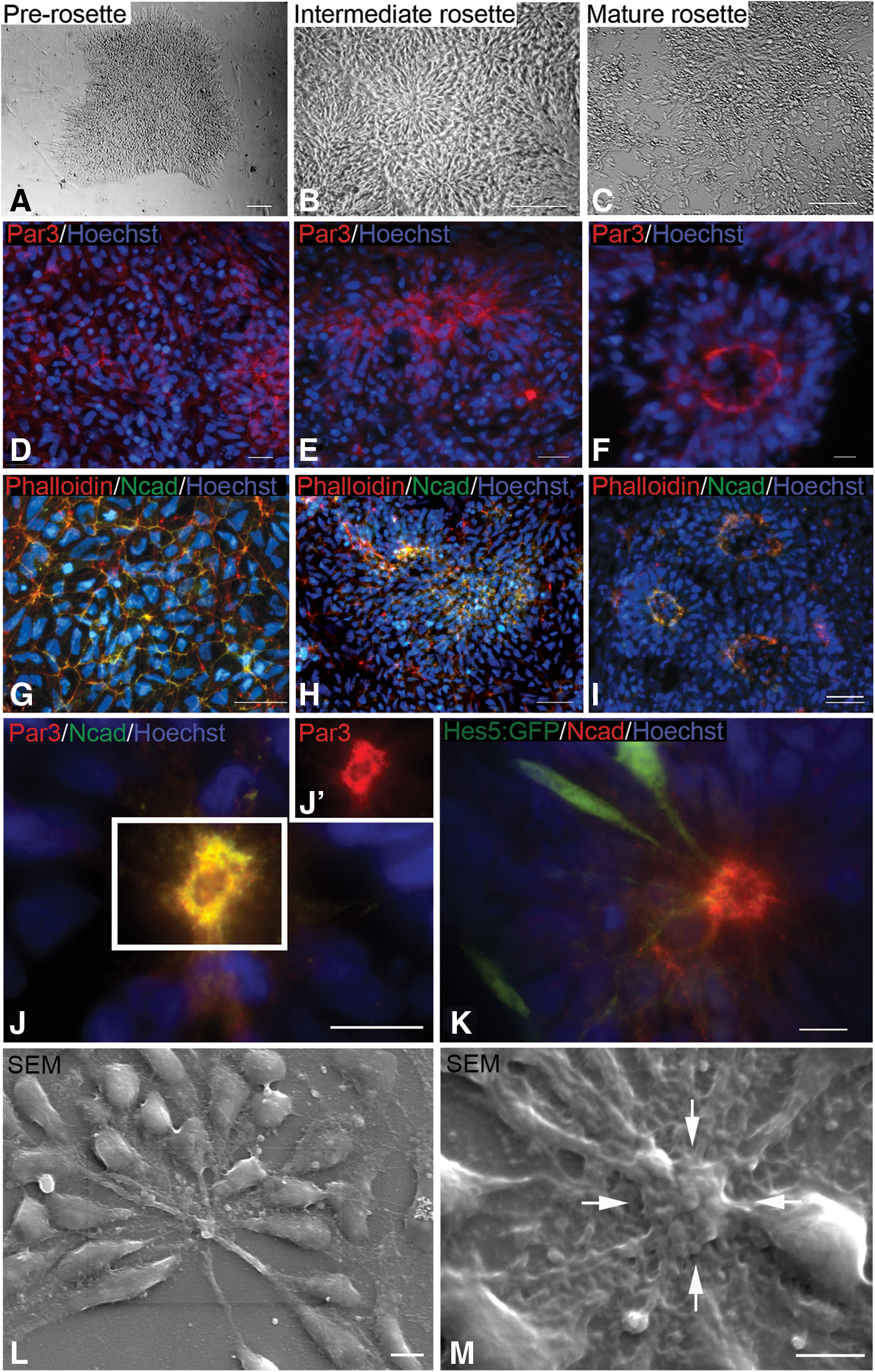
Morphogenesis of apical markers.
Apical processes can be visualized using an hESC reporter cell line that expresses GFP downstream of the HES5 promoter, a downstream target of Notch activity. We chose rosettes with a few GFP-positive cells to more easily image individual cells. In the scattered cells expressing the reporter, GFP fills the entire cell, including the process (Fig. 2K). The processes terminate in the band of Ncad that marks the adherens junctions (see yellow dots in Fig. 2K). Scanning electron microscopy reveals prominent apical processes that coalesce at the lumen (Fig. 2L). They are more extensive when associated with cell bodies located some distance from the central lumen (Fig. 2L). Scanning images often reveal a raised lumenal ring (see arrows in Fig. 2M). The diameter of this ring, 5–10 μm, is similar to the diameter outlined by the ring of Ncad immunostaining, suggesting that it corresponds to the adherens junction ring.
Primary cilia translocate to the lumen during rosette maturation
Primary cilia are present at the apical ends of radial glia and SVZ type B astrocyte neural stem cells, extending into the ventricle [17,63]. We therefore determined the distribution of primary cilia as rosettes assemble in hES3 hESC cultures using an antibody directed against the small GTPase Arl13b, which localizes to the axoneme of primary cilia [63]. Primary cilia are scattered throughout the intermediate rosette (Fig. 3A). In mature rosettes, however, primary cilia are concentrated at the interior of the rosette pointing into the lumen, suggesting they have translocated to the now apical end of the differentiating neural stem cells as they polarize (Fig. 3B). We quantified primary cilia distribution by dividing rosettes into concentric rings of equal area, and then calculating the cilia/nucleus ratio for each ring for immature and mature rosettes. Figure 3C and D demonstrate a clear clustering of primary cilia in the innermost ring, at the lumenal surface, on rosette maturation.

Primary cilia translocate to the lumenal surface of maturing rosettes.
To determine the position of the primary cilium relative to the apical process, we used the HES5:GFP cell line combined with Arl13b immunocytochemistry (Fig. 3E, E’). The apical edge of the GFP-labeled processes terminate in an apparent broadened end-foot structure [13], closely associated with a primary cilium (see arrowhead in inset of Fig. 3E’). These data suggest that apical processes are associated with a single primary cilium, as observed for neural progenitors in the neurepithelium and ventricular zone of the embryo.
Hedgehog signaling in neural rosettes
Given the demonstrated link between primary cilia and both Hedgehog and Notch signaling, we examined the localization of pathway components within rosettes and the role of these two pathways in rosette neurogenesis. The localization of the Hedgehog effector Smo is regulated by ligand addition [38,39]. We therefore examined the association between Smo and primary cilia, under control conditions and in the presence of exogenous SHH. We have previously shown that SHH is produced by differentiating mouse ESC-derived neural stem cells in monolayer culture [51]. Human hES3 ESC cultures were treated with rhSHH after 6 days of neural differentiation as described in the Materials and Methods section. Cultures treated with SHH showed significantly more colocalization of Smo and primary cilia than under control conditions (Fig. 4A, D control, Fig. 4B, E SHH-treated). In control cultures, only 3% of primary cilia contained Smo, whereas about 13% of primary cilia in cultures treated with SHH colocalized with Smo (Fig. 4C). Interestingly, when Smo associated with the primary cilia in the absence of exogenous ligand, it was more frequently located at the base versus within the cilia axoneme (67.7% vs. 32.9%, Fig. 4D, F); whereas with exogenous SHH, Smo localized more frequently to the axoneme than the base (57.4% vs. 42.6%, Fig. 4E, F). Smo was rarely observed simultaneously at the base and within the axoneme of the primary cilium. These data suggest that, within rosette cultures, Smo transport within primary cilia is controlled by the SHH ligand.
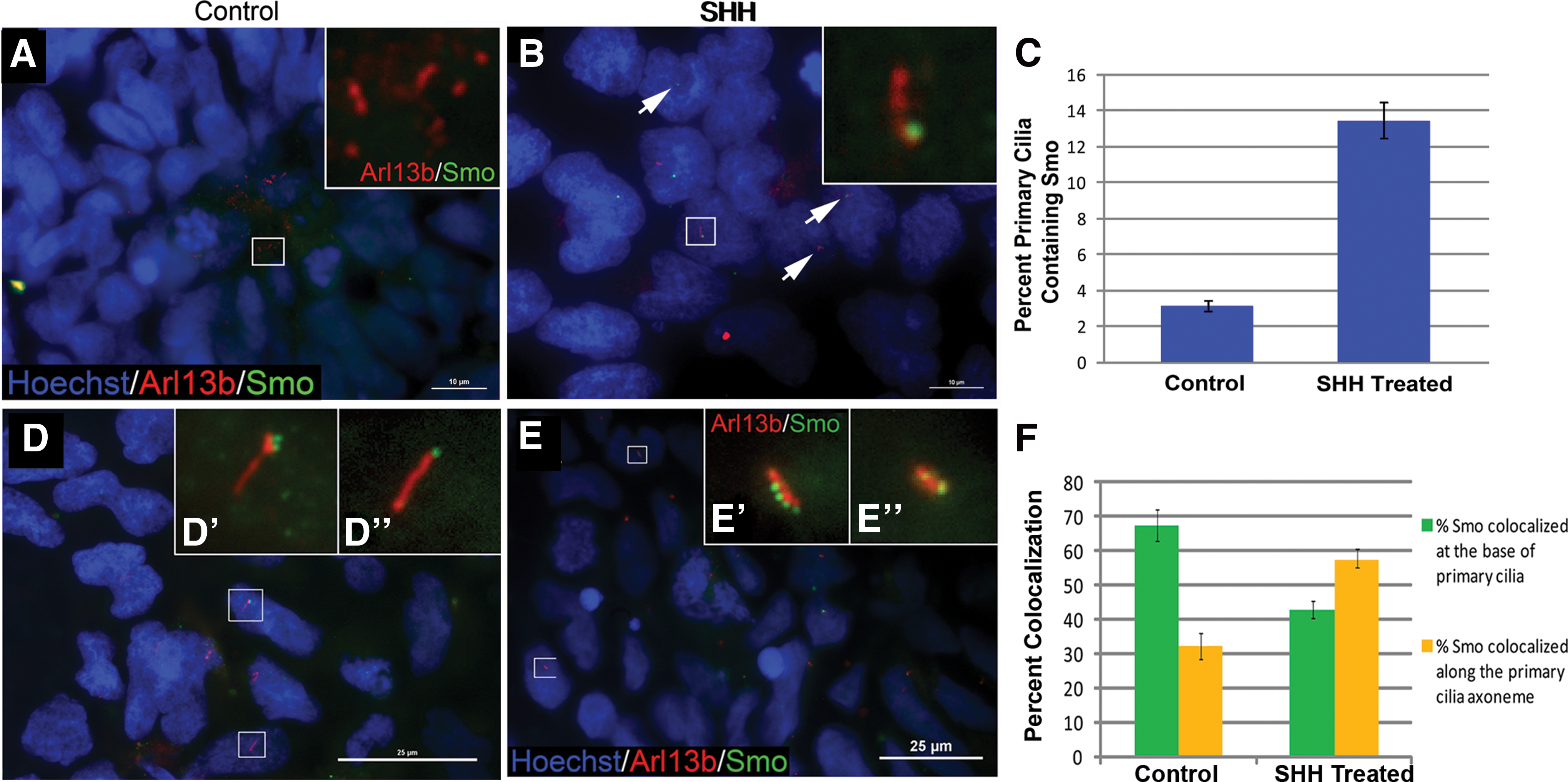
Localization of Smo within primary cilia is altered in response to SHH ligand.
Notch signaling in neural rosettes
Previous studies demonstrated the expression of Notch 1, 2, and 3 in ESC rosette cultures [50], with the receptor localized to plasma membranes and concentrated at the central cells of mature rosettes [64]. To determine whether activation of the Notch pathway is restricted to the apical domain, we examined the localization of Presenilin 2, the catalytic subunit of γ-secretase, relative to two apical domain markers, N-cad and Arl13b. Figure 5A–D demonstrates that in prerosettes, Presenilin 2 is distributed throughout the plasma membrane in a honeycomb pattern, as observed for N-cad. In mature rosettes, however, Presenilin 2 is restricted to the apical domain of the cells, where it forms a lumenal ring, again as observed for N-cad (Fig. 5E–H). Both proteins appear to be distributed in a punctate array surrounding the lumen in a pattern that is somewhat overlapping (Fig. 5H). Notch 3, as expected, is predominantly membrane associated and present in apparent apical processes (Fig. 5I). This pattern is distinct from the Ncad-like distribution of Presenilin 2, which is restricted to the adherens junction ring (Fig. 5J). Examination of the distribution of Presenilin 2 relative to the ciliary protein Arl13b indicates that Presenilin 2 is not localized to primary cilia in mature rosettes (Fig. 5J), an association noted in epidermal cells [47]. With the caveat that Presenilin 2 has multiple substrates [65], these data suggest a mechanism by which Notch is selectively cleaved into its active NICD selectively at the apically localized adherens junction, based on the restricted localization of Presenilin 2 to this domain.
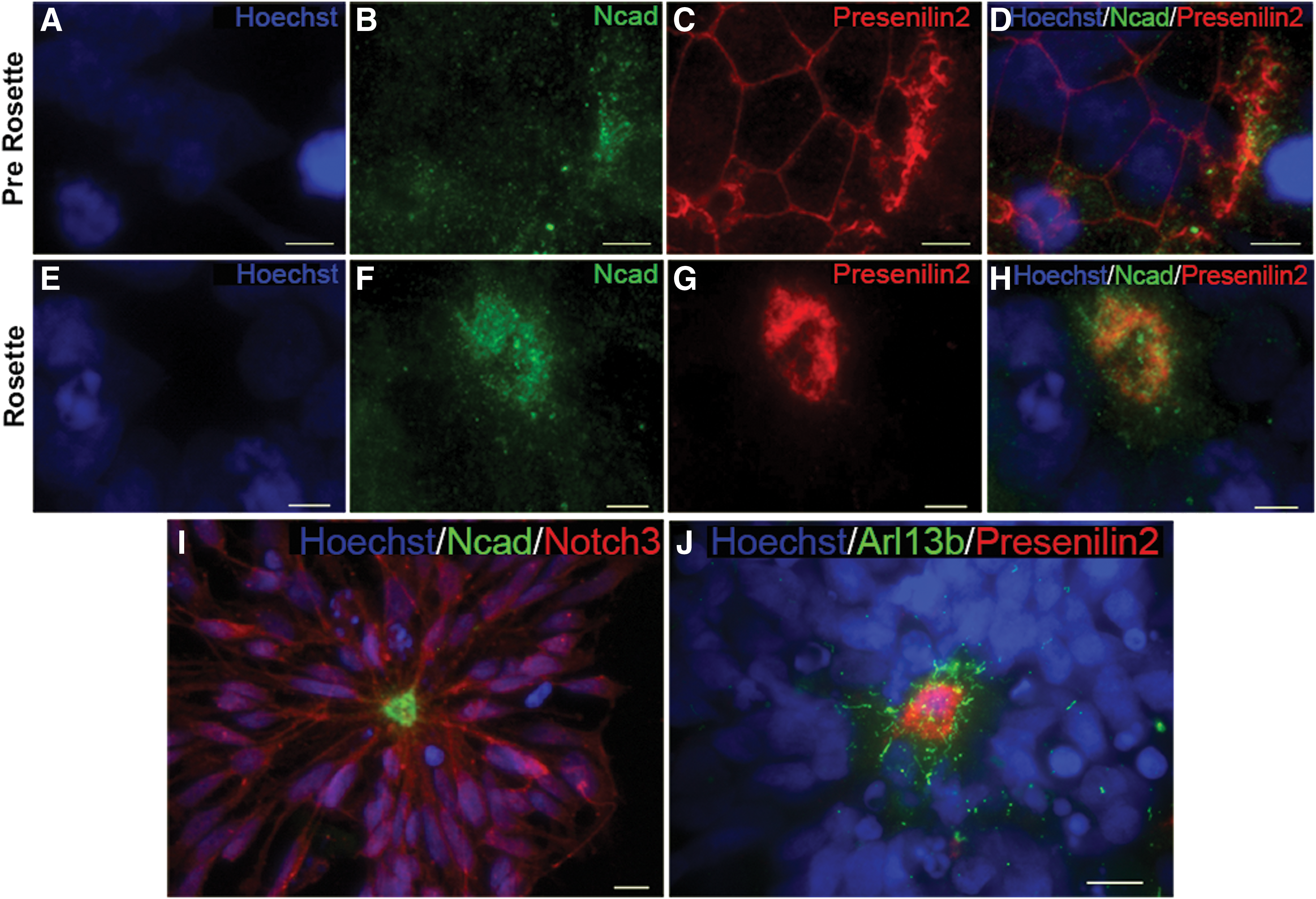
Identification of apical structure association for the Notch effector Presenilin 2.
Hedgehog and Notch regulate neurogenesis
Numerous studies have documented a role for the Hedgehog and Notch pathways in embryonic, adult, and ESC neurogenesis. Our own work demonstrated that SHH produced by neural stem cells in mouse ESC rosette cultures promotes the survival and proliferation of neural stem cells in an apparent autocrine loop [51]. Using rhSHH and purmorphomine or a Hedgehog antagonist [51], as well as the Notch inhibitor DAPT, which blocks the cleavage of the Notch receptor into its active NICD, we examined the role of both pathways in hESC neurogenesis. DAPT was used at a final concentration of 5 μM, a concentration sufficient to inhibit expression of the Notch reporter HES5:GFP in the hESC line used for these studies [57] (data not shown), though additional alternative pathways may also be affected [65]. As a measure of neuron production, we focused on MAP-2-positive cells. Figure 6A–G demonstrates a dramatic increase in neuron production on DAPT addition, apparent under all three Hedgehog conditions: agonist, control, and antagonist. Note the more extensive neurite network visible with DAPT treatment (Fig. 6D–F). Under control Hedgehog conditions (absence of antagonist or agonist), the addition of DAPT doubled the percent MAP2+cells, from 20% to 40% (Fig. 6G). This effect of DAPT alone on neuron production has been previously noted [50]. In the absence of DAPT, addition of Hedgehog agonists reduced production of MAP2-positive cells from around 20% under control conditions to 12% (Fig. 6G). There is a further decline in neuron production with the addition of Hedgehog agonist in the presence of DAPT, but this effect is not statistically significant.
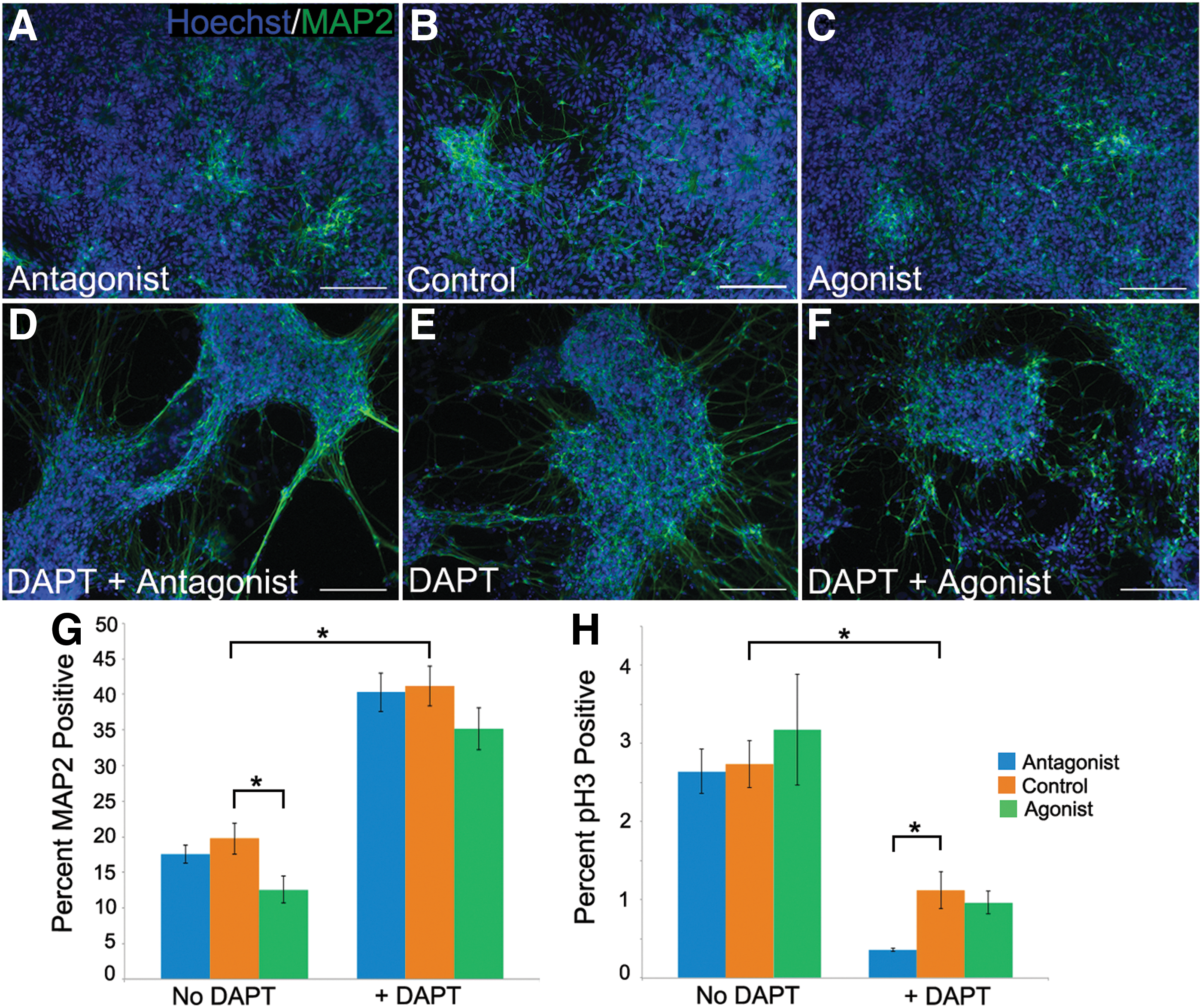
Altering levels of Notch and Hedgehog affect neuron production and progenitor proliferation.
To characterize the neural stem cell population, we examined the proliferation marker phosphohistone H3, expected to mark a subset of the proliferative neural progenitor population and not postmitotic neurons. Again, the most dramatic effect is observed with DAPT addition, which decreases the percent of phosphohistone H3-expressing cells, from around 2.6% under control Hedgehog conditions to 1.1% (Fig. 6H). The ability of Notch inhibition alone to impede neural progenitor proliferation in neural rosette cultures has been previously described [50], and is based on a delay in the G1/S phase transition [59]. We demonstrate here that there is also an additive inhibitory effect when Hedgehog antagonist is present as well as DAPT, with only around 0.35% phospho-histone H3-positive cells (Fig. 6H) present under this condition. Taken together, these data are consistent with a role for both the Notch and Hedgehog pathways in promoting the neural stem cell state at the expense of neuronal maturation, though a more robust role for Notch is observed.
Discussion
Many aspects of neurogenesis are recapitulated using ESC rosette culture, suggesting that this in vitro model can be used to elucidate relevant regulatory mechanisms. We document key structural elements that assemble in rosette culture. We show that ESC-derived neural stem cells within a neural rosette generate a basal, as well as apical surface, based on localization of the extracellular matrix receptor β1integrin. As the cells polarize and rosettes form, we observe a transition from uniform distribution of the adherens junction protein Ncad and the polarity protein Par3 throughout the plasma membrane to restricted apical domains. Apical processes terminate at the Ncad-positive adherens junction and display a primary cilium.
The important functional role of primary cilia is highlighted by the identification of human diseases associated with mutations in cilia proteins [23 –25]. These ciliopathies can be modeled in mouse mutants deficient for cilia components, and the mutants are frequently embryonic lethal with neural tube defects [66]. In addition to defects in patterning due to alterations in SHH-mediated ventralization, brain tumors and phenotypes consistent with defects in neurogenesis are also observed, suggesting a broad role for primary cilia in neural development [15,16,67,68].
During rosette maturation, primary cilia become concentrated at the rosette lumen, associated with adherens junctions. We demonstrate that components key to the activity of two signaling pathways implicated in neurogenesis, Hedgehog and Notch, are restricted to apical structures; Smo to primary cilia and Presenilin 2 to adherens junctions. These observations suggest a means by which these pathways can act locally within a field of cells. The gradient of neural differentiation observed in rosettes, with neural stem cells adjacent to the lumen and mature neurons at the rosette outer boundary, is therefore the likely outcome of concentrating Hedgehog and Notch activity, which promote neural stem cell maintenance and proliferation and inhibit neuronal differentiation, at the rosette core. Consistent with this conclusion, manipulating levels of Hedgehog or Notch signaling in rosette culture alters the proportion of neural stem cells to neurons, with both pathways promoting the neural stem cell state (Fig. 6) [50,59,64]. However, Notch inhibition has a significantly stronger effect than Hedgehog inhibition, resulting in a dramatic decline in cell proliferation tied to a two-fold increase in the percent of MAP2-positive neurons. The less significant role played by SHH can be explained using a model in which SHH and an as-yet-unidentified factor act upstream of Notch. The alternate factor is still active when Hedgehog is inhibited and can promote Notch activity. A cooperative role for SHH and Notch in promoting symmetric versus asymmetric cell division of neocortical progenitors has been reported [36], and supports our observations.
What provides the trigger for cells to begin neuronal differentiation and exit the cell cycle? Many studies have documented the importance of a transition from symmetric to asymmetric cell divisions as neurogenesis proceeds in the developing neural tube [1,60,69]. Elevated levels of Notch are associated with the daughter cell that remains a proliferating progenitor, while lesser levels are associated with the differentiating daughter cell [34,35]. An asymmetric cell division in rosette culture could produce one cell that remains associated with the lumen at its apical surface, thereby still under the influence of Notch and Hedgehog, while the other cell, no longer in contact with the lumen, is not responsive to these signals, and therefore becomes an intermediate progenitor or differentiating neuron. Release from the apical surface is likely regulated by downregulation of Ncad expression, mediated by an increase in the transcription factors Foxp2 and Foxp4 [70].
An intriguing alternative mechanism for initiating neuronal differentiation, based on high-resolution live-cell imaging of the chick and mouse neural tube, has recently been described [71]. Cells with an apical process at the ventricular surface can undergo a subdivision, termed abscission, in which the apical-most end of the cell, containing the ciliary axoneme but not the basal body, is separated from the rest of the cell. This dismantles the primary cilium and thereby blocks SHH signaling, which promotes cell cycle progression. Decreasing levels of Ncad are associated with apical abscission and may facilitate the process by loosening cell–cell adhesions. These detached cells then exit the cell cycle and differentiate into neurons. The buildup of axoneme-associated Arl13b we observe in rosette lumens (Figs. 3 and 5) may include isolated apical domains produced via abscission. In the apical abcission process, atypical protein kinase C (aPKC) [9] is restricted to the portion of the membrane left behind and not present in the withdrawn process [71]. The cell, now detached from the lumen, has lost apical domain information. The distribution of Presenilin 2 that we report here suggests that it could be cast off through abscission, leaving the cell without a means of processing Notch to its active ICD, thereby promoting neuronal differentiation. Future live-cell imaging analysis of differentiating rosette cultures will determine whether the abcission observed in the neural tube also occurs in vitro.
A number of reports have extended the rosette-generating self-assembly of ESC-derived neural progenitors into three dimensions. These cultures recapitulate the complex cytoarchitecture of corticogenesis [69,72,73] or optic cup differentiation [74] with remarkable accuracy. This in vitro morphogenesis is noteworthy given the absence of local in vivo signals and landmarks, including cerebral spinal fluid and blood vessels, both of which are known to regulate neural differentiation [75 –77]. Our observations suggest that pluripotent stem cells provide in vitro models that can be used to study the early stages of neurogenesis as well as the complex patterning associated with later stages of CNS morphogenesis.
Footnotes
Acknowledgments
The authors thank Sandy Becker and Amy Kimble for excellent technical assistance and for reading the article and Jeff Gilarde for assistance with SEM. hES3 NKX2.1 GFP/w cells were the generous gift of Stewart Anderson and Andrew Elefanty, and the HES5:GFP line was the generous gift of Mark Tomishima. The use of human ESC lines was approved by the ESCRO committee of Wesleyan University. This work was supported by grant 11 SCB28 from the Connecticut Stem Cell Fund to LG.
Author Disclosure Statement
No competing financial interests exist.