
Abstract
Human embryonic stem cells (hESCs) exhibiting skewed X chromosome inactivation (XCI) have been reported. The copy number variations (CNVs), loss of heterozygosity (LOH), or single-nucleotide variant (SNV) events in those epigenetically distinct cells remain unknown, and whether such genetic abnormalities will influence the XCI status of hESCs is unclear. In this study, three hESCs with skewed XCI, three with random XCI, and two male hESC lines at different passages were analyzed for CNVs and LOH levels using a high-resolution genotyping microarray. Whole-exome sequencing was used to investigate the potentially damaging SNVs. On average, 17.6 CNVs and 5.3 cases of LOH were identified in the skewed hESCs, which were similar to the rates observed in random hESCs. Five recurrent CNV regions were uniquely identified in the skewed hESCs, but all of them were considered polymorphisms. With the exception of a nongenic CNV, no additional CNVs were detected on the X chromosome in the skewed hESCs. Although the XCI status in two hESC lines was observed to be changed from random to skewed, no significant CNV difference was identified before and after the XCI change. SNV analysis indicated that normal alleles are maintained for most genes within copy-neutral LOH regions. Three types of expression patterns were observed in heterozygous alleles, and the damaging SNVs in skewed hESCs favored the expression of the wild-type alleles. In conclusion, in the present study, we did not find genetic differences in the CNV and LOH levels between hESCs with and without skewed XCI. Wild-type allele expression in the presence of damaging SNVs on the X chromosome in skewed hESCs might alleviate adverse effects in those hESCs.
Introduction
G
XCI is regarded as an effective indicator of the epigenetic patterns in female hESCs because XCI is one of the first measurable epigenetic changes that occur in early mammalian embryos, and this process can coincide with the differentiation of ESCs [14]. Recent studies have described skewed and random XCI patterns in hESCs [15 –17]. Although previous studies have demonstrated that skewed hESCs have classical propagation and differentiation abilities [17,18], the differences in genomic integrity between skewed and random hESCs have not been thoroughly investigated. In addition, it remains unknown whether certain genetic abnormalities in hESCs are correlated with their XCI status, and whether the wild-type allele or mutant allele is more likely to be chosen for inactivation in hESCs is also unclear.
Recent research studies have demonstrated that most of the hESCs with skewed XCI are normal diploid cells [10,15,17]. However, in our previous study, we demonstrated that skewed hESCs can also be karyotypically abnormal; for example, one of our skewed hESCs contained unbalanced Robertsonian translocations of chromosome 13 and the other showed triploidy [17]. These results indicated that it is necessary to monitor the genomic integrity of those epigenetically distinct hESCs. To date, there have been no analyses comparing small abnormalities of less than 3 Mb, especially CNVs, LOH, or SNVs, on the X chromosome itself between hESCs with and without skewed XCI. Considering that previous studies of genomic integrity have elucidated the presence of numerous CNVs, SNVs, and LOH variants in hESCs during prolonged culture [2,7,19 –21], the possibility of small genetic abnormalities in these skewed hESCs cannot be ruled out without detailed investigation.
LOH, which is commonly reported in cancers and tumor diseases [22], was recently used as a sensitive marker to evaluate the genomic integrity and stability of hESCs [2,19]. Although previous research has demonstrated the instability of LOH events in hESCs [2], the research in this field is also limited. Further analysis is required to elucidate whether there are any damaging protein-coding SNVs on recessively inherited genes within the LOH regions and whether those LOH events or SNVs would influence gene expression in hESCs with a skewed XCI pattern.
In the present study, we performed high-resolution, single-nucleotide polymorphism (SNP) microarray analyses on three skewed female hESC lines at different passages to analyze their genomic integrity and to assess whether there is any correlation between skewed XCI and chromosomal abnormalities at the CNV, LOH, and SNV levels. For comparison, three female hESC lines with random XCI and two diploid male hESC lines were used [17,18,23]. For a higher-resolution investigation, we used whole-exome sequencing (WES) and Sanger sequencing to investigate and validate the SNVs. The sequencing and relative expression analyses of mRNA were also performed to investigate the expression levels of genes in the skewed hESCs.
Our data revealed that although hESCs had different XCI patterns and two cell lines were even observed to change their random XCI status to skewed XCI, the genomic integrity of those cells showed no differences at the CNV and LOH levels. Investigation of SNVs on the X chromosome indicated that most of the important genes within the LOH regions can be maintained normally. Our data also indicated that there are three types of expression patterns in both damaging and benign heterozygous alleles in the skewed hESCs, and a skewed choice of which chromosome to inactivate for most of the damaging SNVs in hESCs favored the X chromosome carrying mutations.
Materials and Methods
Ethics statement and culture of hESCs
This research was approved by the Ethics Committee of the Third Affiliated Hospital of Guangzhou Medical University. Embryos of poor quality, which were discarded by an in vitro fertilization center, were obtained for the derivation of hESC lines only after the donors had provided written informed consent. This study conformed to the principles outlined in the Declaration of Helsinki, and the signed informed consent documents clearly stated that all donated embryos would be used only for basic research and not for reproductive purposes.
The protocols for the derivation, culture, and maintenance of hESCs were performed as previously described [23]. In this study, a total of eight hESC lines were investigated for CNV, LOH, or SNV levels. Two female hESC lines were freshly derived (FY-hES-25, FY-hES-35), and six hESC lines were well characterized, including three female hESC lines with skewed XCI (FY-hES-5, FY-hES-8, and FY-hES-11), one female hESC line with random XCI (FY-hES-10), and two male hESC lines (FY-hES-1 and FY-hES-9) [17,18,23]. For dynamically observing genetic and epigenetic instability in the skewed hESCs during culture in vitro, undifferentiated cells at different passages (passage P20, P40, and FY-hES-5, -8 additionally at P60) along with spontaneously differentiated embryoid bodies at days 0 and 20 were cultured and harvested. The three random hESCs were further harvested and analyzed at very early passages (P8–P10) and moderate passages (P20–P40) to investigate whether their random XCI status would change to skewed after in vitro culture and propagation.
DNA extraction and quality evaluation
All the undifferentiated and differentiated hESCs were collected for DNA extraction. The extraction and purification of genomic DNA were performed with a Qiagen DNeasy Tissue Kit, according to the manufacturer's instructions (Qiagen, Hilden, Germany). High-quality genomic DNA, that is, double-stranded DNA that was free of polymerase chain reaction (PCR) inhibitors and not degraded and had an absorption ratio A260/A280>1.8, was required for the performance of the following assays.
XCI pattern analyses
Dynamic analyses of XCI patterns of hESCs were performed as previously described [17]. Briefly, XCI patterns were determined by analyzing methylation patterns using the HUMARA assay, the expression of the XIST gene, and the immunofluorescence of histone H3 lysine 27 trimethylation (H3K27me3). Details of these analyses are described in the Supplementary Materials and Methods section (Supplementary Data are available online at
Genotyping-based microarray analysis
Genome-Wide Human SNP Nsp/Sty 6.0 arrays or CytoScan 750K/HD arrays were used to investigate the genomic integrity of hESCs. Microarrays were prepared according to the manufacturer's protocol (Affymetrix, Santa Clara, CA). The raw data were initially analyzed with Affymetrix Genotyping Console software (version 4.1.2; Affymetrix) or analyzed and viewed using the Affymetrix Chromosome Analysis Suite (ChAS 2.2; Affymetrix). The locations of the CNVs and LOH events were determined based on the human genome assembly from February 2009 (GRCH37/h19). To avoid excess false-positive calls, a minimum number of 50 probes, a loss size value of 50 kb, a gain size value of 100 kb, and a 3,000-kb region for LOH were designated as the thresholds for the first run analysis. Any small positive CNVs that might be filtered out due to the threshold were manually confirmed.
Evaluation of CNVs and LOH
The free online database hosted by the University of California, Santa Cruz (UCSC,
Whole-exome sequencing
WES was performed to investigate SNVs in hESCs with or without skewed XCI; the protein-coding SNVs within the genes located in the LOH regions and on the X chromosome were given special attention. Sequencing libraries were prepared from 3 μg of high-quality genomic DNA using the SureSelect target enrichment system for the Illumina paired-end sequencing libraries kit (V1.5, November 2012; Agilent Technologies, Santa Clara, CA). Hybridization was performed using 750 ng of each prepared sequencing library, followed by exon capture using the SureSelect Human All Exon 50 Mb kit (Agilent Technologies). Paired-end sequencing of index-tagged and pooled samples was performed on a HiSeq2000 instrument (Illumina, Inc., San Diego, CA) according to the manufacturer's protocols. The data were aligned and mapped to the NCBI reference genome (GRCH37/h19) and were analyzed using NextGene software (SoftGenetics, LLC, State College, PA). To ensure high confidence in the results, SNVs that had a coverage depth of less than 30 were not used for the analysis. A validation of WES data was performed by comparison with data extracted from the microarrays or by Sanger sequencing.
Expression analyses of the mutated alleles by complementary DNA sequencing and real-time PCR
Total RNA was extracted using a PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA), and DNase I (Qiagen) digestion was performed to eliminate DNA from the RNA sample. DNase-treated RNA (0.5 μg) was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer's instructions. To clarify the differences in gene expression patterns between skewed and random hESCs, the relative expression patterns of XIST and the predicted damaging alleles and selected heterozygous SNP alleles located at different regions on the X chromosome were analyzed by real-time PCR and sequencing. The primers for sequencing and real-time PCR and PCR conditions are described in the Supplementary File S1 and Supplementary Materials and Methods section.
Results
Characteristics of the hESC cultures
In total, eight hESC lines were derived and cultured in our laboratory [18,23]; all these hESCs can be maintained well in the undifferentiated stages and have full differentiation abilities (data not shown). In our previous study, we demonstrated that the karyotypes of FY-hES-8, FY-hES-10, and FY-hES-11 were 46, XX, and that FY-hES-1 and FY-hES-9 were 46, XY, whereas FY-hES-5 had an unbalanced Robertsonian translocation with 46, XX, +13, der(13;13) (q10;q10) [18,23]. The microarray data showed that the karyotypes of FY-hES-25 and FY-hES-35 were 46, XX (Table 1).
Most of the hESC lines were analyzed at passage 20, but FY-hES-25 was analyzed at passage 8, and FY-hES-35 at passage 9. Dynamic CNV analyses were performed at different passages of these cell lines, and microarray data can be found using ArrayExpress accession number: E-MTAB-1992.
Whole duplication of chromosome 13 was found on unbalanced Robertsonian translocations, which would influence the average size of CNVs calculated in FY-hES-5. Thus, we excluded chromosome 13 and only counted CNVs on the remaining 22 pairs of chromosomes.
Although these two lines changed their XCI status from random to skewed after propagation, CNV comparisons were performed of these two cell lines in their random state.
chr, chromosome, CNV, copy number variation; Del, deletion; Dup, duplication; hESC, human embryonic stem cell; XCI, X chromosome inactivation.
In our previous study, we have demonstrated that FY-hES-5, FY-hES-8, and FY-hES-11 exhibited skewed XCI, whereas FY-hES-10 exhibited random XCI [17]. In the present study, we dynamically investigated the XCI status by analyzing the methylation status of HUMARA and demonstrated that both the newly derived FY-hES-25 and FY-hES-35 had random XCI patterns at very early passages (P8–P9) (Table 1 and Fig. 1). Although a stable skewed XCI pattern was maintained in FY-hES-8 and FY-hES-11, a variable XCI pattern was observed in FY-hES-35 and FY-hES-10, both of which changed their XCI status from random to skewed after propagation and culture in vitro (Fig. 1). The genetic differences between these epigenetically distinct cell lines and even within the same line, but with a different XCI status, were then investigated.
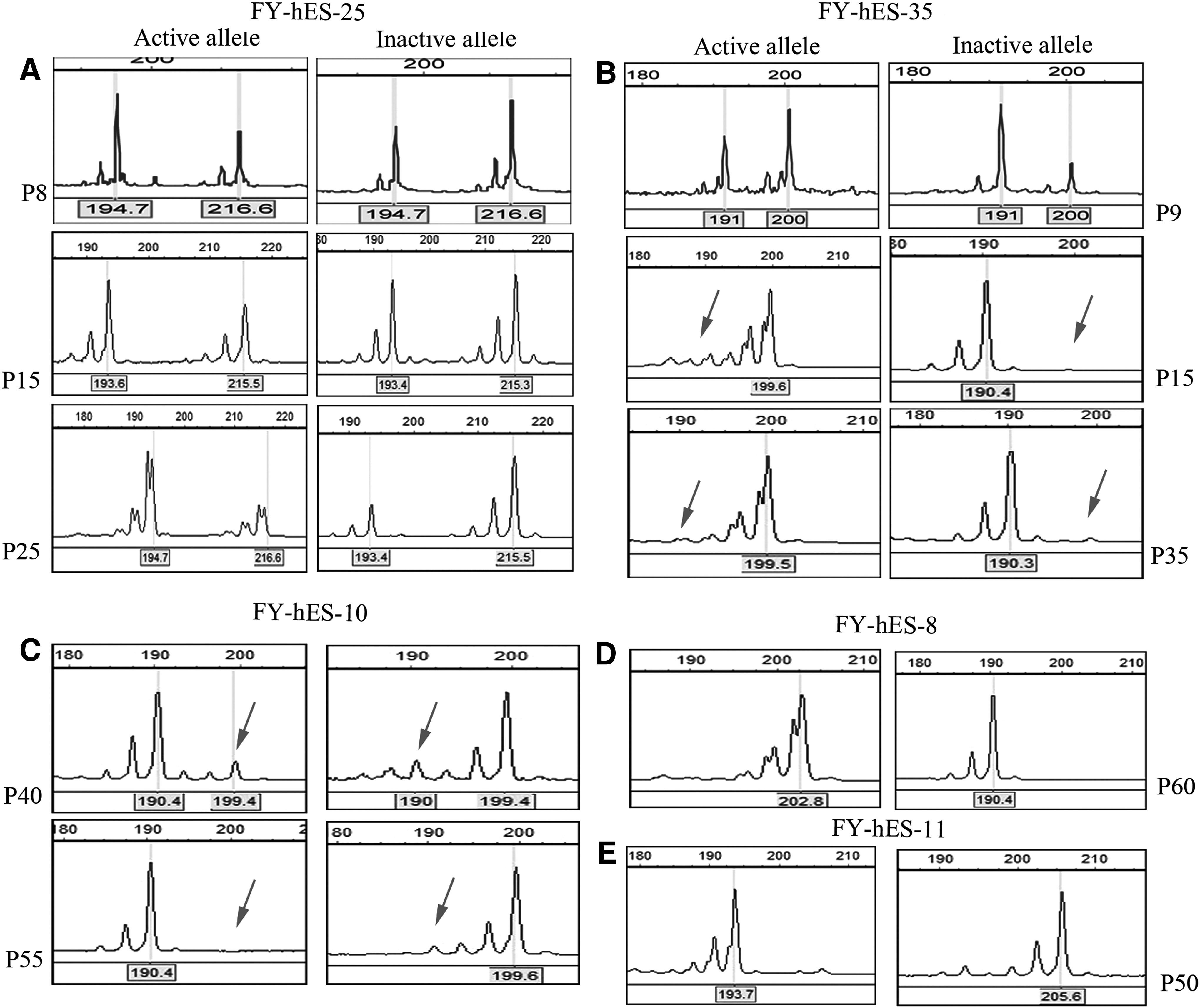
Methylation of HUMARA for X chromosome inactivation (XCI) pattern analyses. For skewed XCI determination, a common criterion of HUMARA methylation results is that the peak area ratio of inactivation alleles to activation alleles must be similar to or no more than 80:20, and an extremely skewed inactivation is defined as an inactivation to activation allele ratio of more than 90:10.
CNV analyses in different hESC lines with or without skewed XCI
The average number of CNVs in the two normal, diploid skewed hESCs at passage 20 (FY-hES-8 and FY-hES-11) was 17.6, with an average size of 246.5 kb. Similar to the number of CNVs in the skewed hESCs, on average, 15.0 CNVs were identified in each of the three random hESC lines (FY-hES-10 at P20, FY-hES-25 at P8, and FY-hES-35 at P9), but with a smaller average size (154.8 kb). For autosomal comparison, the average number of CNVs in two male hESCs was 12.5, with an average size of 253 kb (Table 1 and Supplementary File S2). For FY-hES-5, the unbalanced Robertsonian translocation hESC line, an entire duplication of the long arm of chromosome 13 was found in the array data and this abnormality could be stably maintained after 20 passages of propagation and differentiation (Fig. 2A). Excluding the whole duplication of chromosome 13, the number of CNVs identified on the remaining 22 pairs of chromosomes in FY-hES-5 was 17, which was similar to the CNVs found in all the other female hESCs (Table 1).

Copy number variation (CNV) analyses in human embryonic stem cells (hESCs) with skewed or random XCI.
In this study, we found that the distribution pattern of the CNVs between the hESCs with and without skewed XCI on each chromosome was also similar; our data indicated that chromosomes 1, 4, 16, and 22 were more likely to have deletion CNVs, whereas chromosomes 2, 7, and 14 were prone to duplication CNVs (Fig. 2B–E).
A total of seven CNVs on the X chromosome were found in all investigated hESCs (Table 1). Among the seven CNVs, two were found in male hESC lines (FY-hES-1), and four were found in random hESCs (FY-hES-25 and -35 at early passages) (Supplementary Fig. S1A). Only one 45-kb nongenic deletion CNV was found in one skewed hESC line (FY-hES-5) (Supplementary Fig. S1B); no additional duplication or deletion CNVs were found on the X chromosome in the other two skewed hESC lines (Supplementary Fig. S1C).
In this study, five recurrent CNV regions, including four deletions and one duplication, were identified as CNVs uniquely present in the skewed hESCs (Supplementary Table S1). However, after searching in the UCSC genome browser, we found that all five of these CNV regions overlapped entirely with the regions reported in the DGV database, suggesting that these five unique CNVs identified in skewed hESCs were polymorphisms (Supplementary Fig. S2).
CNV analysis in one hESC line with different XCI statuses
Because two hESC lines (FY-hES-10, FY-hES-35) were observed to have changed their XCI status, we investigated the FY-hES-35 line at three different passages to determine whether any de novo CNVs occurred in this originally random hESC line during culture and which CNVs were favored to be chosen after changing its XCI status. Although methylation results of the HUMARA assay indicated that this cell line changed its XCI status from random (P9) to skewed (P15 and P35) after a short propagation in vitro, the total number and size of CNVs observed in FY-hES-35 at three different passages remained relatively similar (Supplementary Fig. S3A). A slight difference in CNVs was reported by the ChAS software, such as CNV changes on chromosome 19 and chromosome X at different passages; a manual review of these CNVs indicated that these changes were acceptable errors in size classification due to the algorithm itself or that these differences were merely polymorphisms or located on nongenic regions (Supplementary Fig. S3B–D). Relatively stable CNVs were also observed in the other hESCs with or without skewed XCI (Supplementary Fig. S3E, F).
Analysis of copy-neutral LOH regions in hESCs
With a size threshold of 3,000 kb, we identified an average of 5.3 copy-neutral LOH regions in the three skewed hESCs, 4.3 LOH regions in the three random hESCs, and two in the male hESCs (Table 2). The number of LOH regions located on the autosomes was similar between the female and male hESCs (Table 2). In this study, we observed that chromosomes 16 and X tended to have more LOH regions than the other chromosomes (Fig. 3A). An LOH region, which was located at chrX: 76473325-78096142 with a size of 1623 kb, was found in all three skewed cell lines (Fig. 3B). In this region, three different allele lines and only two different alleles were observed in the random hESCs and the three skewed cell lines, respectively, indicating a difference in the LOH patterns between these cells (Fig. 3B). A total of 18 genes, including 6 disease-associated OMIM genes (ATRX, MAGT1, ATP7A, PGK1, FGF16, and COX7B), were identified in this unique LOH region (Fig. 3C).
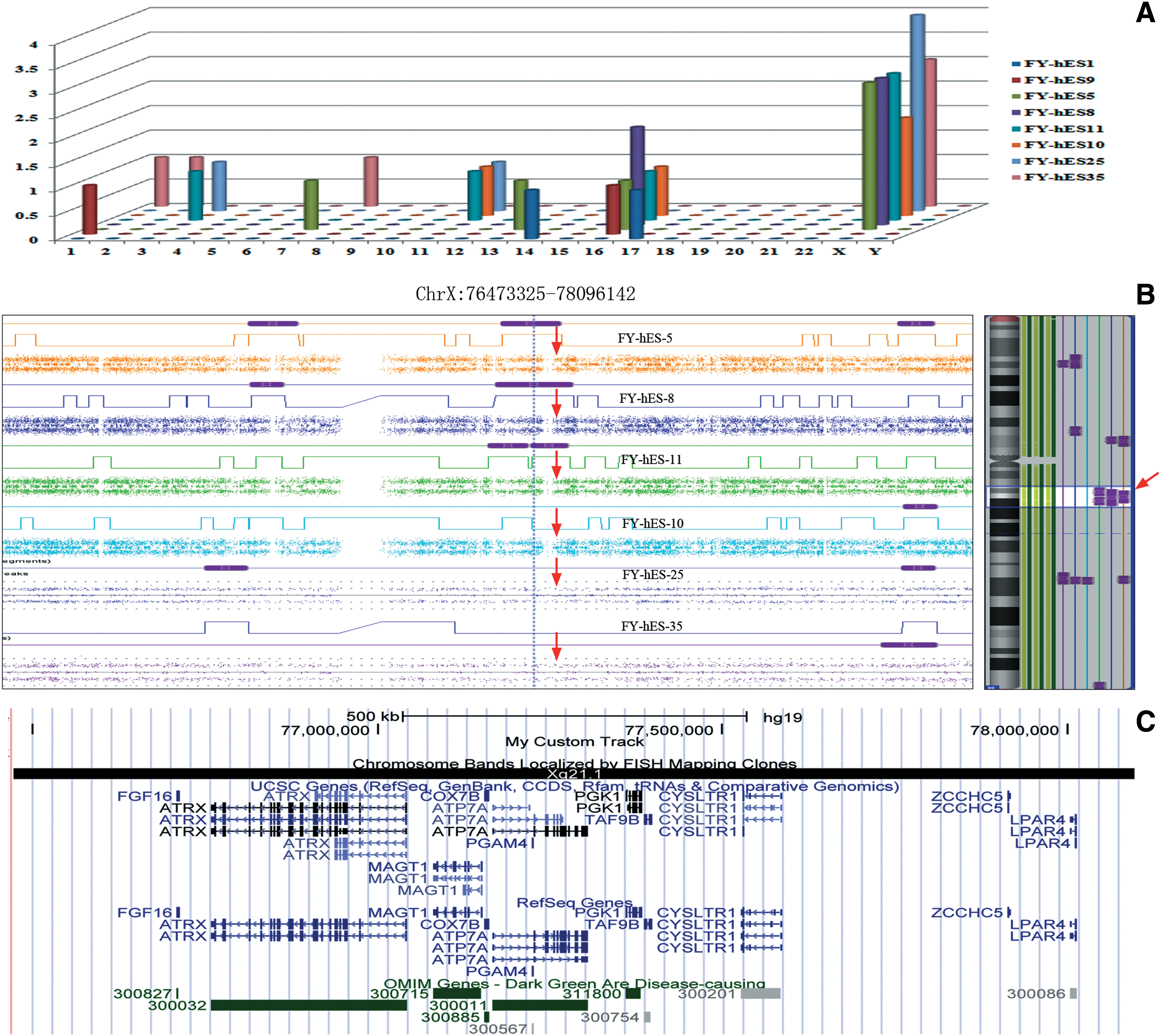
Copy-neutral loss of heterozygosity (LOH) analysis in hESCs.
LOH, loss of heterozygosity.
Evaluation of CNVs and LOH regions in the skewed hESCs
Excluding the whole duplication of chromosome 13 in the FY-hES-5 cell line, a total of 125 CNVs were evaluated. Including all of the unique CNV regions identified only in the skewed hESCs, 120 CNVs entirely overlapped the CNV polymorphism regions reported by the DGV database and some of them were also found to overlap the CNV regions reported in previous studies [2,7]. Therefore, 96.0% (120/125) of the CNVs identified in our hESCs, regardless of the skewed XCI status, were considered to be polymorphisms. The remaining five CNVs, which partially overlapped with the DGV database, were located in random hESCs or male hESCs and none of them were found in skewed hESCs (Supplementary Table S2).
The evaluation of genes within the LOH regions indicated that there was no difference in the total numbers of cancer genes, oncogenes, tumor suppressor genes, and imprinted genes located within the LOH regions between the hESCs with and without skewed XCI (Supplementary Fig. S4). In this study, we found that the number of recessively inherited genes within the copy-neutral LOH regions was higher than the other four types of evaluated genes, prompting the further investigation of the potential variation of those genes at a higher resolution.
Whole-exome sequencing analysis
To investigate any potentially damaging SNVs in the hESCs, we chose three skewed hESCs (FY-hES-5, FY-hES-8, FY-hES-11) and two hESCs with random XCI status (FY-hES-10, FY-hES-25) for higher-resolution analysis. The variant call format (VCF) files from these five female hESC lines were extracted from NextGene software and submitted to the wANNOVAR web server (
An analysis of the predicted damaging SNVs on the entire X chromosome of all female cells indicated that there were 3, 1, and 4 predicted damaging SNVs on the X chromosome in the FY-hES-5, FY-hES-8, and FY-hES-11 lines, respectively. One predicted damaging SNV was also found on the X chromosome in FY-hES-10, and none were identified in FY-hES-25 (Table 3). Among the nine damaging SNVs, three disease-associated SNVs were identified. Two (ARHGEF6 and UBA1) were found in FY-hES-5, and one (GLA) was in FY-hES-10; the SNVs in these genes are associated with mental retardation, spinal muscular atrophy, and Fabry disease, respectively.
AA, amino acid; C, conserved; chr, chromosome; D, damaging; Pred, predicted.
Confirmation of the SNVs and complementary DNA expression level analyses
More than 98% of the SNPs determined by the SNP microarrays coincided with the results from the WES analysis (data not shown). We used Sanger sequencing to validate certain SNP alleles reported by the SNP arrays that differed from the WES data, and these data indicated that the WES results were correct (data not shown).
In this study, nine predicted damaging SNVs were all heterozygous; complementary DNA (cDNA) sequencing was used to elucidate their mRNA expression patterns. In FY-hES-5 and FY-hES-8, most of the mutated alleles were inactivated, with only the wild-type alleles expressed. Meanwhile, three of four damaging, heterozygous mutated alleles in FY-hES-11 and one of three mutated alleles in FY-hES-5 showed expression of both alleles, indicating that these cells may have completely or partially escaped inactivation or may have reactivated these alleles. One allele in FY-hES-11 expressed only the wild-type allele, indicating that a skewed pattern was maintained well in these cells. The expression of both alleles was observed in FY-hES-10 (Fig. 4).

Complementary DNA (cDNA) analysis of heterozygous damaging single-nucleotide variants (SNVs). Three allele expression patterns were identified from a total of nine damaging SNVs.
To address whether biallelic expression was due to reactivation or the escape of inactivation events in skewed hESCs, we further chose several heterozygous SNPs located in different regions of the X chromosome as well as those predicted damaging heterozygous alleles to observe their relative expression patterns. Although the methylation pattern of HUMARA demonstrated a skewed XCI in FY-hES-8 and FY-hES-11, inactivation escape and reactivation events were observed in those cells (Fig. 5 and Supplementary Fig. S5).

Real-time polymerase chain reaction (PCR) for relative gene expression level detection.
Gene expression level analyses
To determine whether the loss of XCI markers happened in the skewed and random hESCs that this study was working with, dynamic relative expression levels of XIST and immunofluorescence staining of H3K27me3 were assessed in these hESCs. At early and moderate passages, all examined skewed and random hESCs had detectable XIST expression and showed distinct H3K27me3 signals (Fig. 6A, B), demonstrating that a distinct XCI status was maintained in these hESCs. After in vitro culture, reduced XIST levels in FY-hES-8 and FY-hES-11 and a complete loss of XIST expression and H3K27me3 signals in FY-hES-10 were observed, indicating unstable XCI marker exhibition in these cell lines (Fig. 6A, B).
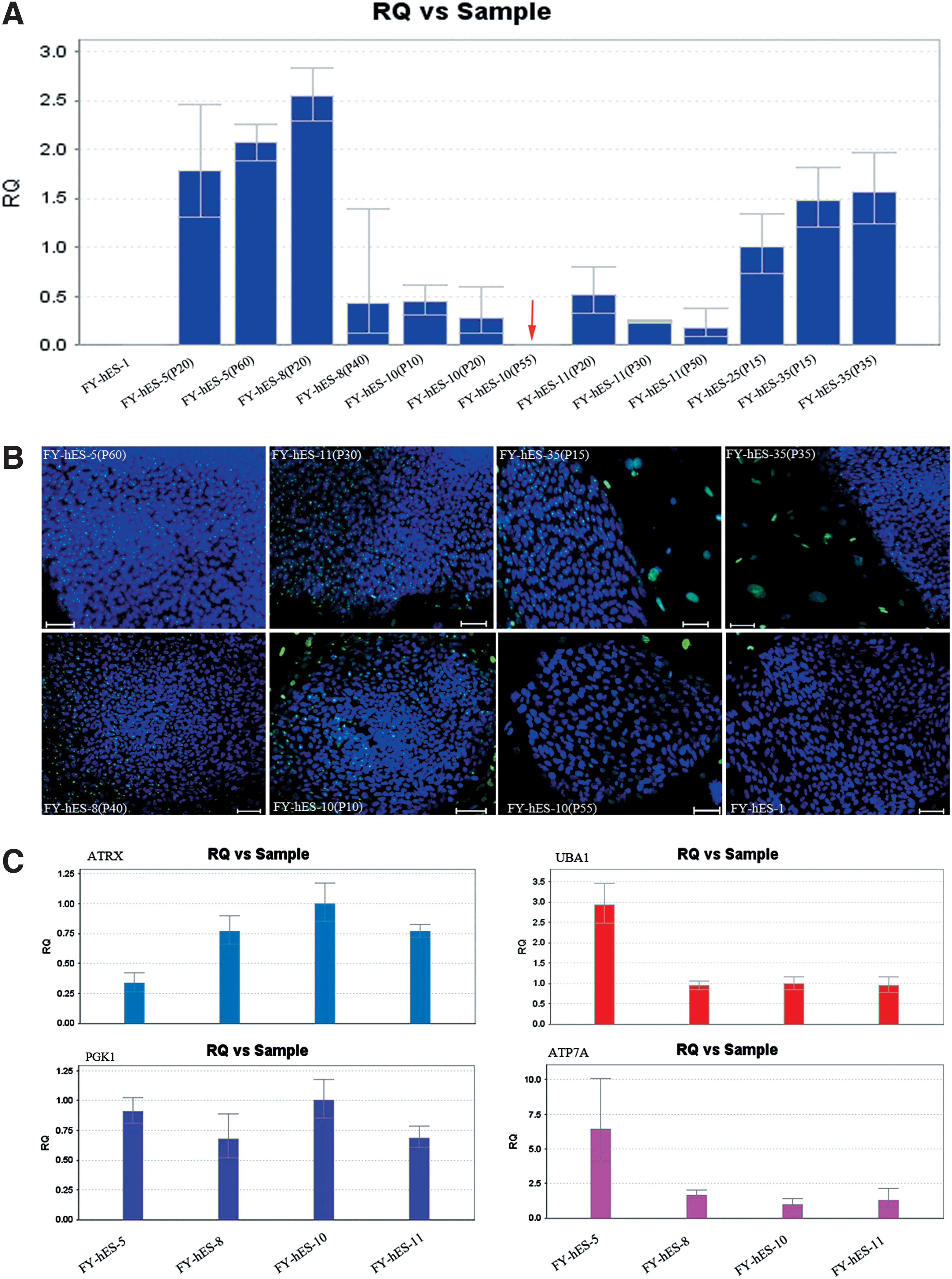
Analyses of XCI status in hESCs and the relative expression levels of genes within the LOH region.
To investigate whether copy-neutral LOH and mutated allele would influence gene expression, we further investigated the expression of four OMIM disease-causing genes and found no significant differences in the expression patterns of ATRX and PGK1 between skewed and random cells. Higher expression levels of UBA1 and ATP7A were found in FY-hES-5, and similar expression patterns of those two genes were also found in the remaining hESCs, whether they showed skewed or random XCI (Fig. 6C).
Discussion
Recent reports have described hESCs with skewed and random XCI [10,15,17]. Previous studies have demonstrated that the ratio of female hESCs exhibiting a skewed XCI is much higher than in female adults [10,15,17]. For example, in our previous study, we demonstrated that four of the five investigated female hESC lines have skewed XCI [17], and Shen et al. [10] observed monoallelic expression patterns for X-linked genes in all three of the hESC lines that they investigated, demonstrating skewed XCI in these hESCs. Although a skewed XCI pattern was observed in so many hESC lines, the genetic integrity and stability of those epigenetically distinct cells is unclear, especially in terms of CNV, LOH, or SNV events. Considering the high prevalence of skewed XCI in female hESCs, it is necessary to clearly elucidate the genomic integrity of these hESCs for their potential safety in therapeutic applications.
In this study, we demonstrated that there was no notable genomic difference at the CNV and LOH levels between hESCs with random and skewed XCI. Because there is no database available for genetic comparisons of pluripotent cells, human genome databases, such as DGV, DECIPHER, ISCA, and OMIM, and CNVs reported in previous hESC studies can be used for the evaluation and interpretation of CNVs in hESCs. The potential influence of all the identified CNVs was evaluated according to the size of each region and its gene content using online resources [26,27], which allowed us to determine whether a CNV was a polymorphism, a variant of unknown significance, or pathogenic. Our data indicate that the average number of CNVs identified in hESCs with skewed or random XCI was consistent with a previous report [2], and some of them also overlapped with the CNV regions that were previously reported [2]. Ninety-six percent of the CNVs identified in our female hESCs, including the five recurrent unique regions found only in the skewed hESCs, can be considered polymorphisms and those recurrent polymorphisms were more likely to be inherited from embryo donors than de novo CNVs appearing during in vitro culture; thus, these polymorphism CNVs would be considered to have no disadvantageous influence on the skewed hESCs.
Why so many hESCs have skewed XCI remains poorly understood. XCI essentially occurs in two steps: counting the number of X chromosomes present and choosing which X chromosome to inactivate [28]. The normal initiation of XCI in females results in a random choice. However, ∼10% of healthy females can have a skewed XCI [29], and the cause of this skewed X inactivation is unclear. Genetic mechanisms, including chromosomal abnormalities and mutations in the XIST promoter, are potential reasons for the skewed inactivation because it would be preferable for an X chromosome carrying a detrimental mutation to be chosen for inactivation; this phenomenon has been observed in carriers of several human X-linked diseases [30 –33]. In hESCs, the reason for skewed XCI is hypothesized to be the result of clonal selection in culture rather than of inheritance from the inner cell mass [15], but additional experiments are needed to examine which X chromosome is selected and whether a skewed choice is favorable or unfavorable for the X chromosome carrying a mutation.
In our previous study, we demonstrated that FY-hES-5, FY-hES-8, and FY-hES-11 exhibited a skewed XCI pattern at very early passages (P10), suggesting that clonal selection happens at a very early stage. In this study, two hESCs were observed to change their XCI status from random to skewed after culture in vitro, including a newly derived random hESC line, FY-hES-35, which quickly changed its XCI status from random to skewed after only six passages of propagation. We hypothesized that this rapid epigenetic variability would probably be due to manual propagation proceeding rather than influenced by culture selection pressure. Because there is a limited number of high-quality clones that can be chosen for propagation at very early passage, if only one clone was chosen for propagation, the definite XCI pattern of this single clone would be kept and maintained throughout the following clonal expansion and a skewed XCI would be observed. This process was often observed in the propagation of derived hiPSCs with skewed XCI from a single clone [34].
Although the skewed XCI may simply be caused by a bottleneck in the propagation of the line, the possibility of chromosomal abnormalities present on the X chromosome itself that would influence the choice during XCI still needs to be examined. Therefore, the CNV events located on the X chromosome were further analyzed. Interestingly, although there was a slight CNV difference observed in FY-hES-35 before and after the XCI status change, all the differences were polymorphisms or were merely located on nongenic regions. Additionally, those differences could contribute to the errors in size classification due to the algorithm itself. With the exception of a single nongenic CNV that was identified in the FY-hES-5 line, there were no CNVs found on the X chromosome in the other two skewed female hESC lines.
From our results, we hypothesized that CNVs located on the X chromosome may be just one of several complex causes of the selection of skewed XCI and not the reason for the skewing in our cells; this hypothesis is supported by a previous study [35]. As in non-ES cells, such as in female human blood samples, skewed XCI is rarely found to be due to X chromosome changes. After analyzing SNP arrays, Jobanputra et al. [35] found only four CNVs located on X chromosomes in 45 females with skewed XCI patterns and five CNVs in 45 normal control females, indicating that there are no significant differences in CNV frequency between females with skewed versus random XCI. In addition, no detectable CNVs were found in two of three women with highly skewed XCI [35], further supporting our findings and suggesting that skewed XCI is unlikely to be affected simply by CNVs on the X chromosome.
Because LOHs are found predominantly as spontaneous mutations in ESCs and always accompany tumor development [36 –38], LOH on the X chromosome was further investigated in this study to clarify the genetic difference between hESCs with or without skewed XCI and to determine whether this is a possible reason for the skewed phenotype. Although an LOH region located on the X chromosome was identified in each of the three skewed hESC lines, most of the oncogenes and recessively inherited genes within LOH regions can maintain the wild-type alleles. XIST, which is critical for the initiation of XCI, was also found within the copy-neutral LOH regions, and no damaging mutations in the XIST gene body or in the promoter region were found in either the skewed or random hESCs (data not shown). Furthermore, we did not find any predicted damaging SNVs of those previously mentioned genes within LOH regions. Therefore, we suggest that the copy-neutral LOH events with no damaging mutations may have no significant adverse functional influence on the skewed hESCs.
To validate this hypothesis, we analyzed the gene expression patterns of ATRX, PGK1, and ATP7A between skewed and random hESCs. Although these three genes are found in copy-neutral LOH regions in skewed cells and are heterozygous in the random hESCs, no obvious differences in expression levels were observed among those cell lines. This suggests that when no damaging mutations were found within copy-neutral LOH regions, those LOH events had similar effects on skewed and random hESCs.
Using bioinformatics tools, we identified nine predicted damaging SNVs in our female hESCs. Interestingly, although those SNVs were predicted to be damaging, they were all heterozygous, which means that these heterozygous mutations may function as carriers and may not have any adverse effects on the function of the hESCs. However, if the female hESCs have a skewed XCI pattern and the wild-type allele was chosen for skewed inactivation, it would be highly possible for the damaging function to affect the hESCs, even if the mutation is heterozygous. Therefore, we further conducted cDNA sequencing and real-time PCR analyses of these mutated alleles to elucidate their expression patterns. Three types of expression patterns were found in our skewed hESCs. In one expression pattern, the skewed hESCs inactivated the X chromosome carrying one or more mutated alleles and stably expressed the wild-type allele. The other two patterns were complete and showed partial biallelic expression of both of the heterozygous alleles, suggesting that an escape from inactivation or reactivation of the inactivated X chromosome occurred for those alleles.
To elucidate whether the expression patterns of the damaging SNVs were unique events in skewed cells or if this was a common phenomenon for both damaging and nondamaging SNVs in each line, we randomly chose several heterozygous alleles for investigation. Similar to the observation in damaging alleles, three expression patterns, which were monoallelic expression, partial biallelic expression, and complete biallelic expression patterns, were observed in the benign alleles. The definite expression of XIST and positive H3K27me3 signals confirmed that the expression of heterozygous alleles is controlled by an XCI pattern.
To determine whether the biallelic expression pattern was a reactivation or escape inactivation event, we dynamically investigated those heterozygous alleles using real-time PCR to evaluate their expression levels. In this study, both reactivation and escape from inactivation events were observed in our skewed hESCs. Three pairs of alleles of CAB5, PTCHD1, and FTHL17, which are located on the short arm of the X chromosome (Xp), were observed to have biallelic expression at early passages. Approximately 15% of X-linked alleles are known to escape XCI in human female somatic cells, and the reactivation of skewed alleles in hESCs has also recently been reported [10,39]. The escape of those alleles is coincident with previous reports that genes on the Xp are prone to escape from XCI [39].
The reactivation of silenced X-linked alleles was reported to contribute to the absence of XIST expression in class III female hESCs [10]. In the present study, although a detectable XIST expression level was observed in FY-hES-8 and FY-hES-11, their expression levels were significantly lower than that in random calculator cells, and reduced expression levels after further culture indicating a gradual loss of XCI markers also occurred in these cells. We think that the reactivation events of some alleles in skewed hESCs may contribute to the gradual loss of XCI markers. Although reactivation and escape from inactivation of both benign and damaging alleles were found in skewed hESCs, a more definitive assessment of whole allele expression patterns using high-throughput RNA-seq is required to clarify this phenomenon.
In this study, the skewed expression of wide-type alleles was observed in skewed hESCs. We hypothesize that the skewed inactivation of mutated alleles, that is, expressing only wild-type alleles of the disease-causing genes UBA1, PLXNA3, and TKTL1, may contribute to nonsense-mediated decay [40]. Alternatively, there are two other possibilities. First, negative selection against the mutated X chromosome may have occurred during culture due to clonal selective pressure because the expression of the wild-type alleles is more favorable in culture conditions. Second, these mutated alleles may have been selected for inactivation during the initial XCI decision process, and multiple mutations occurred on both of the two X chromosomes; therefore, accompanying a loss of XCI markers, an escape or reactivation from skewed XCI may occur.
In conclusion, we found no genetic differences in the CNV and LOH levels of hESCs with or without skewed XCI. Skewed expression, reactivation, and escape from inactivation of alleles were observed in skewed hESCs, and the heterozygous damaging SNVs in skewed hESCs favored the expression of wild-type alleles.
Footnotes
Acknowledgments
The authors thank Yu-Mei Luo, Xin-Jie Chen, and all members of the Key Laboratory for Major Obstetric Diseases of Guangdong Province for hESC line derivation and culture. This work is supported by the National Natural Science Foundation of China (31171229 to X.F.S.), the NSFC-Guangdong Joint Foundation (U1132005 to X.F.S.), Science and Information Technology of Guangzhou Key Project (2011Y1-00038 to X.F.S.), the National Natural Science Foundation of China for Young Scholars (81301184 to W.Y.H.), the Foundation for PhD and Returnees of Visiting Scholars of Guangzhou Medical University (2013C56 to W.Q.L.) and the 973 program grants from the Ministry of Science and Technology of China (2010CB529601 and 2013CB945404 to B.L.W.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.