
Abstract
Umbilical cord mesenchymal stem cells (UC-MSCs) show properties similar to bone marrow mesenchymal stem cells (BM-MSCs), although controversial data exist regarding their osteogenic potential. We prepared clinical-grade UC-MSCs from Wharton's Jelly and we investigated if UC-MSCs could be used as substitutes for BM-MSCs in muscoloskeletal regeneration as a more readily available and functional source of MSCs. UC-MSCs were loaded onto scaffolds and implanted subcutaneously (ectopically) and in critical-sized calvarial defects (orthotopically) in mice. For live cell-tracking experiments, UC-MSCs were first transduced with the luciferase gene. Angiogenic properties of UC-MSCs were tested using the mouse metatarsal angiogenesis assay. Cell secretomes were screened for the presence of various cytokines using an array assay. Analysis of implanted scaffolds showed that UC-MSCs, contrary to BM-MSCs, remained detectable in the implants for 3 weeks at most and did not induce bone formation in an ectopic location. Instead, they induced a significant increase of blood vessel ingrowth. In agreement with these observations, UC-MSC-conditioned medium presented a distinct and stronger proinflammatory/chemotactic cytokine profile than BM-MSCs and a significantly enhanced angiogenic activity. When UC-MSCs were orthotopically transplanted in a calvarial defect, they promoted increased bone formation as well as BM-MSCs. However, at variance with BM-MSCs, the new bone was deposited through the activity of stimulated host cells, highlighting the importance of the microenvironment on determining cell commitment and response. Therefore, we propose, as therapy for bone lesions, the use of allogeneic UC-MSCs by not depositing bone matrix directly, but acting through the activation of endogenous repair mechanisms.
Introduction
B
In this context, we prepared clinical-grade UC-MSCs [6], and we investigated if UC-MSCs could be used as substitutes for BM-MSCs in musculoskeletal regeneration. UC-MSCs clearly display typical MSC features as defined by the International Society for Cellular Therapy [17]. Both UC-MSCs and BM-MSCs are plastic adherent and show similar cell surface marker expression. Interestingly, the only major difference is their differentiation potential [6]. UC-MSCs have a very weak osteogenic [18 –20], chondrogenic [21], and adipogenic [19,20] differentiation capacity compared with BM-MSCs. The reasons for these differences need to be further clarified, but it has been postulated that the reduced differentiation potential of UC-MSCs may depend on their position within the UC tissue they are isolated from [22]. As the cells in the human UC stroma are not uniformly distributed, it may be hypothesized that, depending on the isolation technique used, slightly different types of primitive cells with unequal differentiation capabilities can be obtained [6]. It must also be taken into consideration that whereas BM-MSCs may be more committed to osteogenesis because of their natural density in the bone and in maintaining the self-renewal capacity of hematopoietic stem cells, UC-MSCs having a fetal origin might require stronger inductive stimuli or more time to differentiate into a specific tissue [23,24].
Increasing evidence has also recently suggested a predominant paracrine role of MSCs in promoting tissue regeneration due to their secretome enriched in proinflammatory, chemotactic, and angiogenic factors rather than due to their direct replacement of affected cells at the site of injury [25]. Additionally, since vascularization is a crucial aspect in the tissue regeneration process, this inherent characteristic of MSCs, particularly of UC-MSCs, would be interesting to explore.
Hence, to better clarify the potential of UC-MSCs in regenerative medicine and to compare them with BM-MSCs in an in vivo osteogenic environment, we wanted to answer the following questions: 1. Can an osteogenic environment, whether ectopic or orthotopic, influence the osteogenic regenerative capacity of UC-MSCs expanded according to a clinical-grade protocol? 2. Are the regenerative effects of implanted UC-MSCs attributed to their direct engraftment or could their paracrine/trophic effects be responsible? 3. Can UC-MSCs mediate a therapeutic angiogenic response, thus leading to enhanced bone regeneration?
Materials and Methods
Cell isolation and culture
Human UC-MSC culture
Human UCs were collected from pregnant women after cesarean sections. Informed written consent was obtained. The UC processing was performed in accordance with a GMP-compliant protocol for the isolation and expansion of UC-MSCs [6]. Briefly, the UC was cut into segments, which were split open to expose the inner surface. The UC segments were transferred to an expansion medium, consisting of alpha-minimum essential medium with GlutaMAX™ (Invitrogen) enriched with 5% human platelet lysate (PL) obtained from healthy donors, 2 IU/mL Na-heparin (Epsoclar; Hospira s.r.l.), and 100 μg/mL streptomycin (Sigma Chemical Co.), and minced. Minced pieces were incubated in the expansion medium at 37°C and 5% CO2 for 1 week to allow the cells to adhere, and then they were removed. At 80% confluence, the cells were detached by treatment with Tryple (Invitrogen) and passaged. All experiments were performed using at least three different primary cultures at either P1 or P2.
Human BM-MSC culture
Human BM-MSCs were obtained from iliac crest marrow aspirates of healthy bone marrow transplant donors after informed consent. The BM-MSCs were cultured in complete culture medium consisting of Coon's modified Ham's F-12 medium containing 2 mM glutamine, 100 IU/mL penicillin, 100 μg/mL streptomycin (Sigma Chemical Co.), and 10% fetal bovine serum (FBS; Invitrogen) in the presence of 1 ng/mL human recombinant fibroblast growth factor-2 (FGF-2; Peprotech). At 80% confluence, the cells were detached by treatment with 0.05% trypsin −0.01% ethylenediaminetetraacetic acid (EDTA; Sigma Chemical Co.) and passaged. All experiments were performed using at least three different primary cultures at either P1 or P2.
Conditioned medium preparation
The conditioned medium (CM) was collected from 80% confluent cultures of either UC-MSCs or BM-MSCs in 10-cm tissue culture dishes. Each culture dish received 7 mL of serum-free media for 48 h. The CM was collected by centrifugation at 440 g for 5 min and by a second centrifugation at 1,750 g for 3 min, both centrifugations were at 4°C [26].
Platelet lysate preparation
Whole blood was collected from voluntary donors following current procedures for blood donation. Blood units were screened for the absence of infectious agents in compliance with national regulatory requirements. Buffy coats (BC) were obtained by centrifugation of whole blood donations. Five BC units were pooled, platelet-rich plasma (PRP) separated by light-spin centrifugation, and the platelets concentrated by a second heavy-spin centrifugation. PRP was resuspended in homologous plasma and stored at −30°C. The standard PRP unit had a platelet concentration of 1–2×106/μL. PL was obtained by subjecting PRP to three cycles of freezing and thawing. The PL was aliquoted and stored at −20°C.
Ectopic bone formation assay
All experimental animal procedures were evaluated and approved by the IRCCS AOU San Martino-IST Ethics Committee for animal experimentation (CSEA) and communicated to the Italian Ministry of Health in accordance with article 7 of the D.lgs 27/01/1992 n.116/92.
The in vivo analysis was based on an established model of ectopic bone formation [27]. Briefly, we prepared the implantable constructs by seeding 2.5×106 UC-MSCs onto ceramic scaffolds (Skelite; 4×4×4 mm cubes of 33% hydroxyapatite and 67% silicon-stabilized tricalcium phosphate, Si-TCP) and embedding the scaffolds in a fibrin glue (Tissucol; Baxter). Moreover, nonseeded empty Skelite and Skelite seeded with 2.5×106 BM-MSC constructs were used as negative and positive control groups, respectively (n=4).
Scaffolds were implanted subcutaneously on the dorsal surface of immunocompromised mice (CD-1 Nu/Nu; Charles River). The implants were recovered at 60 days and histologically analyzed.
Histological analysis
Harvested samples were fixed in 3.7% paraformaldehyde, decalcified in EDTA 10%, and paraffin embedded. Sections were cut at 4 μm thickness and stained with hematoxylin and eosin (H&E) and Masson's trichrome (MTC) (Masson's trichrome special staining kit; Bio-Optica) stains. Moreover, chromogenic in situ hybridization (Zytovision kit) to detect human ALU repeat sequences was performed following the manufacturer's instructions. Images of the sections were acquired using a phase-contrast Axiovert 200M microscope (Zeiss). Polarized light images were acquired using a phase-contrast Axiophot microscope (Zeiss).
The number of blood vessels in the ectopic implants (n=3) was manually counted in multiple, representative MTC-stained sections for UC-MSCs and BM-MSC-seeded and empty scaffolds (six per condition per experiment). The bone surface area in the orthotopic implants (n=3) was manually measured in multiple representative H&E-stained sections for all groups. Three sections per condition per experiment were analyzed. The ratio between the neoformed bone area and total defect area was calculated for each section.
To perform the histologic quantification analysis, ImageJ (NIH) software was used.
In vivo live cell-tracking analysis
To perform live cell-tracking experiments, UC-MSCs and BM-MSCs were transduced using a replication-incompetent amphotropic bicistronic retrovirus encoding the firefly luciferase gene (Luc) [28]. Retroviral vectors and infection protocols were performed as previously described [29]. Luc-infected UC-MSCs and BM-MSCs were selected by adding G418 (Gibco) at 0.5 mg/mL to the medium for 10 days. MSC-transduced cells (2.5×106) were seeded onto Skelite scaffolds and implanted as reported above.
The in vivo bioluminescence reaction induced by the substrate luciferin (150 mg/kg; D-Luciferin Firefly Xenogen) injections in mice was detected using the IVIS® Imaging System LUMINA II (PerkinElmer) (n=3 in BM-MSC implants and n=6 in UC-MSC implants). Mice were imaged for 5 min after 1, 7, 21, and 30 days postimplantation.
Metatarsal angiogenesis assay
The metatarsal angiogenesis assay was performed as previously described [30]. Briefly, metatarsals of 17-day-old, wild-type mouse embryos (FVB/N; Charles River) were transferred to culture dishes to allow their attachment for 72 h. Bones were then cultured for 13 days with either UC-MSC CM or BM-MSC CM, both implemented with 10% FBS. Fresh complete medium containing 10% FBS was used as the experiment control. After 13 days of culture, bones were fixed and stained for PECAM-1/CD31. Quantification of the number of CD31 positively stained pixels and the outgrowth area from bone were performed using ImageJ (NIH) software (n=3).
Proteomic identification and bioinformatics analysis
Cytokine screening of UC-MSC secretome was performed on CM using a human cytokine array (Panel A; R&D Systems) following the manufacturer's instructions. The cytokines identified in the CM of UC-MSCs were uploaded into the web-based DAVID platform (
Assessment of bone formation in the mouse calvarial defect
Discs of nanohydroxyapatite/poly (ester urethane) (nHA/PU) with a diameter of 5 mm and height of 2 mm were provided by the AO Research Institute (Davos, Switzerland). These scaffolds have been shown to support orthotopic bone formation in a rabbit model [2]. Immediately before surgery, 2.5×106 UC-MSCs were loaded onto the scaffolds and embedded in a fibrin glue (Tissucol; Baxter). Moreover, nonseeded empty nHA/PU discs and discs seeded with 2.5×106 BM-MSC constructs were used as negative and positive control groups, respectively (n=3).
The constructs were implanted in 5-mm critical-size defects in the calvaria of immunocompromised mice (CD-1 Nu/Nu; Charles River).
The critical-sized calvarial defects were made as previously described in the literature [31]. Mice were anesthetized with ketamine (80–100 mg/kg) and xylazine (5–10 mg/kg). A 5-mm defect was prepared in the center of the parietal bones of each mouse calvarium using Mectron Piezosurgery® instruments (Mectron s.p.a.). The construct was then carefully placed and the flap was repositioned and sutured using 4.0 vicryl (Ethicon) suture material. The implants were recovered 90 days postoperatively, decalcified, and then histologically analyzed.
Statistical analysis
The results are expressed as mean and standard error of the mean. Statistical significance was determined using the ordinary one-way ANOVA and the value of P<0.05 was considered to be statistically significant. Statistical analysis was performed by Graphpad software.
Results
UC-MSC behavior in an ectopic bone formation model
Histological assessment of the tissues formed within the ectopically implanted scaffolds revealed the formation of mature bone in the pores of the scaffolds seeded with BM-MSCs (Fig. 1E), whereas UC-MSC-seeded constructs showed a compact fibrous tissue ascribable to an immature bone-like structure (Fig. 1F). In the nonseeded implants, a loose connective fibrous tissue was observed (Fig. 1D). Polarized light examination confirmed the presence of highly organized collagen fibers in the BM-MSC-seeded implants (Fig. 1H) compared with the weaker signal observable in the UC-MSC-seeded scaffolds (Fig. 1I). In the nonseeded scaffold samples, no polarized light signal was detected (Fig. 1G).
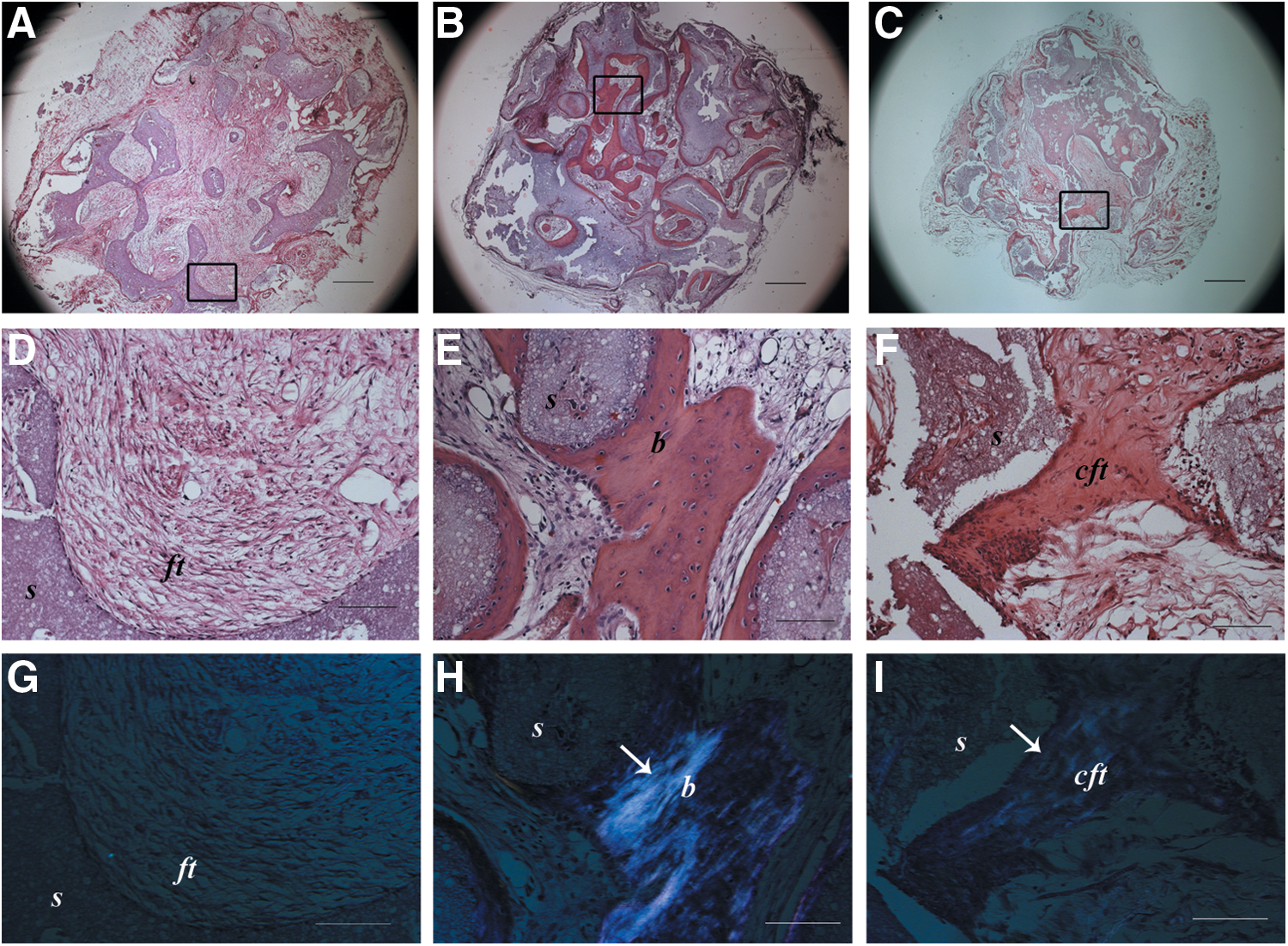
Histological images of ectopic implants after 2 months and stained with hematoxylin and eosin
Low-magnification micrographs (Fig. 1A–C) showed that the bone tissue in BM-MSC-seeded scaffolds was formed in most of the area of the implant (Fig. 1B), whereas in UC-MSC-seeded scaffolds, the immature bone-like matrix was sparsely present (Fig. 1C) with the majority of the area filled with a loose connective tissue similar to that seen completely filling the nonseeded scaffolds (Fig. 1A).
UC-MSC angiogenic effect in vivo
Although only a minimal amount of immature bone-like matrix could be detected in the UC-MSC-seeded scaffolds, an accurate analysis of these implants revealed an abundance of blood vessels (Fig. 2C) compared with the BM-MSC-seeded (Fig. 2B) and empty implants (Fig. 2A). The characteristic gold staining of the MTC stain revealed the presence of red blood cells in most of the vessel-like structures (Fig. 2A–C). Quantitative analysis confirmed the presence of a significantly higher number of blood vessels in UC-MSC implants than in either BM-MSC-seeded (P<0.0001) or empty scaffolds (P<0.0001) (Fig. 2D).
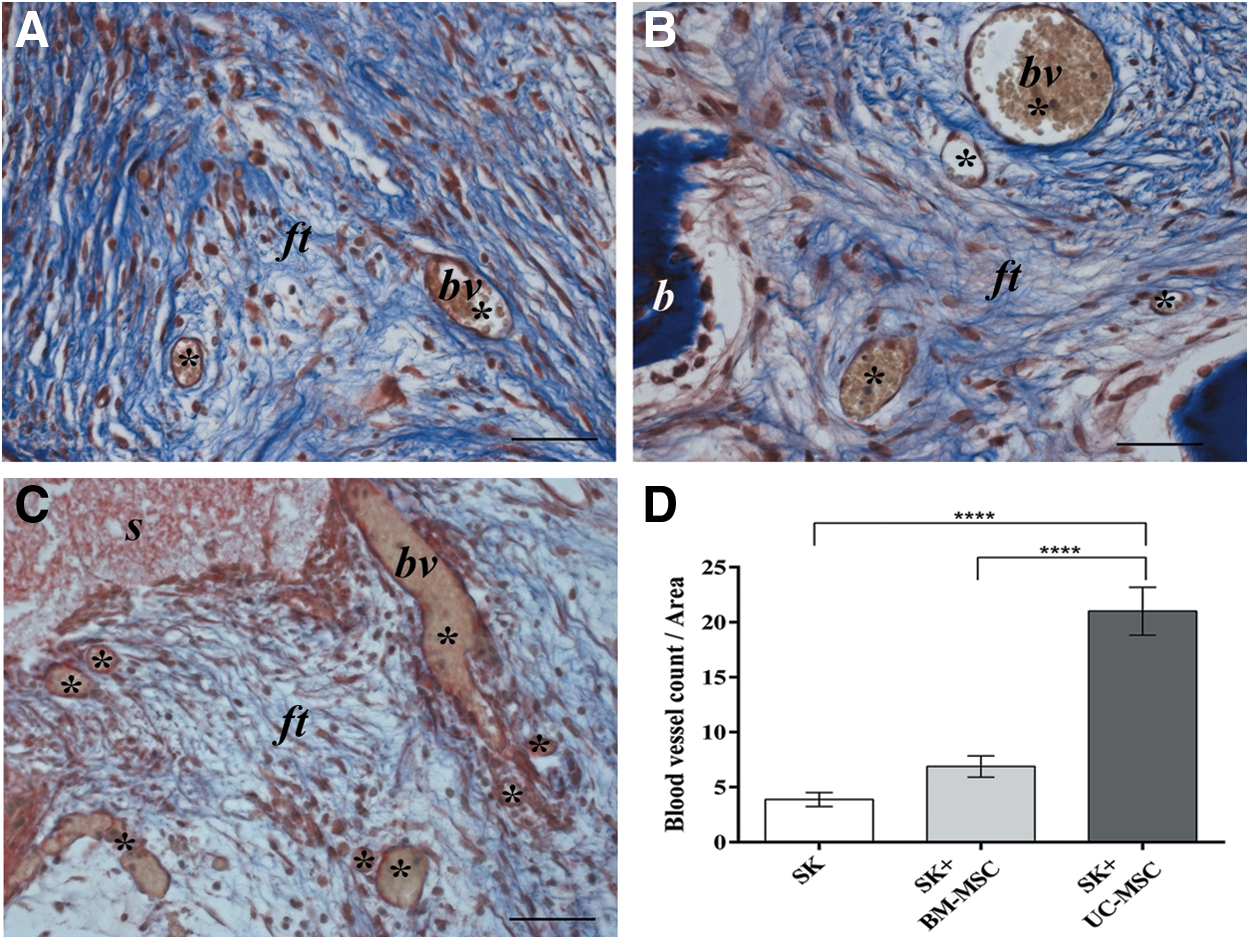
Histological images of ectopic implants recovered after 2 months and stained with Masson's trichrome showing a high number of blood vessel-like structures in the UC-MSC-seeded scaffold
In situ hybridization analysis showed that there was no positive signal for human ALU repeat sequences detected in any area of the 2-month UC-MSC implants, including the fibroblasts forming the compact fibrous tissue and the endothelial cells lining the vessel structures. Therefore, based on this observation, we classified the immature bone-like structure (Fig. 3E, F) and the neoformed vessels of murine origin (Fig. 3G, H). On the contrary, the mature bone formed in BM-MSC implants was undoubtedly of human origin. Cells derived from implanted human BM-MSCs were evident both in the newly formed bone as osteoblasts and osteocytes and in the fibrous tissue as fibroblast-like cells (Fig. 3C, D). As expected, no signal for ALU sequences was detected in the nonseeded implants (Fig. 3A, B).
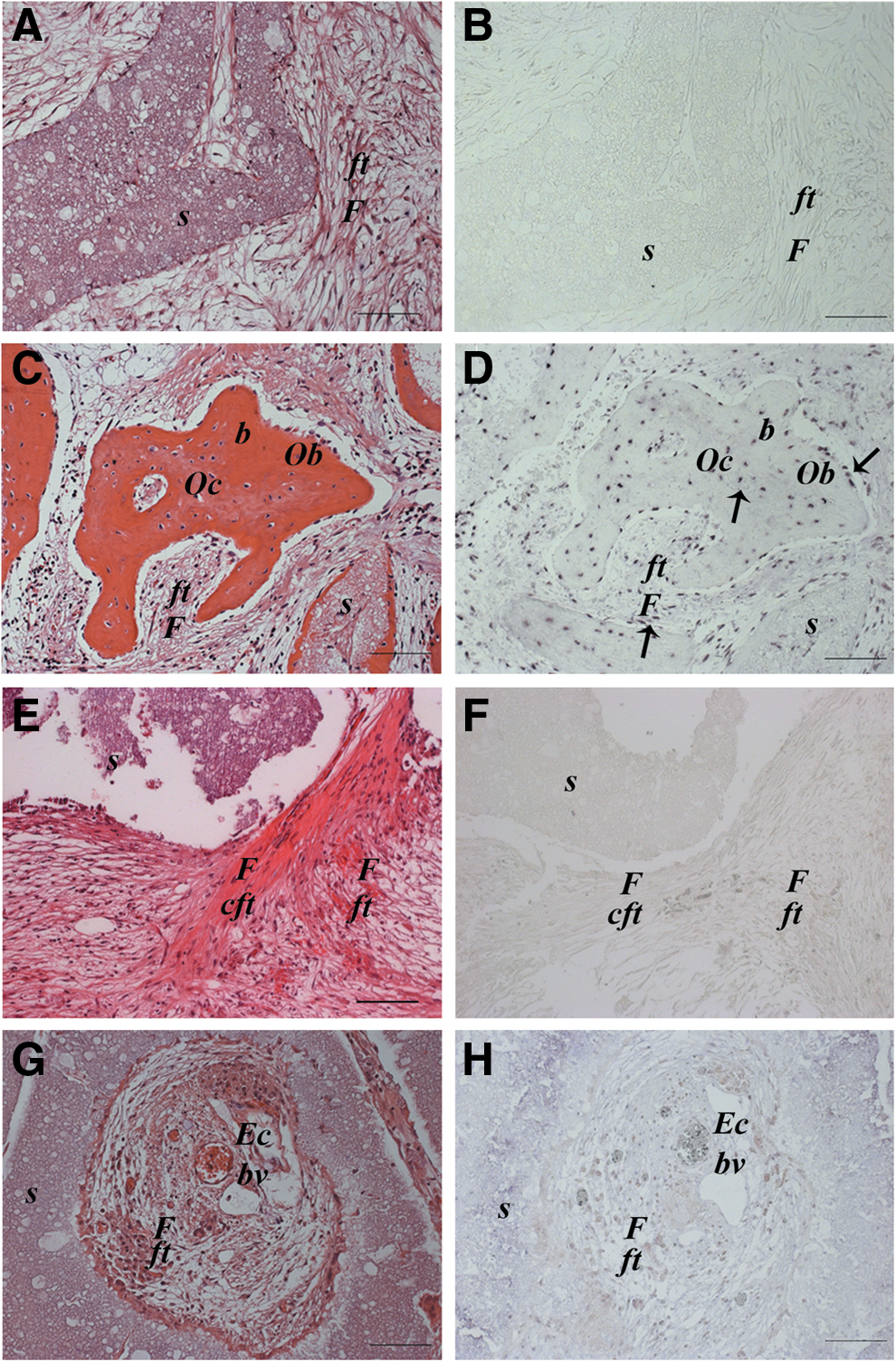
Histological images of ectopic implants recovered after 2 months and stained with hematoxylin and eosin (
In vivo permanence of UC-MSCs
In light of the previous evidence, we performed live cell-tracking analysis by detecting the light signal produced by transduced MSCs. The bioluminescent signal produced by UC-MSCs drastically decreased over time and completely disappeared by 1 month (Fig. 4B). On the contrary, the BM-MSC bioluminescent signal remained detectable throughout the entire testing period (Fig. 4A).
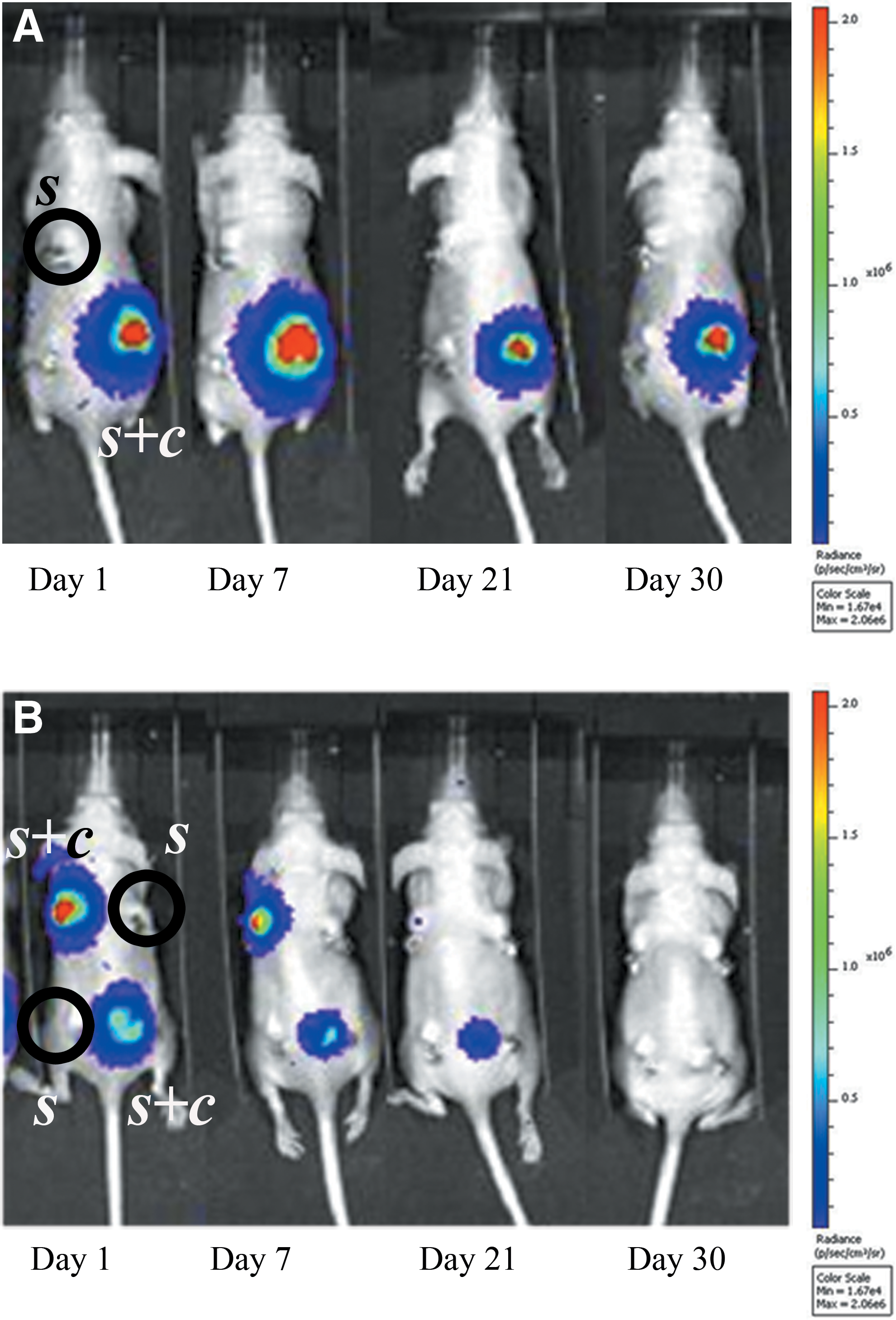
In vivo bioluminescence imaging, at different time points after surgery, of mice ectopically implanted with scaffolds seeded with cells (s+c) or not seeded (s). A decreasing signal is noticeable for UC-MSC-seeded scaffolds
UC-MSC secretome induces angiogenesis in vitro
In light of the previous data, we investigated whether the UC-MSC secretome (CM) alone could induce angiogenesis in a functional in vitro assay. Indeed, a widespread sprouting of blood vessels from fetal bone was induced by UC-MSC CM (Fig. 5C). On the contrary, BM-MSC CM showed a poor effect on inducing the formation of new blood vessels and behaved similar to the fresh medium controls (CTR) (Fig. 5A, B). UC-MSC CM significantly enhanced the number of PECAM/CD31-stained cells (vs. CTR P<0.05; vs. CM BM-MSC P<0.05) (Fig. 5D) as well as the area of vessel outgrowth when compared with the other conditions (vs. CTR P<0.05; vs. CM BM-MSC P<0.05) (Fig. 5E).
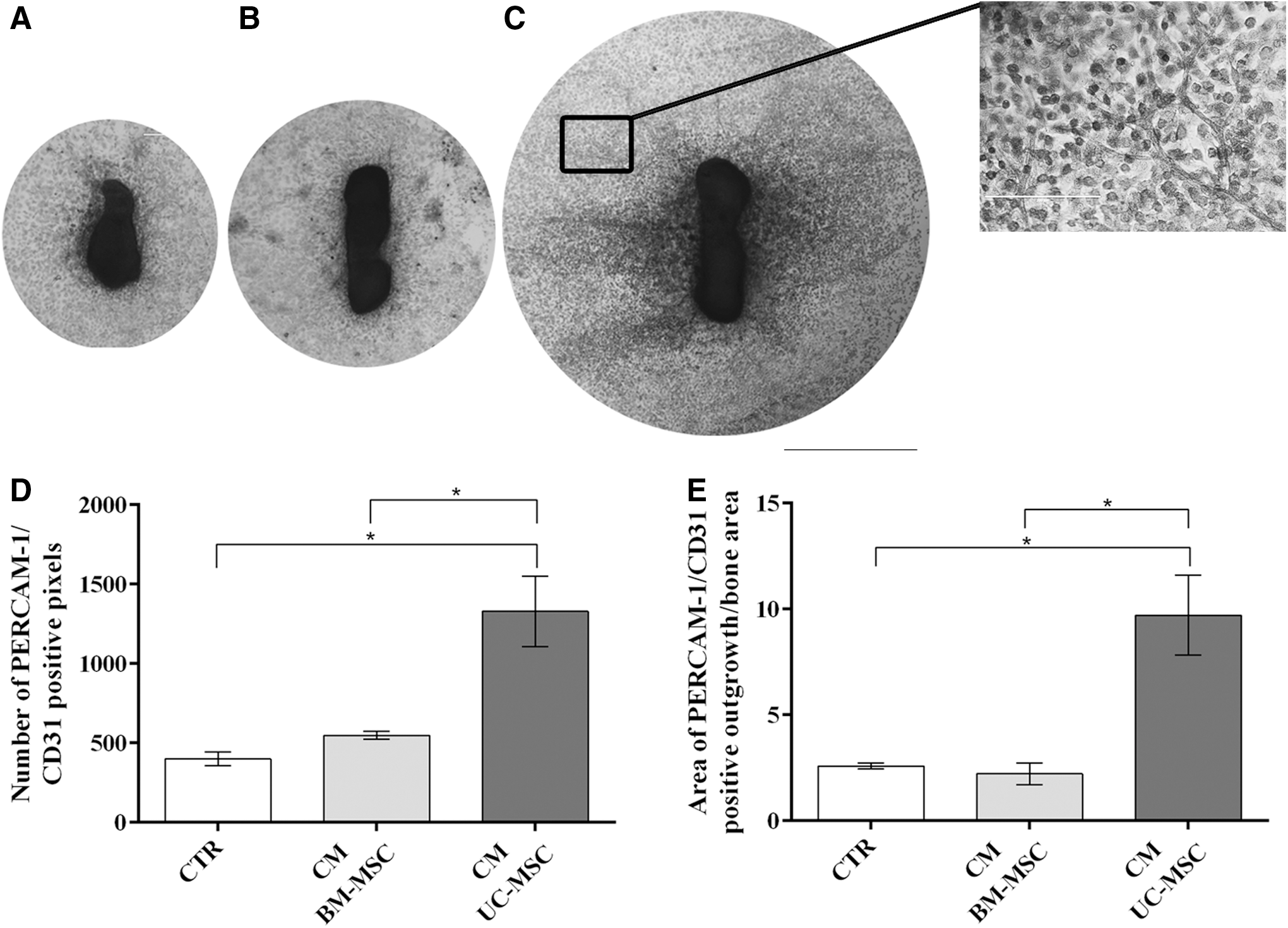
Blood vessel sprouting in explants of mouse fetal metatarsal bones maintained in different culture conditions. Vessels were stained with antibodies against PECAM-1/CD31. Explants were cultured in the presence of
UC-MSC secretome is rich in proinflammatory, proangiogenic, and chemotactic cytokines
We addressed the possible paracrine effects of UC-MSCs by investigating their secretome. Numerous cytokines were secreted by UC-MSCs (Fig. 6B). Most of the ones present in a high amount (IL-6, IL-8, CCL2, PAI-1, CXCL1, MIF, and G-CSF) (Fig. 6C) are known to be involved in the modulation of the inflammatory process. These cytokines were not secreted by BM-MSCs or their level in the CM was much lower than in UC-MSC CM. PAI-1 was the only cytokine secreted in a relatively significant amount by BM-MSCs in the same conditions (Fig. 6A, C).
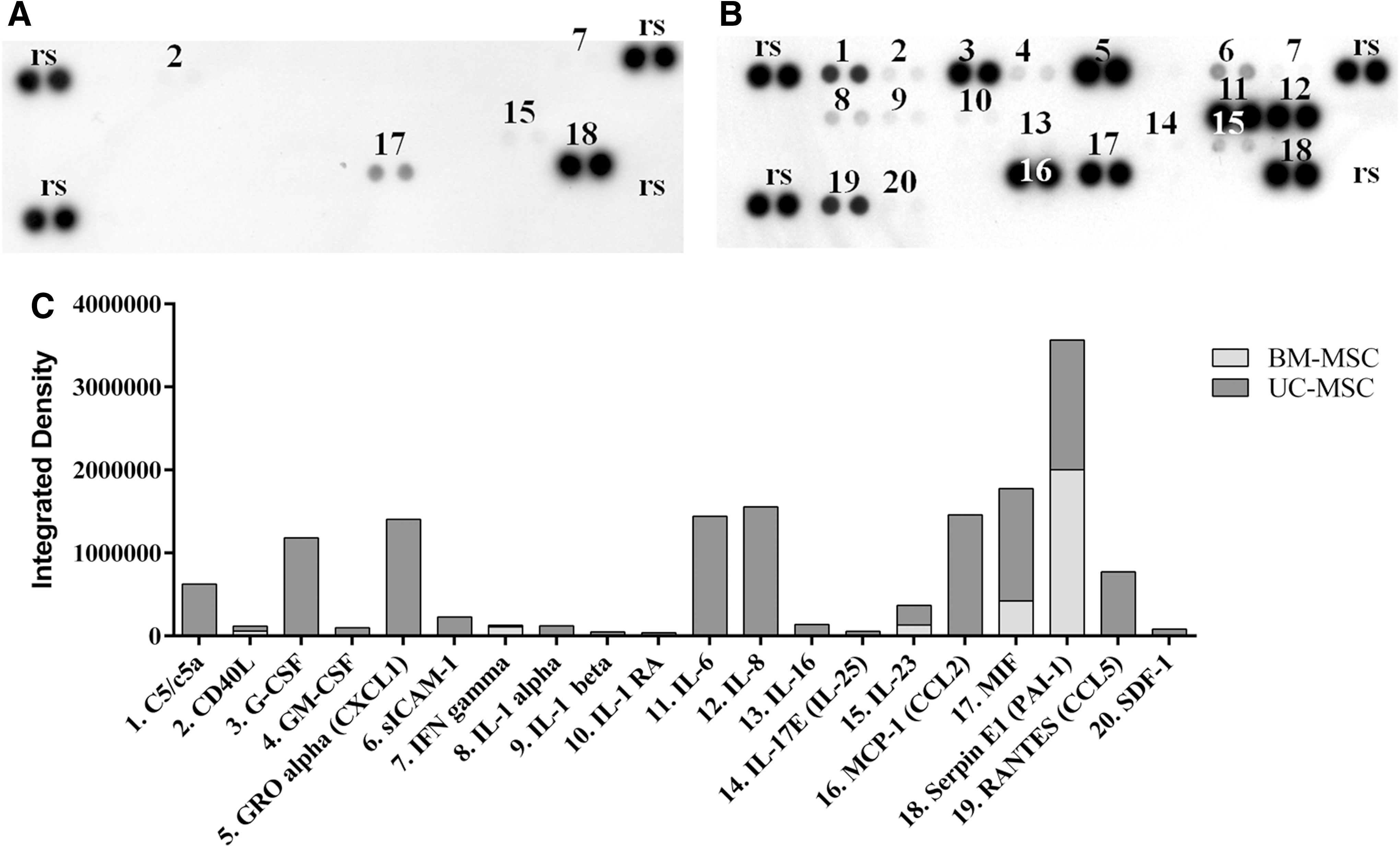
Cytokine array membrane assay of MSC CM;
Bioinformatic analysis performed in GO gave information about the biological processes involving the screened cytokines. The prevalent biological functions recorded concerned the activation of angiogenesis, cell signaling, cell proliferation, chemotaxis, tissue repair/regeneration, modulation of leukocytes, and inflammation. Each of these biological processes involved two to nine of the screened cytokines.
The first four biological processes that emerged by the analysis are strictly related to generic cytokine roles; therefore, we excluded the first four GO term scores (gray rows) from the evaluation (Table 1).
Biological processes identified by the cytokine clusterization (activation of angiogenesis, cell signaling, cell proliferation, chemotaxis, tissue repair/regeneration, modulation of leukocytes, and inflammation) involved from two to nine of the selected cytokines. The first four GO term scores (gray rows) were excluded.
GO, gene ontology.
UC-MSC-induced bone formation by the host cells in an orthotopic mouse model
Histological evaluation of the 90-day recovered orthotopic implants showed that none of the bone defects from any of the three groups had completely closed (Fig. 7A–C). However, abundant, newly formed mature bone tissue was observed in the defects that received BM-MSC-seeded scaffolds (Fig. 7B). Nevertheless, a good amount of mature bone was also observed in the defects filled with UC-MSC-seeded scaffolds (Fig. 7C). In the defects that received noncell-seeded scaffolds, only a small amount of new bone tissue was noted (Fig. 7A, D). An analysis using polarized light highlighted the presence of abundant bone tissue with highly organized fibers in BM-MSC-seeded implants (Fig. 7B). In UC-MSC-seeded implants, the polarized light revealed less extended and less defined fibers (Fig. 7C). A negligible signal was detected in nonseeded implants (Fig. 7A). There were no significant differences in the amounts of new bone filled between BM-MSC and UC-MSC groups (P=0.97). However, it was significantly higher in both groups with respect to the nonseeded controls (CTR vs. UC-MSC P<0.0001; CTR vs. BM-MSC P<0.001) (Fig. 7D).
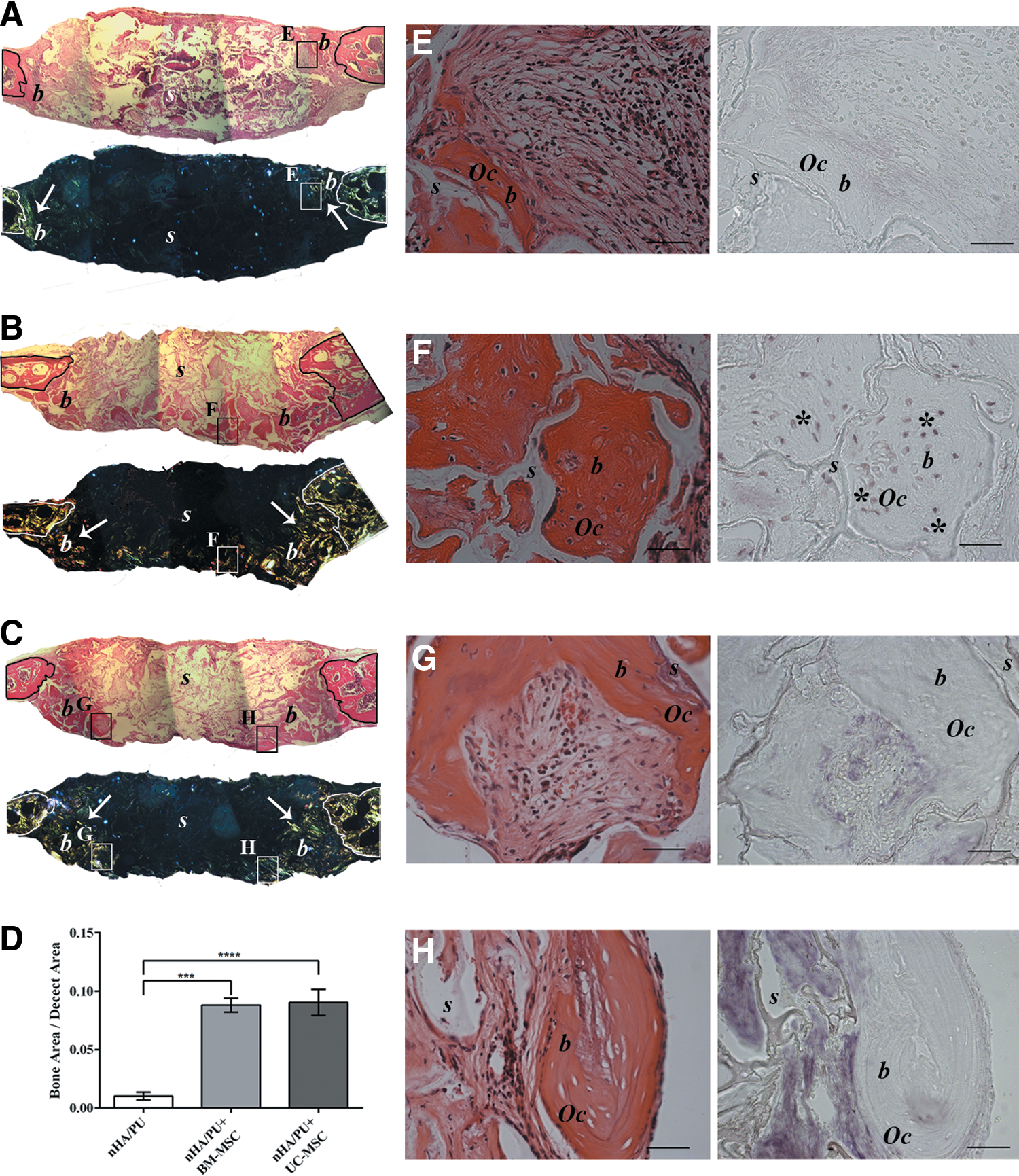
Histological images of orthotopic implants in mouse calvaria recovered after 3 months, stained with hematoxylin and eosin, and analyzed by polarized light at low magnification
To determine the origin of the regenerated bone, in situ hybridization analysis of human ALU sequences was performed. Similar to what was demonstrated in the ectopic implants, in the BM-MSC-seeded scaffolds, osteocytes of human origin were clearly detectable (Fig. 7F), indicating that the seeded MSCs remained in the scaffold for 3 months and successfully differentiated into viable osteoblasts that could lay down new bone matrix. On the contrary, we could not detect any signal for human ALU sequences in osteocytes within the newly formed bone in the UC-MSC implants, neither in the center (Fig. 7H) nor at the periphery (Fig. 7G) of the defect. This confirmed the murine origin of the regenerated tissue and again that UC-MSCs did not physically remain in the scaffolds. As expected, no signal was detected in the nonseeded implants (Fig. 7E).
Discussion
Despite several articles that have reported similar phenotypic characteristics for UC-MSCs and BM-MSCs, controversial data exist on their respective roles in the body's physiological tissue environments and their fate after in vitro expansion and in vivo implantation. In this work, we were mainly concerned with the osteogenic potential of in vitro-expanded UC-MSCs compared with BM-MSCs [30 –32].
To obtain the cells required for the comparison of the osteogenic potential of the two MSC populations, we relied on culture protocols authorized for use in cell therapy applications (
On the contrary, the classical expansion protocols for BM-MSCs, accepted for clinical use by the regulatory agencies [4], still employ FBS with FGF-2 as the medium additional supplement, which provides excellent results [37,38]. However, with regard to the present study, it should be noted that no major changes were observed in the phenotype characteristics and properties of BM-MSCs expanded in the presence of PL or FBS [34,36,39].
Although UC-MSCs are considered MSCs since they fulfill classical criteria [17], they do not appear as efficient as BM-MSCs for ectopic bone formation. UC-MSCs failed to deposit a well-developed bone structure, being limited to the promotion of the deposition of a dense collagen matrix. Similar results were obtained also by other research groups [32,40]. Furthermore, preinduction of UC-MSCs by osteogenic differentiation media, or BMP-2, did not significantly enhance their ossification potential in vivo [32,41]. The bone lack was even not influenced by the in vivo stay time (data not shown) or by the ceramic scaffold itself [42].
On the contrary, ectopically implanted BM-MSCs underwent osteogenic differentiation and deposited a true bone tissue. Interestingly, when orthotopically implanted in the mouse calvaria, both MSC populations promoted the formation of new bone; however, while bone was directly deposited by BM-MSCs, UC-MSCs did not directly deposit a bone matrix and instead triggered the recruitment of osteogenic host cells.
We showed by in situ hybridization and by flow cytometry (data not shown) that UC-MSCs remained in vivo at the implantation site only for a brief time. This observation is consistent with other published data where implanted human amniotic fluid stem cells were detected only for a few days within the implants [43]. Similarly, implanted UC-MSCs remained detectable in a skin lesion model only up to 11 days [44]. However, others have reported that ectopically implanted UC-MSCs could be detected in newly formed bone tissue after 8 weeks of implantation [45].
At variance with UC-MSCs, it is widely accepted that BM-MSCs are able to directly engraft, differentiate, and deposit new bone [40], although only 10% of the adult implanted BM-MSCs remain in the ectopic implants after 14 days [46].
Recent studies suggest that the benefits of MSC transplantation may be associated with a paracrine modulatory effect and not only with a direct engraftment potential [43,47]. In particular, fetal MSCs have gained much attention as tools capable of activating the endogenous repair mechanisms of the host by furnishing a burst of paracrine factors [44,46]. Such an effect has been demonstrated despite their rapid clearance from the site of implantation and confirms that their physical existence at the site throughout the entire time required for repair to take place is not necessary to ensure complete healing. Furthermore, it has been shown that the secretome of these cells alone is capable of bringing about the same response [23,48]. The degree of stemness or level of lineage commitment of an MSC population appeared to directly influence this phenomenon. UC-MSCs, expressing low levels of typical embryonic stem cell (ESC) markers, such as POUF1, Nanog, Sox-2, and Lin28 [49], show a gene profile closer to ESCs [50], whereas BM-MSCs express an osteogenic gene profile [24]. We observed that UC-MSC paracrine activity played a major role in the in vivo angiogenic effect elicited by implanted cells. The origin of the blood vessels was murine and no human cells differentiated into endothelial cells, indicating an indirect angiogenic effect rather than a direct differentiation of the cells. This was also confirmed by the fact that the UC-MSC secretome (CM) alone was capable of significantly inducing angiogenesis [49,51]. This effect was mediated only by UC-MSCs, excluding the possible role of endothelial progenitor contaminants as previously demonstrated [6].
Indeed, UC-MSCs have a secretome enriched with growth factors and cytokines related to angiogenesis, tissue repair, and wound healing processes when compared with BM-MSCs [52]. Many growth factors linked with angiogenesis were identified in the UC-MSC secretome, such as VEGF-D, PDGF-AA, TGF-β2, b-FGF, and HGF [53]. Interestingly, we also found that UC-MSCs secreted proinflammatory (CD40L, C5, and IL-6), proangiogenic (IL-6 and PAI-1), and chemotactic (CCL2, CCL5, IL-6, and IL-8) cytokines in high quantities [49,54]. IL-6 was recently associated with MSC pluripotency and immunoprivilege. The combination of BM-MSCs and IL-6 was shown to be much more effective in attenuating liver fibrosis than BM-MSCs alone [55]. These results suggest that UC-MSCs alone could be a more appropriate candidate for cell therapy since these cells secrete higher IL-6 levels [56].
Recent studies have highlighted the essential roles of proresolving M2 macrophages in tissue repair and regeneration [57]. We showed that UC-MSC secretome was enriched in M2 inducers, GM-CSF [58], CCL2, and IL-6 [59], thus suggesting a crucial role of the UC-MSC secretome in activating M2 proresolving macrophages, thereby modulating the wound healing process [26].
Despite their poor osteogenic capability when implanted ectopically, UC-MSCs were able to sustain the formation of new bone matrix in an orthotopic model in an amount comparable with that of implanted BM-MSCs [30,31]. It remains unclear if the in vivo paracrine effects of UC-MSCs are merely mediated by the UC-MSC secretome itself or also by an intercellular cross talk between the exogenous stem cells and the host endogenous progenitors; a new concept recently hypothesized in the literature [25].
In summary, the therapeutic potential of UC-MSCs appears to be vast and diverse and we are just beginning to discover their true potential. Although additional experiments are required to investigate further UC-MSC secretome-mediated angiogenesis in vivo, it can already be deduced from our study that UC-MSCs could have an effective potential use as an angiogenic tool. This is in addition to the immunomodulating role of the UC-MSC secretome making these cells, and/or their secretome, attractive therapeutic candidates for treatment of chronic wounds such as diabetic ulcers or nonhealing fractures.
Footnotes
Acknowledgments
The authors would like to thank Dr. Michele Cilli for the surgical assistance and Dr. Mauro Alini for providing the poly (ester urethane) scaffold. This work was partially supported by funds from Regione Liguria, Italy (POR-FESR 2007–2013).
Author Disclosure Statement
No competing financial interests exist.