
Abstract
There is great need to develop more predictive drug discovery tools to identify new therapies to treat diseases of the central nervous system (CNS). Current nonpluripotent stem cell-based models often utilize non-CNS immortalized cell lines and do not enable the development of personalized models of disease. In this review, we discuss why in vitro models are necessary for translational research and outline the unique advantages of induced pluripotent stem cell (iPSC)-based models over those of current systems. We suggest that iPSC-based models can be patient specific and isogenic lines can be differentiated into many neural cell types for detailed comparisons. iPSC-derived cells can be combined to form small organoids, or large panels of lines can be developed that enable new forms of analysis. iPSC and embryonic stem cell-derived cells can be readily engineered to develop reporters for lineage studies or mechanism of action experiments further extending the utility of iPSC-based systems. We conclude by describing novel technologies that include strategies for the development of diversity panels, novel genomic engineering tools, new three-dimensional organoid systems, and modified high-content screens that may bring toxicology into the 21st century. The strategic integration of these technologies with the advantages of iPSC-derived cell technology, we believe, will be a paradigm shift for toxicology and drug discovery efforts.
Disease Modeling
D
Developing rapid and effective therapies for CNS diseases requires the availability of in vitro models that accurately recapitulate disease phenotypes and predict patient treatment response. A proper model must be both sensitive and predictive while reflecting both normal and disease processes. Equally important, these models should enable the investigation of genetic and environmental risk factors contributing to diseases in a rapid and economical way. Currently used models often do not reflect a typical human response [2 –4], despite efforts underway to better characterize these models and increase their preclinical value in predicting safety and efficacy in the clinic [5,6]. Therefore, there is a great need to develop disease- and patient-specific models from cells directly affected in CNS disorders. These cell-based models, we envision, could either replace or supplement current animal models and enable the efficient translation of basic research into the clinical setting.
Limitations with current CNS models
Currently, drug discovery relies on the use of animal-based or cell-based models, which are not human or disease specific. This has limited the translation of the target to the clinic [4]. Screening systems using different species, such as worm, fruit fly, and zebrafish, have proven extremely useful for basic science insights and, on occasion, repurposing previously approved drugs from the Food and Drug Administration [7 –9]. This is because, in some instances, these models have enabled high-throughput relatively inexpensive screening whose utility can be extended by genomic engineering methodology [7]. Indeed, Drosophila-based models, for example, were used to identify therapies for Fragile X. However, in many instances, the results are species specific and many of the in vivo models are not truly amenable to high-throughput screening or, in some of these species, cell lines and in vitro analogues are simply not available.
The issue of results being species specific is of importance to in vitro assays as well, and the lack of fidelity of rodent results with human results in ALS has been well reviewed [10]. Even in vivo models such as genetically engineered mice do not always faithfully model CNS disorders. Although navigating current limitations with in vivo models can be achieved in some regard by combining different model systems, it adds an added level of uncertainty to the results. Both the in vitro and in vivo models suffer from an additional limitation, which is the issue of assessing allelic variability. Mouse models, which have a different phenotype in different strains, have been described, and it is reasonable to assume that predicting human response to the effects of a drug in a single inbred mouse strain may be difficult.
Thus, models may suffer from one or many deficiencies and these are summarized in Table 1. These limitations include limited supply, lack of patient/disease specificity, and, in the case of tumor cells, genomic changes that may make them very dissimilar from a physiologically normal neural cell. Additional limitations may include not recapitulating the appropriate in vivo disease phenotypes or not expressing appropriate cell-specific metabolic enzymes for drug metabolic studies (eg, lack of cytochrome P450 family). For additional considerations for cell-based models, readers are referred to this comprehensive review on cell-based models for pharmacogenomic discovery [11].
Modeling disease using human cells in general and PSC-derived cells in particular
There is considerable evidence that the current approaches to drug development used by the pharmaceutical industry are not translating to new therapies and that using primary cells in in vitro scalable models might yield better results [12 –16]. One can imagine developing disease models using human cells that would be translatable into high-throughput, in vitro drug screens that are rapid, cost effective, and more accurately reflect human cellular responses [17]. Primary human cell models, likewise, could be used to identify new compounds or repurpose approved medications, as well as provide accurate tissue-specific toxicology endpoints. While this is attractive, it is important to note, however, that using any type of human cell is not sufficient. Cells lines or transformed cells may have undergone changes during culture that reduce the fidelity of their response, thereby making them unreliable predictors of drug activity (Table 1). A suggested improved approach would be to use primary CNS cells that are disease-stage, patient-type, and cell-type specific. While this was possible in some systems, this has been virtually impossible in the CNS as CNS cells rarely divide and tumor lines were unstable in culture. Thus, for example, in PD, PC12 cells, which are a peripheral nervous system line, have been used by some groups as a surrogate for virtually impossible to obtain CNS neurons [18,19].
The recent development of induced pluripotent stem cell (iPSC) and embryonic stem cell (ESC)-derived cells could theoretically enable this improved approach as multiple groups have shown that CNS derivatives can be derived from these cell populations [20,21]. One can envision using iPSC-derived cells to make these disease models both cell-type and patient-specific lines so that the models express the cellular responses while also incorporating the patient's unique genetic profile. Although human ESCs offer many of these same advantages, they are harder to obtain from a technical standpoint and also have ethical issues associated with their use.
It is perhaps worthwhile for industry to evaluate an iPSC-based approach in their drug discovery process for screening large compound libraries given recent documented successes [22 –26]. Zhao et al. provide a noteworthy example supporting iPSC-based screens for novel drug discovery [27]. They developed a high-throughput compatible Wnt/beta-catenin signaling reporter system using neural progenitor cells derived from human iPSCs. This screening system was then able to survey ∼1,500 from an approved Food and Drug Administration library to identify novel small-molecule candidates that target Wnt/beta-catenin signaling. Suffice to say, an iPSC-based approach for screening incorporates disease, cellular, and patient specificity into high-throughput screening in a manner previously not possible [28,29]. Control, disease-specific, and engineered iPSCs and their secondary derivatives could be used in numerous types of screens, including large-scale small molecules, mechanism of action, repurposing of FDA-approved drugs, toxicity-related, high-content, and genomic types [30,31]. Each of these screens utilizes distinct screening agents, including siRNAs, natural compounds, biologics, small molecules, and even FDA-approved drug libraries [27]. These can further include measurement approaches (Fig. 1), such as label-free screening, sophisticated functional readouts, promoter–reporter strategies, and high-content screens, which will improve both the sensitivity and biological relevance of these iPSC-based models. Collectively, these advantages have the potential for improving the drug discovery process by enabling a more efficient and effective screening tool to deliver the cures of tomorrow based on cellular-, disease-, and patient-specific iPSC-based models.

Screening opportunities for induced pluripotent stem cells (iPSCs). Illustrated in this figure is a summary of the opportunities that iPSCs have in screening. Cell sources for deriving iPSCs are numerous and include skin, hair, dental tissue, urine, blood, and postmortem brain tissue. Cell groups can consist of controls, panels, ethnic diversity, and adverse events. These cells could be further modified using engineering approaches or differentiating the iPSCs to specific cellular lineages. Then, these cells could be placed in any number of screens (eg, repurposing screens) where various screening agents could be used and, likewise, numerous output measurements could be made to provide inferences on how a cell responds to a given agent. In addition, numerous cell labels can be used to provide quantitative data for cellular responses.
Although PSC-derived cells are attractive, one has to be careful about assuming that they are the best choice in every instance. For example, many neurodegenerative disorders are often associated with aging where neurons differentiated from patients with several of these diseases do not reproduce the same degenerative phenotype [32 –34]. This is likely to be the result of iPSCs being reset to a fetal developmental profile and they need to mature appropriately. This prolonged period in culture may vitiate their utility. The process of iPSC generation may introduce changes to the very systems one wants to study, and while developing models with patient-specific samples is attractive, the cost associated with screening in multiple lines and assessing the effect of a change in lines with wide allelic variability may make the potential utility of the lines moot. Furthermore, iPSCs are not immune to genetic and epigenetic changes that can occur during culturing. It is also important to note that it is still either difficult or not possible to differentiate iPSCs to some neural cell types (oligodendrocytes and a myelination model, for example) and there is a need to harmonize protocols, which successfully differentiate to specific lineages, to make it easier to compare results in various disease models across research groups [35].
Some progress has been made in addressing these issues. For example, this issue of accelerated maturation has been partially resolved by mimicking aging in iPSCs by overexpressing progerin [36]. This process introduces an added variable and is not applicable to all cell types. Genomic imprinting, whereby epigenetic modifications in the DNA (eg, DNA methylation or histone modification) can influence gene expression without altering the genomic sequence, may also prove difficult to model in some diseases using an iPSC-based approach. For example, in Fragile X syndrome (FX), there are distinct differences from FX-ES and FX-iPSCs with regard to the expression of the FMR1 gene where it is expressed in FX-ES and inactive in FX-iPSCs [37]. Therefore, modeling imprinting using an iPSC-based model may not be possible for FX.
However, there are reports illustrating that imprinting is maintained following iPSC reprogramming in diseases such as in Prader-Willi and Angelman syndrome [38]. Finally, modeling diseases, in which the mitochondria are affected, particularly those resulting from mutations in the mitochondrial genome, could be challenging as this genome is much more susceptible to mutation and may be more likely to succumb to mutational events during the process of iPSC reprogramming [39 –43]. In summary, PSC-derived primary cells for modeling are no panacea and these limitations should be taken into account in developing ESC or iPSC-based models.
How iPSCs Can Enhance the Utility of In Vitro Models of Disease
Despite the limitations discussed above. ESCs (identified through preimplantion genetic diagnosis), and iPSCs in particular [44 –46], can resolve some of the limitations mentioned in the previous section by their ability to be disease and patient specific, the decrease in the cost of deriving such lines, and the concerted effort to make well-characterized lines widely available. Since iPSCs can be differentiated into most cell types (or cocultures consisting of multiple cell types) to investigate healthy and disease processes in a cell-type-specific manner, one can assess the effect of a gene or an allelic background in thoughtfully selected mix-and-match experiments (see eg, Xu et al. [47]).
Table 2 illustrates several types of iPSC-based physiological models, in which iPSCs may be used to recapitulate neural cell interactions with other cells and their environment. Several functional interactions can be assessed [48 –54]; these include modeling (1) blood–brain barrier, (2) synaptic networks, (3) neuron–astrocyte interactions, (4) oligodendrocyte myelination, (5) peripheral myelination, (6) retinal processes, (7) developmental processes, and (8) immunological interactions [55 –58]. With the advent of numerous protocols for deriving iPSCs from multiple patient-derived cell sources, academic and industry partnerships should be used to derive large and comprehensive repositories for iPSCs from well-characterized sibling controls and patients suffering from a range of CNS indications. These repositories will supply the field with much needed control, engineered, and disease-specific iPSC lines that will permit easier entry into the field to investigate disease mechanisms and develop screening assays for drug discovery and toxicology applications.
Each of these cell types can be generated from iPSC-derived normal or affected individuals to assess various physiological models, including electrophysiological function, myelination, trophic support by astrocytes, eye function, neural cell migration, and neuroimmunological interactions. For details, please see text.
iPSC, induced pluripotent stem cell.
Modeling rare CNS diseases with iPSCs
While there are limitations and roadblocks to consider, modeling rare CNS diseases using an iPSC-based approach offers many unique advantages. While individual rare diseases are uncommon in their totality, diseases in this classification affect ∼25 million people in North America alone and pose a significant burden on society [59]. iPSCs offer an excellent opportunity to model these diseases and identify therapeutic options. As there are a limited number of patients with rare disorders, using an iPSC-based approach provides the opportunity to generate panels of iPSCs for each rare disease and thereby enables many investigators to interrogate disease mechanisms with the same models. Another advantage of rare diseases in the context of iPSCs is that they are often monogenic (single gene mutation) and therefore amenable to genomic engineering. Therefore, isogenic controls (controls having the same genetics as the diseased sample minus the disease mutation) can be created for many of these rare diseases to enable more precise disease modeling by removing secondary genetic effects from mixed genetic backgrounds.
This strategy has been employed for Niemann-Pick disease, type C1, a rare disorder resulting from lysosomal cholesterol accumulation to identify a compound that can correct the disorder in iPSC-derived neurons and hepatic cells [60]. Another study found that a chemical screen could identify a compound reversing the effects of the rare disease, ataxia-telangiectasia, in neural cells derived from patient iPSCs [61]. An additional benefit of modeling rare diseases with iPSCs is that the identification of the mechanisms of action in these disorders may facilitate the development of new therapeutics and the elucidation of fundamental processes such as survival and proliferation, which may be applied to other neurodegenerative diseases. Table 3 highlights some of the work currently being done at the National Institutes of Health (NIH) on rare diseases using an iPSC-based approach.
BEST, Best's vitelliform macular dystrophy; CNS, central nervous system; GD, Gaucher disease; LCA, Leber congenital amaurosis; L-ORDS, late-onset retinal degeneration; NIH, National Institutes of Health; NIH CRM, NIH Center for Regenerative Medicine; NPC1, Niemann-Pick disease type C1; SLOS, Smith-Lemli-Opitz syndrome.
New Technologies May Further Enhance the Utility of iPSC-Based Screens
In this section, we discuss how various novel technologies (summarized in Fig. 2) have been incorporated with iPSC-based screening methodology to develop advanced iPSC-based model systems.
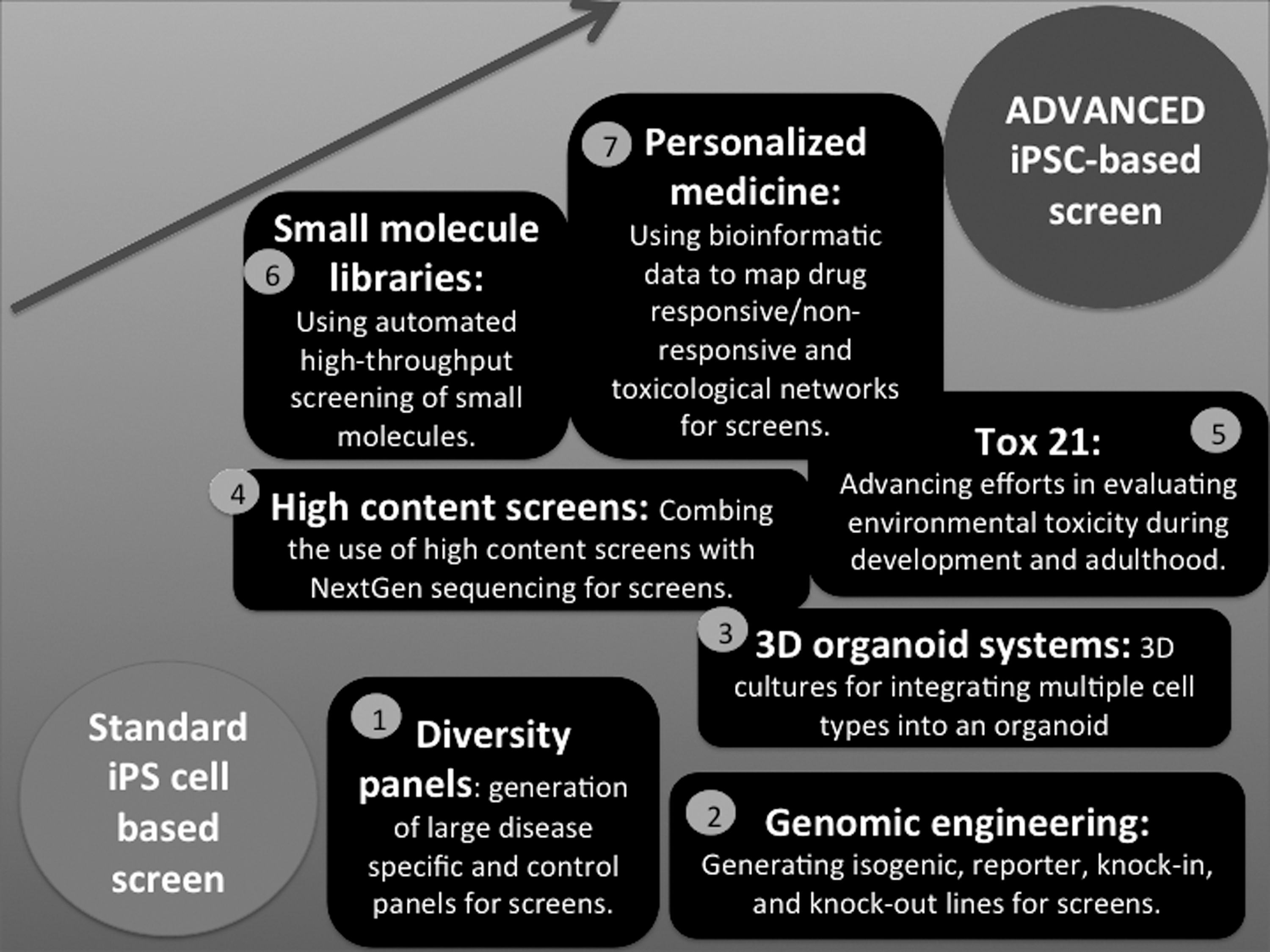
Integrating seven technologies for advancing iPSC-based screening. Standard iPSC-based assays for screening can be further improved leading to advanced iPSC-based screens by integrating technologies that serve as building blocks to make incremental improvements upon current screening assays. The figure summarizes several technologies that will lead more complex iPSC-based screens that will result in exponential advances: (1) ability to develop diversity panels, (2) modern genomic engineering tools, (3) novel 3D organoid systems, (4) high-content screens combined with NextGen sequencing, (5) Tox 21-type large-scale programs with associated database, (6) focused and novel small-molecule libraries, and (7) ability to rapidly combine patient data with in vitro IPSC-based screening to develop personalized drug cocktails for cancer therapy.
Diversity panels
iPSC-based screens may be a solution for accounting for allelic variability illustrated in pharmacogenomics [62]. The problem, simply stated, is that humans are outbred and the allelic variability reflected in gene expression and variation is comparable across species. This has been the basis for reluctance in using primary cells as there is uncertainty whether the response from any given line is representative of a large population or peculiar to a specific subpopulation. In this context, iPSCs afford the ability to generate numerous lines while covering the entire allelic spectrum across a disease. That is, this approach provides immortalized cells without the typical disadvantages. Already, disease-specific panels incorporating sporadic and familial cases from patients with the same disease have been obtained using a standardized methodology and permit high-throughput screening to identify unique and uniform responses and dissect stochastic differences from biologically relevant ones [63,64]. For instance, panels for Parkinson's disease and Huntington's disease are already being developed and are available through the National Institute of Neurological Disorders and Stroke (NINDS) repository. These patient diversity panels along with others in development will prove instrumental in lowering the attrition rates for phase II and III clinical trials [38] by providing a layer of sophistication to cell-based assays for drug safety and efficacy that reflects patient diversity and can identify subgroups (eg, responders vs. nonresponders) critical for success in the clinic. The ability to use genomic engineering technologies to further modify these iPSC lines offers another method to better understand diseases through modeling.
Genomic engineering
Genomic engineering offers up numerous opportunities for engineering iPSCs and their derivatives to use in both screening and cell therapy approaches. For instance, genome engineering has been used in numerous preclinical Alzheimer's disease mouse models to modify neural stem cells before transplantation to make them more therapeutically effective by targeting disease pathologies [65]. There are numerous methods for genomic engineering, including zinc finger nucleases, transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR/Cas9). Interested readers are referred to this review on genome-editing technologies [66]. Figure 3 illustrates genome engineering of iPSCs using some of these methods where these engineered lines can either be differentiated into various cell types for research purposes or propagated and banked for future experiments.
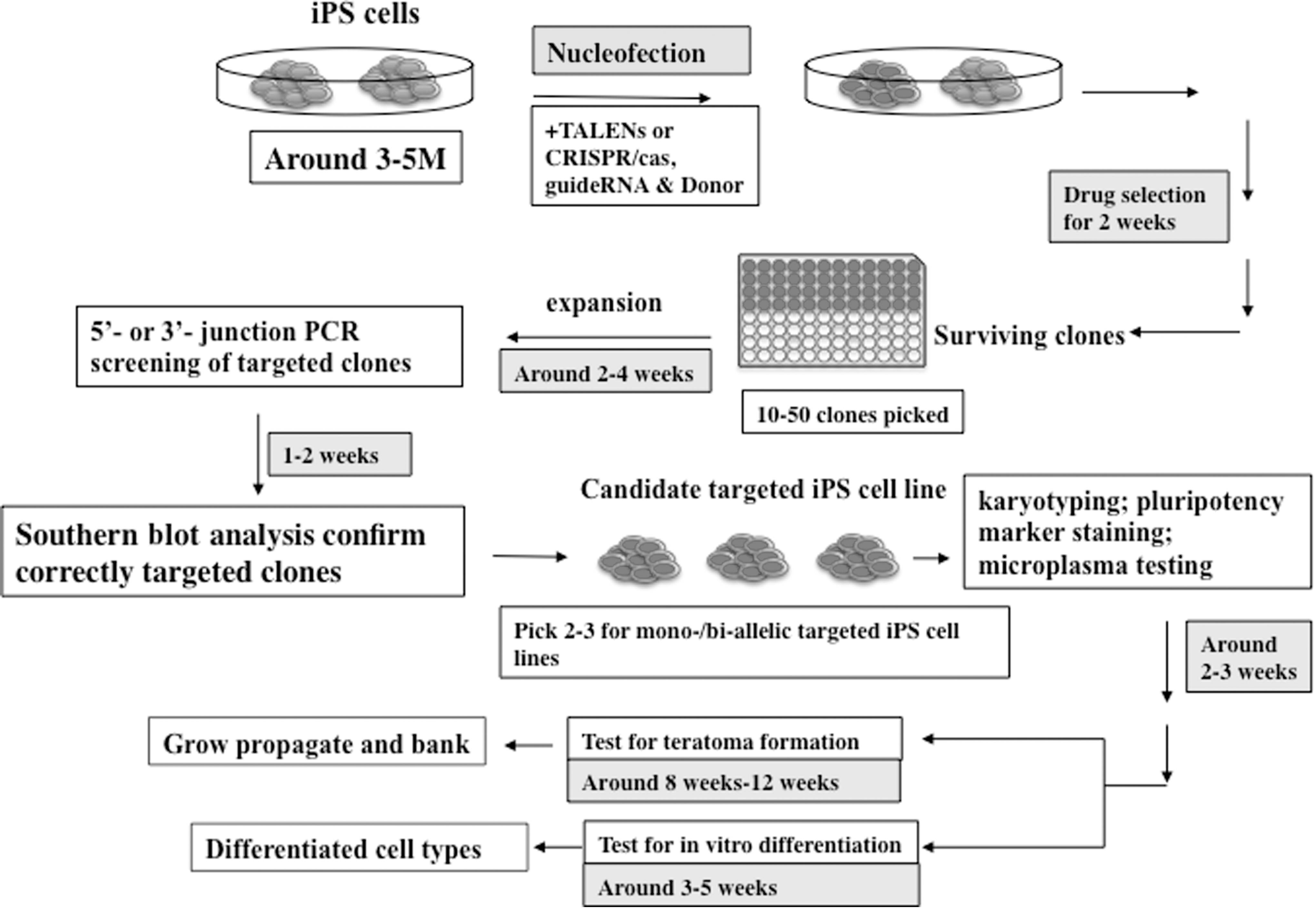
Genome engineering of iPSCs. The process for engineering iPSCs is shown above where iPSCs are reprogrammed (3–5 months), then through nucleofection, the transcription activator-like effector nucleases (TALEN) or CRISPR/cas, etc., and donor are inserted into the iPSCs, and iPSCs that are successfully integrated are selected through drug selection. The surviving clones (∼10–50 clones picked) are expanded over 2–4 weeks where PCR is used to screen for mutations followed by Southern blot analysis to confirm correctly targeted clones. Then, two to three targeted iPSC lines are picked and then assayed for pluripotency, mycoplasma contamination, and correct karyotype. The iPSC lines that pass this assessment are then further assayed for teratoma formation and in vitro differentiation. Finally, these engineered iPSC lines can either be differentiated into various cell types for research purposes or propagated and banked for future experiments. Contributed by Dr. Jizhong Zou, NIH Center for Regenerative Medicine (NIH CRM), National Institutes of Health (NIH).
Therefore, genomically engineered iPSCs offer up tremendous opportunities for modifying current assays to be more sensitive, specific, and versatile for high-throughput screening and other applications. For instance, a recent report developed a high-throughput assay in human cells using a CRISPR/cas-based method to knock out genes systematically to determine their function in bacterial toxicity [67]. Additionally, CRISPR/cas and TALENS have been shown to have low rates of off-targeting following whole-genome sequencing of their modified iPSCs [68]. Genomic engineering also offers the ability to create lineage-specific reporter lines for identifying differentiation-inducing compounds or compounds that target specific pathway reporters, which are not possible using terminally differentiated cell lines. In addition to genomic engineering, creating three-dimensional (3D) organoid systems will enable numerous advantages such as allowing for the formation of more complex structures and cell interactions, allowing for increased functionality.
Three-dimensional organoid systems
Developing culture systems that can integrate multiple cell types into a complex organoid structure represents additional advances for disease modeling and screening. The ongoing development of 3D organoid culture models that better recapitulate human physiology holds great promise for the future of disease modeling and drug screening (Fig. 4). Culturing cells in 3D allows for a microenvironment that supports the formation of cell–cell interactions and cell–extracellular matrix interactions and also readily supports the addition of multiple cell types, allowing for the formation of more complex organoids and more accurate physiological responses than in two-dimensional culture [69 –71].
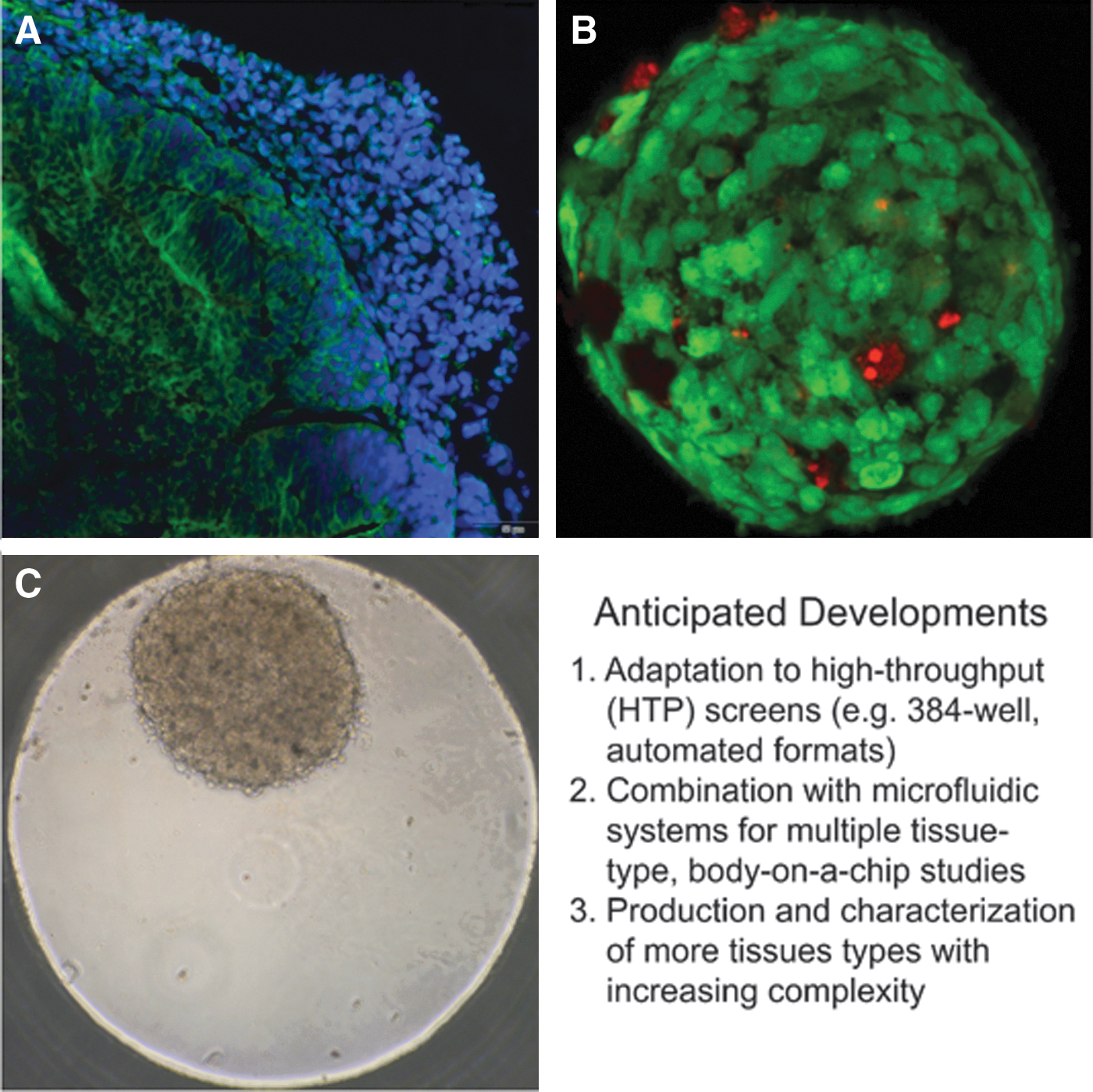
Examples of 3D organoids designed to recapitulate human cerebral, hepatic, and cardiac tissues, important targets for drug toxicity testing.
Culture of iPSC-derived differentiated cells in 3D organoid cultures will mimic the in vivo environment more faithfully, particularly in the context of the CNS, and thereby allow the disease model to offer a more accurate picture of the pathologies resulting from the disease state. While technological advancements are needed before this system can realize its potential in high-throughput applications, it has been effective in some lower throughput screening studies. Three-dimensional spheroid cultures using a Nanopillar Plate have already been used for drug toxicity using hepatocyte-like cells derived from human ESCs and human iPSCs [72]. Another example of developing 3D organoid systems for modeling is demonstrated by developing human gastric organoids (hGOs) [73]. With no current experimental model of normal human gastric mucosa, hGOs can be used to model both human stomach development and disease. The formation of the optic cup from human ESC self-assembles in a dish and could be used to investigate retinal diseases and potential therapies [74]. Cerebral organoids have also been developed to model developmental processes in the brain using human pluripotent stem cells [75]. These investigators also used the approach to model microcephaly using RNA interference paired with patient-specific iPSCs. Focusing on Alzheimer's disease, a 3D culture approach has also recently been applied to a neural cell culture model, which accurately recapitulated neuropathological hallmarks of the disease [76]. Moreover, these types of 3D culture models should be further expandable and applicable to many other CNS diseases and used for both toxicology and drug discovery in addition to investigating developmental processes that may be critical contributors to both healthy and disease statuses. Moreover, advances in genomic engineering have also recently been applied to 3D organoid systems where this opens up the possibility of introducing genetic manipulations into these systems [77]. Another consideration is the use of high-content screens with NextGen sequencing.
High-content screens with NextGen sequencing
High-content screens include any multiparameter methodology used to analyze cells or cellular components. Advances in DNA and RNA sequencing (termed next-generation or NextGen sequencing) have now made it possible to quickly sequence billions of nucleotides at a relatively low cost [78,79]. Combining high-content screening and NextGen sequencing in panels of iPSCs allows large-scale analysis of both cellular phenotypes paired with gene expression and the epigenome to evaluate the drug response over a diverse panel. This paradigm could be realized in a high-throughput approach to first identify classes of drugs that are effective, then segregate patients who may be responders or nonresponders, and finally analyze the entire genome profile to determine the underlying basis for the difference. This is now possible once one generates the appropriate diversity panels and avails all of the sequencing advances to develop an appropriate database. More ambitious proposals have suggested that a similar panel-based approach can be successfully used for polygenic disorders, provided large panels are obtained and screening is combined with whole-genome, exome, and epigenome sequencing. This is an ambitious idea, which, if successful, could completely change the way early drug discovery is tackled. In addition, transcriptional profiling can be a sensitive endpoint for developmental toxicity screens using ESCs or iPSCs to identify differences between differentiation states during development [80]. These developmental toxicity screens can provide data on the mechanism of action and also be scaled up. These high-content screens can be tested using well-defined small-molecule libraries.
Small-molecule libraries
Using iPSCs in high-throughput screening of small-molecule libraries represents both opportunities and challenges. Recent advances in technologies for conducting automated screening assays, along with improvements of detection methods, are making screening of large collections (eg, one million compounds) using complex cell-based protocols a routine undertaking. Using fully automated robotic systems in combination with miniaturization to 1,536-well density, an organization can complete a one million-compound screen within the span of 5–10 days. Such streamlining of screening processes has driven further innovation in the field as in the example of quantitative high-throughput screening (qHTS), dubbed qHTS [81], where each compound in the collection is tested as a dilution series spanning the concentration range from single-digit nanomolar to double-digit micromolar, essentially producing a concentration–response curve for each molecule tested. By doing so, a significantly lower rate of false positives and false negatives is encountered along with the ability to derive nascent structure–activity relationships directly out of the primary screen.
Such advances in the scale and speed of screening, however, require that the system being tested, that is, the assay, be stable and the materials required for the large-scale experiment be available at said scale. Inevitably, this produces challenges for iPSC-derived cellular assays screened against large compound collections. Two primary challenges are that of scalability and cost. For example, ∼50 million cells are needed to screen a library of 5,000 known drugs and tool compounds, and 2 to 10 billion cells are needed for a 400,000-compound screen. Furthermore, the necessary breakdown of cell production into multiple batches leads to poor reproducibility of differentiated cells as expressed in day-to-day variations of cell morphology and properties, as well as variation of cell yields. These as well as other factors conspire to make large-scale screens utilizing iPSC-derived cellular models of disease a very difficult undertaking. It is anticipated that further advances in technologies supporting automatic cell propagation, sorting, and quality control, as well as improvements in differentiation protocols will make the dream of using stem cell models of disease in early stage drug discovery a reality.
In an interesting twist of the process, disease-in-a-dish screens are frequently preceded by screens for small molecules that aid in the reprogramming of an iPSC to the lineage relevant to the particular disease. Identifying small molecules that replace protein-type reprogramming factors carries many benefits, including making the reprogramming protocol cheaper to execute, more reproducible, and scalable, thereby fulfilling the needs for both large-scale disease-in-a-dish screening and potential corrective cell therapy.
Toxicology in the 21st century
Bringing toxicology into the 21st century will require replacing standard animal toxicology studies with new models that more accurately and precisely assess, characterize, and group chemical hazards. The Toxicology in the 21st Century (Tox21) is a unique collaboration among several US Federal Government partners created to develop a vision and devise an implementation strategy to shift the assessment of chemical hazards from traditional experimental animal toxicology studies to target-specific, mechanism-based biological observations largely obtained using in vitro assays [82]. In 2008, in response to the National Academy of Sciences' (NAS) report, Toxicity Testing in the 21st Century: A Vision and a Strategy [83], a collaboration was established between the National Institute of Environmental Health Sciences' (NIEHS) National Toxicology Program (NTP), the US Environmental Protection Agency's (EPA) National Center for Computational Toxicology (NCCT), and the National Human Genome Research Institute's (NHGRI) NIH Chemical Genomics Center (NCGC), presently part of the National Center for Advancing Translational Sciences (NCATS) [84]. In mid-2010, the US Food and Drug Administration (FDA) joined the collaboration, which is now known informally as Tox21. Initial screens within the Tox21 collaboration focused necessarily on engineered cell lines utilizing simple readouts, such as nuclear hormone receptor assays with beta-lactamase and luciferase reporters, as well as assays for various cell stress responses.
Recently, efforts within Tox21 have expanded to include medium-throughput high-content screens and assays using alternate animal models, such as Caenorhabditis elegans, Drosophila melanogaster, and Danio rerio (zebrafish), to complement the quantitative, high-throughput screening assays. Another exciting effort that is underway is the EPA ToxCast project across 331 cell-free enzymatic and ligand-binding, high-throughput screening assays [85]. This effort investigates 976 chemicals that can be classified as failed pharmaceuticals, alternative plasticizers, food additives, and pesticides. This project is amassing copious toxicity data on ∼1,000 chemicals to identify, characterize, and build toxicology pathways that can be used in the future to make predictions about other chemicals based on similar chemistries and also identify the mechanism of action.
To advance our understanding of CNS disorders during development and adulthood, iPSC-derived cells are currently being evaluated by the NTP and its collaborators using an 80-compound library, which comprises drugs and environmental compounds with known (eg, deltamethrin, n-hexane, valproic acid) and unknown (eg, flame retardants, polyaromatic hydrocarbons) neurotoxic potential. Some of the endpoints being assessed include the ability of chemicals to affect neuronal proliferation and differentiation [86], neuronal crest migration [87,88], and neuronal firing [48,51,52,89]. In addition, iPSC-derived differentiated cell populations from patients with neurological disorders such as Parkinson's disease are being used to test for the possibility of differential chemical sensitivity [90,91]
Through the advancement of human-based iPSC models, we are now able to capture, to some extent, genetic variability and susceptibility of response to drugs, pollutants, and other environmental chemicals, which is currently not possible using classical, animal toxicological studies. Importantly, there is the recognition and pressing need for more efficient testing methods that can provide hazard information for the thousands of untested chemicals in commerce with unknown neurotoxic potential while reducing the use of animals in toxicity testing [92,93]. To address global effects of chemicals on other target organs/cell types in addition to the CNS, Tox21 partners are also conducting studies using human iPSC-derived cardiomyocytes, endothelial cells, hematopoietic cells, hepatocytes, and mesenchymal cells.
Efforts are underway to combine the use of iPSC models along with high-throughput screening assays using reporter gene assays [82], 3D organotypic cultures [94], human on a chip (
Personalized medicine
The vision of a personalized approach using a patient's own cells to perform prospective screens is just over the horizon. A current example of a personalized medicine approach is cancer treatment therapy provided in [100]. Cancer biologists have suggested that tumor recurrence likely occurs because a rare tumor repopulating population escapes standard therapy. If these cells are isolated and treated with approved drugs, a tailored optimized cocktail of drugs could be rapidly developed to administer to a patient who has failed therapy. This strategy has shown some success and is a novel approach to screening with immediate benefits.
Adverse event screening during drug trials is also possible. One of the major benefits of conducting this type of screening is the ability to identify harmful, unexpected, and rare complications from a drug that would otherwise go undetected using standard clinical evaluations. Indeed, drugs have been withdrawn, despite their efficacy, in a large population due to risk to a rare population. An iPSC-based approach offers a solution to this problem and has been used in iPSC-derived cardiomyocytes to identify the adverse effects of cardiac glycosides [101]. Patients with adverse events can be identified and their iPSCs generated, and then these iPSCs can be used to extensively map the drug response and compare it with patients who had a beneficial effect. This type of testing may allow industry to salvage drugs as biomarkers and could further be developed to identify patients who should not be administered such a drug.
Likewise, in most clinical trials, some patients respond and others do not. Comparing responders with nonresponders may provide novel insights and assist with stratifying patients into different subgroups for therapy. There is no technical issue in performing such screens, and the size of the screens is not large. A recent study differentiated neurons and astrocytes and neurons from several Alzheimer's disease patients and found that cells from patients were differentially responsive to docosahexaenoic acid [102]. Therefore, it is simply a matter of time and a decision to implement. From here, one can imagine developing panels of cell lines that are genetically diverse and that can be tested routinely with any candidate drug to reduce the risk of unexpected events when a drug is widely used. Such a panel does not exist presently and would require a public–private partnership, but certainly is technically feasible and, in our opinion, its benefits far outweigh the cost associated with such an endeavor. Indeed, efforts along these lines are being initiated, and with careful attention to ensure adequate diversity, we may soon have a panel of iPSC lines for such a purpose.
Concluding Remarks
In summary, iPSC-based approaches to model aspects of CNS disease hold tremendous potential. This review has described classical drug discovery models that have already been used to evaluate CNS diseases with discussions of their limitations, including limited supply or lack of disease specificity. The review has also illustrated how an iPSC-based approach offers unique advantages in CNS disease modeling by providing an unlimited supply of disease- and patient-specific cells that can be differentiated into many different cell types for further interrogation. Additionally, the review has discussed some of the current limitations of this approach and which diseases may prove difficult to model using an iPSC-based approach such as mitochondrial diseases. Finally, the review concluded with incorporating new technologies into current screens to develop advanced iPSC-based screens (Fig. 2) that the authors envision will revitalize the pharmaceutical industry by lowering the attrition rates and accelerating the drug discovery process.
We feel it is important to note that new results acquired from allogeneic drug screens will not entirely supplant the information already gathered from xenogenic screens. Instead, there is a critical need to develop a database that will allow researchers to compare data gathered from xenogenic models with new data generated from iPSC-based models. The ability to quickly and easily compare results will be an essential component to move drug discovery forward in a more efficient way. This strategy also has important implications in better predicting the toxicity of the large numbers of diverse chemicals with poorly characterized exposures and mechanisms of toxicities.
Footnotes
Acknowledgment
The authors would like to thank Dr. William Mundy for reviewing the manuscript.
Author Disclosure Statement
No competing financial interests exist.