
Abstract
Industrial sectors perform toxicological assessments of their potential products to ensure human safety and to fulfill regulatory requirements. These assessments often involve animal testing, but ethical, cost, and time concerns, together with a ban on it in specific sectors, make appropriate in vitro systems indispensable in toxicology. In this study, we summarize the outcome of an EPAA (European Partnership of Alternatives to Animal Testing)-organized workshop on the use of stem cell-derived (SCD) systems in toxicology, with a focus on industrial applications. SCD systems, in particular, induced pluripotent stem cell-derived, provide physiological cell culture systems of easy access and amenable to a variety of assays. They also present the opportunity to apply the vast repository of existing nonclinical data for the understanding of in vitro to in vivo translation. SCD systems from several toxicologically relevant tissues exist; they generally recapitulate many aspects of physiology and respond to toxicological and pharmacological interventions. However, focused research is necessary to accelerate implementation of SCD systems in an industrial setting and subsequent use of such systems by regulatory authorities. Research is required into the phenotypic characterization of the systems, since methods and protocols for generating terminally differentiated SCD cells are still lacking. Organotypical 3D culture systems in bioreactors and microscale tissue engineering technologies should be fostered, as they promote and maintain differentiation and support coculture systems. They need further development and validation for their successful implementation in toxicity testing in industry. Analytical measures also need to be implemented to enable compound exposure and metabolism measurements for in vitro to in vivo extrapolation. The future of SCD toxicological tests will combine advanced cell culture technologies and biokinetic measurements to support regulatory and research applications. However, scientific and technical hurdles must be overcome before SCD in vitro methods undergo appropriate validation and become accepted in the regulatory arena.
Background
O
Animal models are, despite generally accepted limitations in several aspects, still regarded by many as the most relevant physiological systems and this is reflected in the majority of regulatory guidelines for toxicological evaluation worldwide. However, ethical, cost, and time concerns associated with in vivo studies, as well as uncertainty regarding the translation of the results to man highlight the opportunities that physiologically relevant in vitro models will bring to the toxicology field [1,2]. Currently, a major challenge for the identification and characterization of adverse events in target organs is the lack of suitable in vitro systems with adequate cells in a physiologically relevant culture environment [3]. Also, specific, qualitative, and quantitative endpoints (including measures of exposure to the assessed substances), which identify adversity, are needed to enable safety scientists to go beyond hazard identification and move toward risk assessment [4]. Advances in molecular biology and imaging technologies have substantially improved our understanding of the mechanisms of toxicity after exposure of animals to specific substances and, therefore, relevance to human safety. However, the lack of physiologically relevant in vitro systems remains a major obstacle for the successful implementation of in vitro toxicology in some areas of industry. Commonly used in vitro systems such as primary cell cultures and cell lines often fail to adequately reproduce the in vivo situation, as primary cells often lose differentiated functionalities and cell lines are prone to phenotypic and genotypic changes during the transformation process [5]. Stem cells are capable of dividing indefinitely and have the potential to differentiate into any cell type, offering an unlimited and consistent source of cells for in vitro toxicology. The rapid development of reprogramming protocols for the generation of iPSC that enable adult differentiated cells to regain pluripotency has eased the way forward for the application of stem cells in drug development [2,6,7]. This technology not only allows circumventing ethical issues associated with the use of human embryonic stem cells (hESC), but also allows the selection of relevant donors, the minimally invasive collection of material, and the production of pluripotent cells that can differentiate into theoretically any cell type. In addition, the use of SCD 3D organotypic microtissue models for toxicological assessment has given researchers access to an increasing portfolio of cells, which can be used to engineer predictive in vitro models (Table 1), and recapitulate native tissue cell composition and structure. These scientific advances have thus opened the door to expand in vitro research to include in vitro toxicology assessments.
Several challenges inherent to all in vitro systems also apply to SCD systems: mainly the selection and phenotypic characterization of the cells, the cell culture conditions, the dosing regimens, the composition and architecture of the cell culture systems, the duration of the experiments, and the measurement and interpretation of endpoints in the context of potential human risk. In particular, related to SCD cells, the extent of differentiation, the commonly observed fetal properties, and the stability of the differentiated phenotypes add an additional layer of complexity. Also, industrial use of SCD cells will require not only the control of the expansion and differentiation processes, but also the production at an industrial scale and at reasonable costs. Several technical hurdles have been identified when using reprogrammed stem cells (iPSC). The scalable production of iPSC and the maintenance of their pluripotent status have proven time-consuming and often suffer from batch-to-batch variability. This has been partly overcome by using stirred suspension bioreactors that facilitate large-scale expansion studies at lower costs and hydrogel microencapsulation which promotes cell expansion and maintains the pluripotent status [8 –11]. Another specific challenge inherent to transcription factor-based reprogramming is the epigenetic memory of iPSC: their epigenetic landscape is related to the tissue of origin and limits directed differentiation [12]. Thus, developing methods to stably establish embryonic stem cell-like epigenetic circuitry during the reprogramming process will be mandatory. It has been described that the residual DNA methylation observed in low-passage pluripotent iPSC is largely overcome when using higher passage numbers [13] Despite the current technical limitations, the potential of SCD cells (in particular iPSC) and tissues in toxicology is unprecedented and will certainly contribute to in vitro safety assessment in the future.
A gap analysis performed during a scientific workshop hosted by the European Partnership for Alternative Approaches to Animal Testing (EPAA) showed that substantial research needs to be performed to enable the successful implementation of SCD systems in toxicology in industry and academia. The scientific discussions among experts in toxicology, cell biology, stem cell research, pharmacokinetics, 3D culture systems, and industry, focused on the current status and identified specific research areas that should be promoted and financially supported by the European Commission or other funding bodies in the future. The outcome of the scientific workshop is summarized in this article.
Overall Goals and Vision
Despite differences in the specific requirements inherent to each area of industry, there is a shared interest in applying SCD cells for the accurate and early detection of toxicity. The main advantages and long-term goals of the application of SCD systems in toxicity assessment will be the reduction and/or replacement of animal studies, the improved prediction of specific adverse effects for selected human populations, and the reduction of costs and time required for the toxicological assessment of molecules. To optimize the use of SCD systems in toxicology, the robustness and differentiation capacity of stem cells needs to be demonstrated and more complex organotypical cellular models (able to mimic the in vivo situation) need to be developed. Realistically, the implementation of these systems will be gradual, paralleling the scientific advances and the accompanying increased regulatory acceptance. Moreover, the implementation will be adapted to the needs of each industry branch.
In the pharmaceutical industry, toxicity testing is highly regulated to maximally ensure human safety and accurately assess risks associated with new drugs. However, animal studies are poor predictors of the human situation, often resulting in doubtful early (preclinical) or necessary late (clinical) termination of drug development programs [14] (Fig. 1). To avoid costly and dangerous false negative and false positive predictions more accurate extrapolation from toxicity studies to the clinical situation is of utmost importance. Accordingly, more relevant and more predictive test methods with regard to human safety are needed [15]. The successful implementation of SCD systems will support the current pharmaceutical R&D strategy of testing for toxicity endpoints in earlier phases of the drug development process, to select drug candidates with better toxicity profiles before animal studies are performed [16]. In addition, these assays will enhance the extrapolation of toxicological findings to human safety and, therefore, decrease the attrition rate.

From Arrowsmith and Miller, [14].
The safety assessment of agrochemicals is also a rigorously regulated process requiring an evaluation of their intrinsic hazards in animal species, a quantitative assessment of potential exposure in these species (and man) and an overall assessment of risk. In addition to exposure calculations that are often overestimated, safety factors are applied to the lowest no effect levels obtained in relevant toxicology studies to account for the potential sensitivity of the human population. For example, a minimum arbitrary 100-fold safety factor is applied owing to the species differences and genetic homogeneity of the animal models when extrapolating to the human population. One of the major challenges for the invention of new agrochemical molecules is to obtain an earlier and more accurate prediction of both hazard and exposure to increase the likelihood that those molecules will pass the necessary rigorous risk assessments in development. At this point, validated human and animal SCD models may prove beneficial for candidate selection, but may also help develop a better understanding of species sensitivity and human relevance until the regulatory paradigm of animal testing is changed for this industry sector.
The consumer goods and personal care industries, for regulatory and ethical reasons, are no longer able to use animal models to test for toxicological safety of ingredients used in products marketed in Europe (Cosmetics regulation 1223/2009), and other countries such as India are starting to implement similar restrictions. Due to the restriction on performing toxicology studies on animals, this industry sector will need to rely solely on data from in vitro and in silico studies to identify human-relevant hazards. Several endpoints, particularly organ specific toxicity, either do not currently have an in vitro alternative or are based on rodent rather than human cell systems. This makes evaluating human safety a particular challenge, approached in two different ways: (1) Direct replacement of the in vivo endpoint with an in vitro alternative for example, 3D cultured skin micronucleus to replace in vivo bone marrow micronucleus test for genotoxicity. (2) Departure from attempting to predict rodent outcome to a focus on toxicity pathways in human relevant cell models.
Direct replacement of the same endpoint in an in vitro model has been the traditional approach used to define alternatives to animal tests, but the same problems of using rodent models to assess risk to human health still remain. Using perturbations in toxicity pathways potentially allows quantitative risk assessments to be performed using human relevant response pathways with a detailed mechanistic understanding. The problems regarding extrapolation of in vitro data to whole organism are inherent to both approaches; however SCD models may allow more realistic predictions of toxicity and thus facilitate more accurate and pragmatic decisions for protecting consumer and occupational health.
Applying a New Paradigm
A paradigm change in toxicology is needed to better address the prediction of human-relevant effects necessary for the safety assessment performed in several industrial sectors and submitted to regulators, moving away from animal studies. In vitro approaches currently used often fail to recapitulate the mechanisms of toxicology, and cannot capture the influence of underlying genotypes and phenotypes or determine the threshold between adaptive and adverse responses [17]. The problem becomes more convoluted when the toxicological effects of long-term (chronic repeat dose) exposures need to be determined and when several cell types or organ systems are involved. The use of SCD systems taking into account genetic background, dose, exposure, multicellular and multiorgan effects, together with high-content and high-throughput technologies, will enable such a paradigm change by addressing the following points: (1) Human iPSCs will provide relevant model systems which are easily accessible and will enable the generation of a variety of terminally differentiated cells. It should be possible to generate dozens of different tissue-like organ structures from the human body and combine them in multiorgan systems. In this study, specific pathological changes affecting organ-specific functions will be detectable, thus allowing human toxicity assessment with an understanding of the underlying cellular and molecular mechanisms involved. (2) SCD systems will also greatly support personalized healthcare and the risk assessment for subpopulations by utilizing SCD cells derived from specific human populations enriched for selected characteristics, such as genotypes, ethnicity, health status, and age. (3) Human iPSC are expected also to provide a robust and reproducible system which can be controlled and manipulated using molecular biology tools (eg, by genetic manipulation or by using reporter gene constructs) and physiological stimuli. This will enable detailed mode of action investigations under appropriate study design conditions with information-rich endpoints (eg, omics and imaging technologies). (4) Due to the highly proliferative state, iPSC can provide a nonending amount of cells and tissue-like organ structures for automated testing systems and potential high-throughput testing. This would greatly support robust advances in predictive computational and informatic approaches.
Overall the relevance of SCD systems for toxicity assessment in several areas of industry is apparent (Fig. 2). Therefore, in addition to the active academic research, many companies see promise in this area. However, considering the state of the science described in the next sections, more work needs to be done for the successful implementation of SCD systems in toxicology.
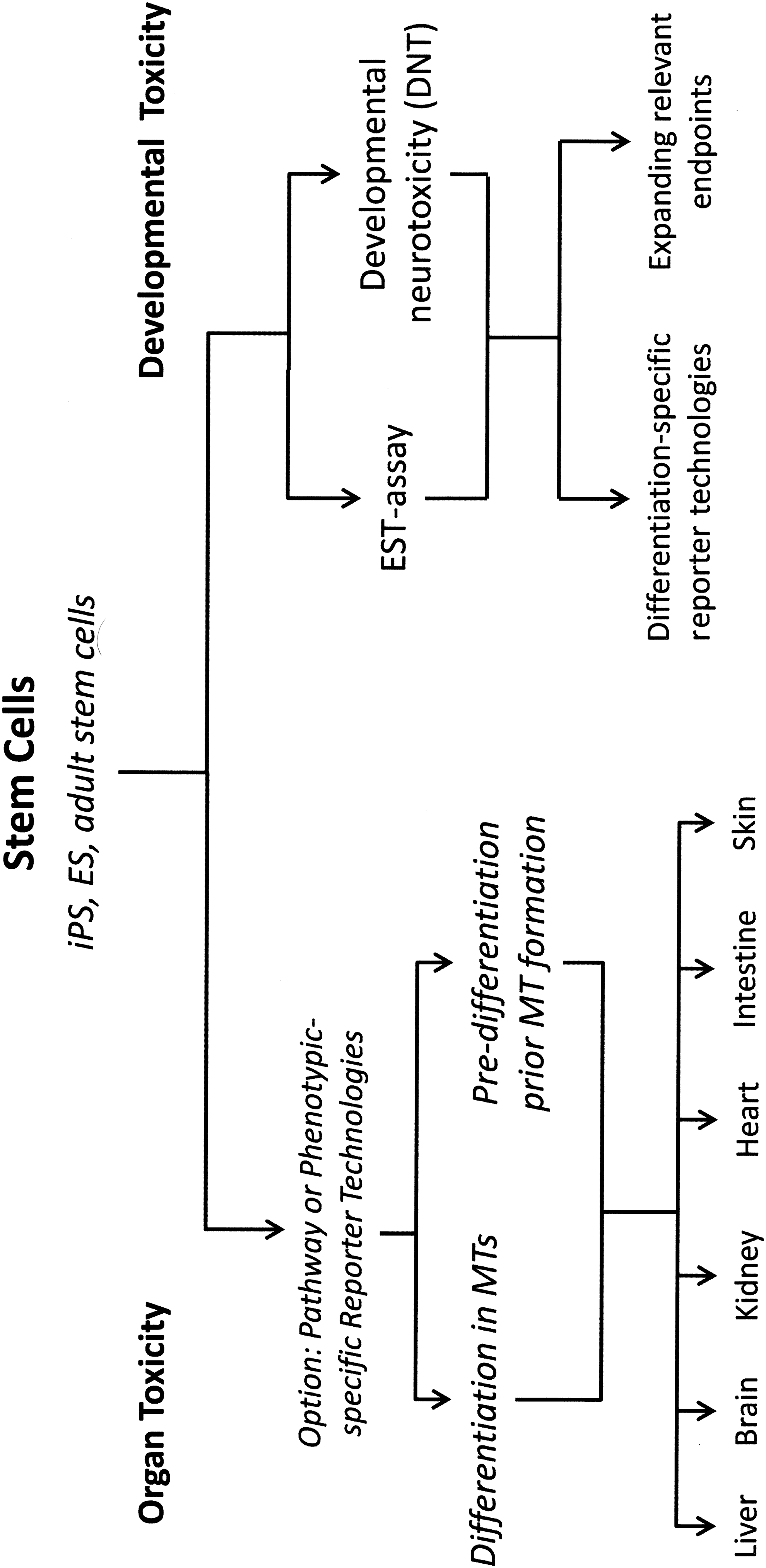
Potential application of embryonic and adult stem cell in the design of toxicologically relevant organotypic microtissue (MT) models.
Current Status and Future Needs of SCD Cellular-Based Systems
SCD cardiomyocytes
SCD cardiomyocytes can mimic inherited diseases, the effects of pharmacological treatment of the heart as well as drug-induced cardiotoxicity despite their immature phenotype [18 –20]. The main differences between immature and adult cardiomyocytes include differences in morphology, the use of metabolic substrates, and resting membrane potential and upstroke velocity [20]. Also, the SCD cardiomyocytes do not form organized T-tubules, a network of intracellular structures involved in cardiomyocyte action potential propagation and calcium influx [21]. Furthermore, the cultures obtained are usually quite heterogeneous with respect to composition of atrial, nodal, and ventricular cardiomyocytes [22]. Attempts to increase the adult phenotype of the cardiomyocytes include prolonged cultivation for more than 120 days [23]. However, because of the low throughput using such protocols many laboratories are developing novel systems, including change of substrate stiffness, cultivation in the presence of different laminins, electric and hormonal stimulation of the cells, and cocultures with for example, smooth muscle cells, fibroblasts, and endothelial cells [20,24]. Other cell types, like subendocardial mesenchymal stem cells that express immunomodulatory molecules may also improve the differentiation of the cardiomyocytes [24].
Despite their shortcomings, SCD cardiomyocytes have proven useful for the exploration of mechanisms of cardiovascular diseases as they can recapitulate disease phenotypes and mimic clinical pharmacological interventions [25]. For example, cardiomyocytes displaying long QT syndrome upon exposure to isoproterenol can be treated with propranolol, which abrogates the increase in the duration of the action potential [22]. Also, mutation in KCNH2 can be recreated in vitro and treatment of iPSC-derived cardiomyocytes with pinacidil or nifedipin can successfully restore the proper action potential [26]. The prolonged action potential in patients with a point mutation in Cav1.2 can similarly be recapitulated and treatment with roscovitine corrects the anomalous calcium transients associated with this mutation [27]. Furthermore, iPSC-derived cardiomyocytes from patients with long QT syndrome (LQT) or familial hypertrophic cardiomyopathy (HCM) exhibit a high incidence of arrhythmia including early after depolarization in LQT cells and delayed after polarization in HCM cells [28]. These examples highlight that SCD cardiomyocytes can effectively recapitulate the disease phenotype associated with channelopathies and mimic many of the drug-mediated restorations as utilized in vivo.
In cardiotoxicity screening, iPSC-derived cardiomyocytes appear more suitable than standard recombinant systems to elicit a correct prediction of verapamil and alfuzosin effects on the hERG channel [28,29]. In addition, cardiomyocytes derived from LQT and HCM patients exhibit increased susceptibility to pharmacological blockade of hERG than cardiomyocytes obtained from control individuals [28]. However, for toxicity screening purposes the systems are not completely developed and more work on characterization, thorough validation, and in vitro–in vivo comparison is required before these systems can constitute an established alternative to the current cardiotoxicity testing [30,31].
SCD hepatocytes
Similarly to the situation described for SCD cardiomyocytes, hepatocytes derived from stem cells are generally fetal in their phenotype. One major additional problem inherent to hepatocytes in culture is that they rapidly dedifferentiate losing their ability to perform basic hepatic functions [32,33]. The key differences between SCD hepatocytes and adult human hepatocytes have hitherto limited the use of the latter for in vitro applications.
Several protocols have been used for the differentiation from stem cells into hepatocyte-like cells. A key component is a first step of differentiation of stem cells into definitive endoderm using Activin A [34], although Activin A-independent protocols have been described for the differentiation of Wharton's jelly mesenchymal stem cells into hepatocyte-like cells [35]. Further improvements include the combination of specific chemical factors with 3D culture systems, either in hollow fiber bioreactors [36] or in spheroids [37]. These protocols lead to enhanced expression of key genes involved in albumin production and drug metabolism. A noteworthy approach involves cocultures of iPSC-derived hepatocyte-like cells with endothelial cells and mesenchymal stem cells on a presolidified matrix forming 3D spheroids (liver buds). These liver buds are then transplanted into immunodeficient mice where highly vascularized human liver tissue displaying many hepatic functions develops, although bile ducts are lacking [38,39]. Another interesting approach is the use of small molecules to enhance the differentiation of stem cells to hepatocytes that express specific markers as discussed by Shan et al. [40]. It will be interesting to see if this or similar protocols can be successfully reproduced in other laboratories and developed further.
Regarding suitability of SCD hepatocytes for toxicity assays, experiments have been performed using known hepatotoxins and high-content imaging-based determination of toxicity [41] or ATP-based toxicity assays [42,43]. The results show that in some cases the sensitivity for drug toxicity in these SCD hepatocytes is similar to that observed in hepatocytes cultivated for 48 h, but does not necessarily reflect drug-induced liver injury (DILI) in man. Arguably, DILI can only be reproduced in cell systems amenable to chronic drug toxicity testing, as the onset of injury often requires 4–12 weeks exposure. Currently, SCD hepatocytes are stable only for a maximum of 2 weeks, hampering the use of such cells for chronic drug exposures. However, using a 3D collagen matrix culture (3D clump cultures) relevant expression of CYP3A4 could be maintained for 75 days [44]. In addition, the introduction of immune cells and nonparenchymal cells together with the SCD hepatocytes into 3D in vitro systems, although challenging, would allow mimicking idiosyncratic reactions that often depend on specific HLA class I or II antigens. In particular, for the study of rare hepatotoxicity it would be meaningful to use cells derived from patients susceptible to drug-induced liver toxicity as well as from controls.
In conclusion, the technology as it stands leads to the production of SCD hepatocyte-like cells that have a fetal phenotype, are only partly differentiated, and thus cannot yet replace human primary hepatocytes for screening of drug-induced hepatotoxicity. Some approaches mentioned above, involving complex culture systems or in vivo transplantation lead to promising liver functionality, but are complex and very tedious. Further development and simplification of such protocols and additional knowledge about the key components necessary for the maintenance of hepatocyte phenotype in vitro would be necessary before iPSC-derived hepatocytes become a competitive alternative to primary human hepatocytes.
SCD renal cells
In the kidney, most toxicities are recorded in the proximal tubule (PT) and in the glomerulus, therefore, current in vitro methods to study the nephrotoxicity are primarily focused to PT cells and podocytes. While animal and human primary cell culture is possible for these regions, most studies rely on permanent cell lines such as the PT cell lines ciPTECs, HK-2, and RPTEC/TERT1 that have been extensively characterized [45 –48]. Human podocyte cell lines have also been developed by transfection of primary cells with the SV40 large T antigen (SV40TAg) or the temperature-sensitive variant (SV40tsA58) alone or in combination with hTERT [49 –51]. Some of these cell lines may be useful for benchmarking SCD lineages.
A number of studies have demonstrated the potential to differentiate hESC into renal progenitor-like cell lineages [52 –54]. Activin A and retinoic acid (RA) seem to be crucial drivers of differentiation as in other tissues [53]. In addition, bone morphogenic factor 7 (BMP7), which is critical for renal development in vivo [55], is also an important factor for renal differentiation of stem cells [53]. Using these factors, Takasato et al. could capture the major steps in renal development, namely development of the primitive streak, the intermediate mesoderm, and the metanephric mesenchyme leading to cell expressing specific proximal and distal tubule markers and suggesting a coordinated nephron development [56]. An alternative protocol utilizing the GSK3α/β inhibitor CHIR-99021, followed by FGF9 and removal of growth factors gave similar results [56,57]. A more direct route to produce proximal tubular cells from hESC has been demonstrated by stimulating differentiation with BMP2, BMP7, activin A, and RA [58]. The authors report a yield of close to 40% AQP1 (aquaporin 1)-positive cells. In addition, the differentiated cells were shown to express cadherin-16, glutamyl transferase (GGT), aminopeptidase N (CD13), and several proximal tubular specific transporters. Functional characterization was carried out by measuring water transport, ammonia production, and response to parathyroid hormone [58]. While there is, as of yet, no reported differentiation protocol for proximal tubular cells, it is expected that iPSC-derived PT would differentiate similarly to ESC-derived lineages, although a recent report indicates a higher efficiency of differentiation of ESC than of iPSC for the differentiation into intermediate mesoderm and nephron progenitor cells [59]. Differentiation of iPSCs into podocyte-like cells has been achieved using a combination of activin A, BMP7, RA, and beta mercaptoethanol [60].
In summary, there is clear evidence from several independent groups that derivation of PT and podocyte phenotypes is possible using temporal application of specific growth factor cocktails, even though these methods are less well established than differentiation of hepatocytes or cardiomyocytes from stem cells. The further development and optimization of differentiation strategies for these and other renal lineages will, however, be required for the successful application of these cells to toxicity testing.
SCD skin models
For personal care/consumer goods products, where the skin is often the primary organ of initial exposure, toxicological evaluation of effects in the skin is important for a number of endpoints. Currently available reconstructed tissue models containing a keratinocyte and an epithelial cell layer are commonly utilized. These models were primarily developed for the treatment of burnt skin and chronic wounds, but also used for toxicity testing and more recently for research on skin diseases [61]. However these primary skin models are often sourced from multiple donors and, therefore, suffer from large interindividual variability and a limited life span. Also, skin models lack many of the critical structural features that are important for normal skin function, as they focus on two main cell types: keratinocytes and epithelial cells. The skin itself is a complex organ containing hair follicles, sweat glands, and pigment-generating cells and studies have shown that much of the structure of the skin that occurs during fetal development is under control of several of the developmental genes such as wnt, shh, and bmp [62].
Recent research has led to the development of in vitro models with pigmented skin [63] and vascularized skin [64]. Petrova et al. report a process whereby reprogrammed iPCS epidermal cells are cultured in an air–liquid interface driving the formation of keratinocytes that maintain an epidermal skin barrier that is very close to normal human skin [65]. Guo et al. take this a step further and report plans to generate a basic skin model comprised of epidermal cells and keratinocytes, gradually increasing the complexity of the model, incorporating hair follicles, sweat glands, and a functional immune response system to resemble the complexity of normal skin [66].
Functional SCD-derived human skin models would allow the dermal route of exposure to be considered in greater detail than is currently possible without the use of laboratory animals. In addition to exposure assessments, SCD skin models could serve for the in vitro evaluation of dermal toxicity. However, further evaluation will be necessary to assess their performance in comparison to currently available skin models generated using differentiated skin cells.
SCD systems for the study of toxicity mechanisms
For any future SCD system to be integrated in toxicity testing it will be essential that it captures critical (organ-specific) molecular mechanisms. This would include the analysis of breaking points of toxicity, that is, at what exposure levels adaptive responses are no longer sufficient to cope with the new environmental conditions, resulting in cell death. In vivo and in vitro transcriptomics analyses largely focused on liver, heart, kidney, as well as skin have revealed many of the cellular stress response pathways that are associated with and precede the onset of adverse outcome in different target organs [67]. Candidate cellular stress response pathways involve the oxidative stress-related KEAP1/Nrf2 signaling, NF-kB signaling, the endoplasmic reticulum stress or unfolded protein response, as well as the related heat shock response, osmotic stress signaling, hypoxic stress signaling through HIF1alpha signaling, and DNA damage responses [68,69]. For these canonical pathways, reporter assays have been established that allow the evaluation of cellular stress responses [70]. A similar approach could be integrated in SCD systems, to allow the investigation of the activation of cellular stress response pathways. A prerequisite for the establishment of such reporter assays would be a careful comparison of the functionality of the cellular stress response pathways in primary cells and SCD systems, including a detailed assessment of the relationship between stress pathway activation and onset of adversity.
General cellular stress response pathways do not provide information on organ-specific toxicities. Hence, additional organ-specific reporter systems are needed. For SCD liver systems, the molecular networks that are involved in liver-specific toxicities should be present, preferably to a similar extent as in the human liver. This includes the phase I, II, and III biotransformation enzymes that are critical in the determination of liver toxicity as well as nuclear hormone receptors that are essential in liver toxicity, including PPARs, PXR, FXR, CAR, and AhR [69]. Evaluation of the gene regulatory networks by these nuclear hormone receptors should also be similar as in primary human hepatocytes. Since liver toxicity in vivo depends on multiple cell types, including Kupffer cells and stellate cells [71], the integration of these cells with liver hepatocytes and the derivation of these cells from SC will also be essential in the context of the mechanisms of hepatotoxicity.
As indicated before, the PT of the kidney is a major target of drug-related adverse events. Careful analysis of the expression and function of uptake and efflux transporter family members in SCD renal epithelial cells will, therefore, be critical. A similar essential determinant of renal toxicity in vivo involves the dependency of the proximal tubular cells on oxidative phosphorylation. Therefore, SCD renal cells should be characterized with respect to the composition and function of the mitochondria as well as the ability to mediate gluconeogenesis. Moreover, renal toxicity is not limited to PT; often the glomerulus is affected and can also involve the influx and activation of immune cells, only an integrated SCD multicellular renal culture system can reflect this.
Regarding the mechanisms involved in cardiac toxicity, SCD cardiomyocytes from healthy and diseased/susceptible patients can be used for the study of hERG channel blocking. SCD cardiomyocytes obtained from patients with mutation in relevant ion channels have now also been established [72]. In addition, targeted CRISPR/Cas9 or TALEN-based genetic modification of healthy cells as well as the rescue of mutant cells is an additional powerful tool to assess the contribution of genetic traits in the individual susceptibility to chemical-induced cardiomyopathies. Besides hERG channel inhibition, direct cytotoxic events can also occur in cardiomyocytes, which include classical biochemical perturbations, including mitochondrial injury and oxidative stress [73]. Such cellular perturbations impact the normal beating function of the cardiomyocyte. Anticancer drugs are likely to modulate various complex kinase-dependent survival pathways [74], which may also interfere with cardiomyocyte function. To assess the adverse effects of such novel compounds on cardiomyocytes, a careful evaluation of SCD cardiomyocytes with regard to the expression and activity of various critical kinases in comparison to the primary cardiomyocytes will be important.
Biokinetic considerations for SCD systems
As it was amply delineated in the previous paragraphs, we have now the ability of obtaining SCD cells that can be maintained over prolonged periods of time in culture and used for the performance of repeated dose toxicity studies in vitro, one of the more challenging areas when devising nonanimal test methods. From in vivo and also in vitro data it is clear that the toxicity profile of compounds may differ between single and repeated dosing for a number of reasons. One reason can be the accumulation of the compound over time at the site of the toxicological target, that is, in or at the surface of cells. A second reason can be the fact that repeated dosing may lead to a depletion of the cellular defense mechanisms against the toxic effects. Both situations would lead to an increased sensitivity toward the toxic insult over time. Yet another reason might be the ability of the biological system to adapt by upregulating cellular protection mechanisms, which would decrease the sensitivity to the toxic agent over time. These factors play a role in the intact organism, and likewise in in vitro toxicity test systems, provided that the culture conditions reflect the in vivo exposure regarding time and dose/concentration. Thus, also in in vitro systems attention needs to be paid to the biokinetics and the freely available concentration of the compound in the culture medium are better determinants of toxicity than the nominal concentration [75,76]. Data on in vitro exposure measurement can help overcome challenges associated with compounds of differing physicochemical properties, influencing the extent of adsorption to tissue culture materials such as extracellular matrices and scaffolds. This becomes more difficult for complex, 3D in vitro systems since due to their inherent heterogeneity, cells in different parts of the microtissues may be exposed to different amounts of compound.
Using the outcome of in vitro-derived toxicity data for the purpose of safety or risk assessment, for the human population will imply the quantitative extrapolation of the concentration-effect relationship in vitro toward a dose-effect relation in vivo [77]. Such Quantitative In Vitro–In Vivo Extrapolations (QIVIVE) make use of physiologically based biokinetic modeling in the process of reverse dosimetry [78]. A number of examples of such exercises have been published, the first one being the evaluation of acrylamide toxicity [79,80]. One of these studies used the embryonic stem cell cardiomyocyte system for the evaluation of the embryotoxicity of glycol ethers and their metabolites [81]. An example of the incorporation of in vitro-derived toxicity data with computer-aided biokinetic models was the exposure of kidney cell cultures to cyclosporine A for a period of 14 consecutive days [82]. A more comprehensive overview of the implementation of cell culture-derived toxicity data in a risk assessment is detailed by Blaauboer et al. [83]. A prerequisite for the establishment of such extrapolations is the accurate measurement of in vitro compound exposures over time, and analytical capabilities need to be optimized to address exposures in complex, organotypical coculture systems.
Advantages of 3D systems in SCD cells for toxicology
Stem cell proliferation, cell fate determination and maturation of differentiated phenotypes are highly dependent on extracellular cues, including extracellular matrix and soluble factors, as well as biophysical and physicochemical factors [84,85]. Several groups have established that 3D cultures are more suitable for long-term cell culture as well as for differentiation of iPSC into populations of interest and multiple engineering approaches have been developed to recapitulate those chemical and physical cues in multicellular 3D configurations [86,87]. There are a large variety of 3D systems displaying different degrees of complexity: bioreactors, lab chips, and microtissues are among the most commonly used systems.
Bioreactors are designed to provide efficient mass transfer in controlled systems with online monitoring and automated control of culture variables. At present, there is a myriad of bioreactor types available for iPSC bioprocessing, including an array of custom-made designs; the most widely applied being microfluidic devices, rotary cell culture systems, and stirred culture vessels [84]. Bioreactor design and development focuses on the improvement of accurate control of the cellular microenvironment with reduced shear and working volumes and increased parallelization. In addition, the mode and frequency of media replenishment can be monitored and adjusted in real-time. Due to this tight control, bioreactors can contribute greatly to the development of automated, standardized, and reliable processes for SCD long-term toxicology testing systems.
Liver has been one of the target organs that has benefitted the most from engineering efforts from multiple communities (tissue engineering, cell therapy, drug efficacy, and safety), with an array of bioreactors being applied to the generation of microphysiological models of the human liver [88]. The modular extracorporeal liver support (MELS-BAL) perfusion bioreactor based on hollow fiber perfusion technology with internal oxygenation, initially developed by Gerlach et al. [89] has been recently miniaturized [90] and validated for hepatic differentiation and maturation of hESC [36]. However, this bioreactor does not allow withdrawal of cells during the culture time and hollow fiber systems may present limitations in terms of accurate control of pH and dissolved oxygen as well as adsorption of compounds to the scaffold. Other scaled down perfusion hepatocyte bioreactors have been developed based on micropatterning and microfluidic techniques [91], which impose a physiological oxygen gradient and recapitulate liver zonation, incorporating homo- and heterotypic cell–cell interactions that can be maintained for up to 2 weeks [92]. Also, Griffith and coworkers developed a multiwell microscale device that can be multiplexed and maintain the viability and phenotype of rat hepatocytes and liver nonparenchymal cells in 3D cocultures for a week [93]. Stirred-tank bioreactors in perfusion operation mode have also been applied to 3D cultures of human hepatocyte spheroids (of 80 μm in diameter) in physiologically relevant oxygen concentrations for 3–4 weeks [94]. Spheroids in this system display a functional phenotype displaying bile canalicular networks, phase I and II enzyme activities, and inducibility of CYP P450s. The main advantages of stirred-tank bioreactors are the tight control and homogeneity of physicochemical conditions, the scalability of the system, the maintenance over extended culture periods and the sterile sampling along culture time [84,95]. However, such bioreactors need to be further miniaturized to be amenable to high throughput experiments.
Bioreactor technologies seem to be an ideal environment to promote and maintain differentiation of SCD cellular systems, although their use for directed differentiation of iPSC into hepatocyte-like cells has been hindered by the limited efficiency of available hepatic differentiation protocols [96]. Recent improvements in this field have been rapidly followed by reports on directed differentiation of hPSC in bioreactors [36,97]. It is expected that further integration of these approaches with the previously developed bioreactor-based strategies for primary cultures of functional hepatocytes will facilitate and accelerate the implementation of iPSC-derived hepatic models for toxicology testing in the near future.
Microtissues are 3D structures that can be maintained based on passive diffusion and without the need of complicated and cumbersome pumping devices. In contrast to bioreactor technologies, microscale organotypic microtissue and organoid models are designed to generate a high number of data points [98,99]. They retain a high capacity of cellular self-organization [100], require only small numbers of cells (approximately 500–5000 per microtissue leading to sizes of 150–400 μm in diameter), and are, therefore, perfectly suited for use in automation-compatible multiwell formats. Thus far, the favored cell source to generate organotypic microtissue models are still terminally differentiated primary cells but terminally differentiated SCD cells are currently explored as alternatives, since they should allow the design of multicell-type organotypic testing models. The 3D microenvironment can be an important determinant in the differentiation process and/or the maintenance of differentiated state. Also, microscale tissue engineering technologies enable rational testing of SCD models, as only small cell numbers are required.
Potential Impact and Implementation
To expand the use of stem cells beyond screening and into a regulatory environment, robust and reliable methods need to become available alongside data demonstrating the relevance of such test systems for the toxicological testing of the future [101]. Standardization of SCD used for toxicological studies will require attention to specific characteristics of SCD systems, as well as to the fundamental issues which apply to all cell cultures. The characterization of SCD systems should reduce uncertainties in their use in future regulatory applications by encouraging greater international harmonization, rationalization, and standardization of stem cell work. Appropriate handling practices, quality controls, and compliance with safety procedures and laws, regulations, and ethical principles should be clearly defined. The good characterization of SCD systems based on scientific evidence and adequate performance standards as well as the implementation of Good Cell Culture Practices [102] are necessary. When applied to in vitro toxicology, a detailed description of the SCD test system is essential and should include all relevant information to be able to monitor any changes that might occur during toxicological studies. Information on SCD assay systems should include: (1) how the stem cell was obtained (origin, collecting procedures, processing), (2) the media and growth conditions, (3) the storage conditions, (4) the recovery procedure, (5) the authenticity check, (6) the evaluation of metabolic competence, if applicable, (7) a description of its morphological appearance, (8) the viability acceptance criteria, (9) the standard growth rate, (10) the passage number window of reliable use or the time in culture maintaining its specific performance and/or functionality, (11) a description of the differentiation characteristics, (12) the definition of performance controls of the stem cell test system specific to the application, and (13) specific controls for contamination and cross-contamination. Also, SCD cells should be evaluated at periodic intervals for the expression of critical functional markers during passage to define the timeframe for which they remain usable (eg, stability of the SCD system) for specific toxicological applications.
Concluding Remarks
The outcome of the scientific discussions affirmed the value of SCD systems in toxicology assessment and promoting this key technology, to enable its swift implementation. To this end, specific SCD models need to be defined by assessing their phenotypic characteristics, their stability over time, and their transferability (laboratory-to-laboratory variability). Moreover, additional research should focus on the development of organotypic, multicellular, hence physiologically relevant models, including biokinetic aspects to investigate the relationships between substance exposure, free local concentrations, and response. Taking into consideration the most problematic areas for both predictive models and regulatory needs, priority should be given to liver, kidney, and heart as well as skin as major target organs for toxicity. The future success in this area will require the coordinated and multidisciplinary efforts of scientists from different disciplines, including developmental biologists, chemists, toxicologists, cell biologists, engineers, experts of xenobiotic metabolism and kinetics, and mathematical modelers.
Footnotes
Acknowledgments
The authors would like to acknowledge the support from the EPAA and the significant contributions made by the additional participants of the Workshop held in August 2013: Jonathan Crozier, Reyk Horland, Karl-Heinz Krause, Gaby Küsters, Robert Passier, Koichi Saito, Stefan Schildknecht, Helga Stopper, Lars Sundstrom, Paul Walker, Renate Weissenshorn, and Lorraine Young.
Author's Disclosure Statement
The opinions expressed in this article are based on the own scientific views of the authors and do not necessarily reflect the official positions of their corporate or academic institutions. Laura Suter-Dick acted as a scientific consultant to EPAA on behalf of the University of Applied Sciences Northwestern Switzerland in accordance with its research policy.